
The Role of Apoptosis and Autophagy in the Hypothalamic-Pituitary-Adrenal (HPA) Axis after Traumatic Brain Injury (TBI)
 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
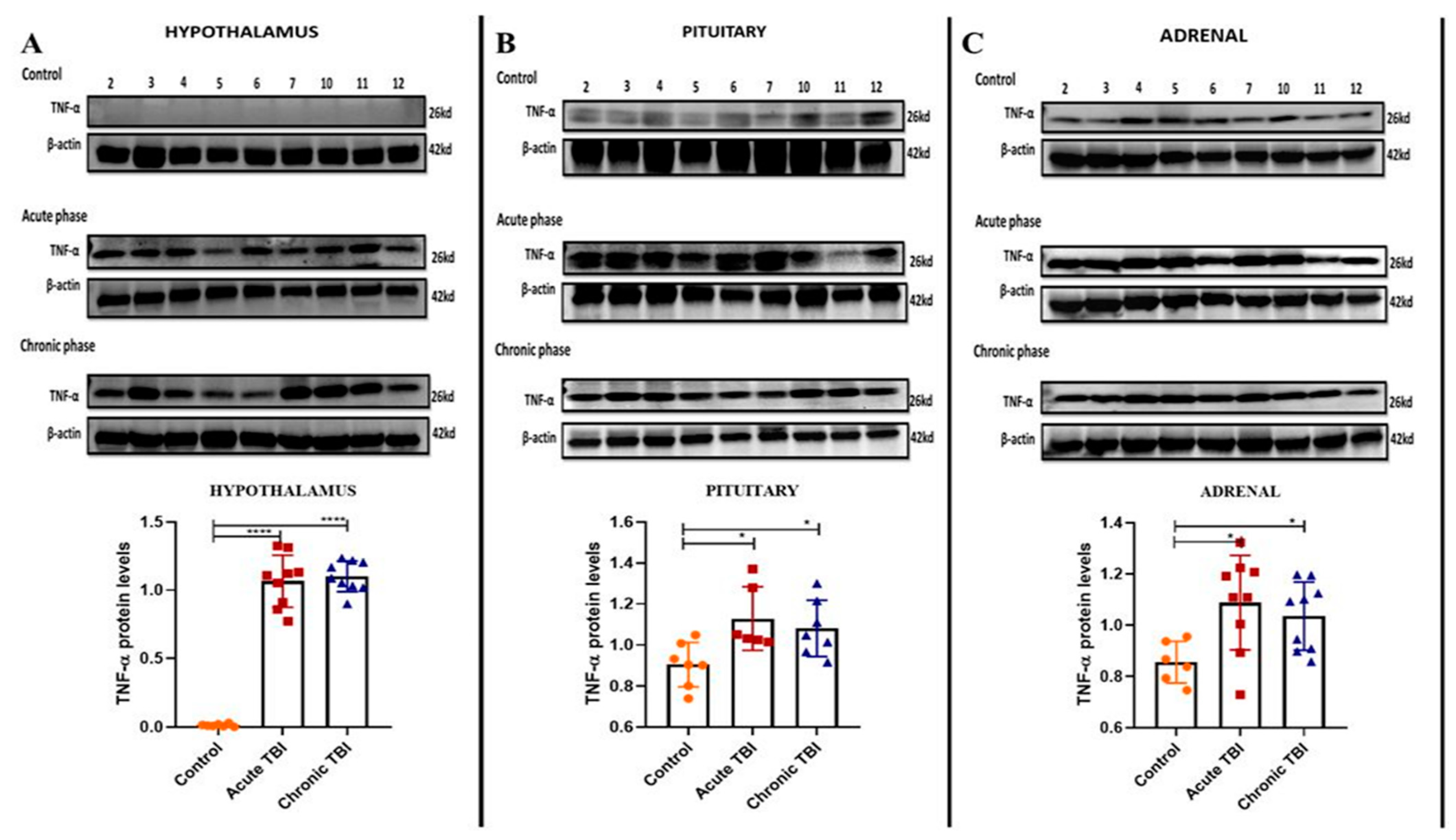
2.1. mTBI-Induced Stress Increases TNF-α Protein Levels in the Adrenals
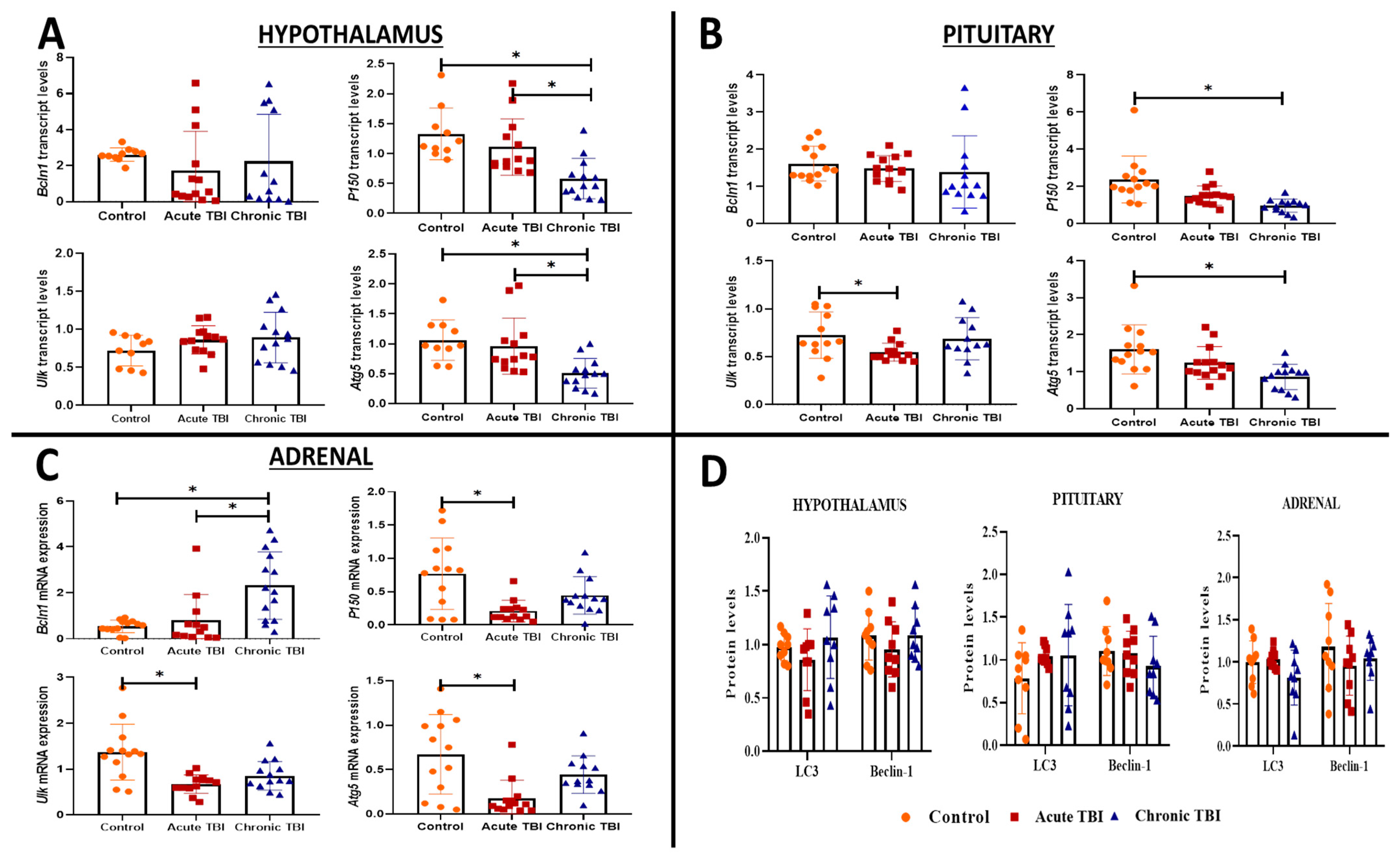
2.2. mTBI Induces Alterations in the Transcript Levels of Autophagy Markers (Lc3, Bcln1, P150, Ulk, and Atg5) Distinctly in the Hypothalamus, Pituitary and Adrenals from the Acute Phase to the Chronic Phase
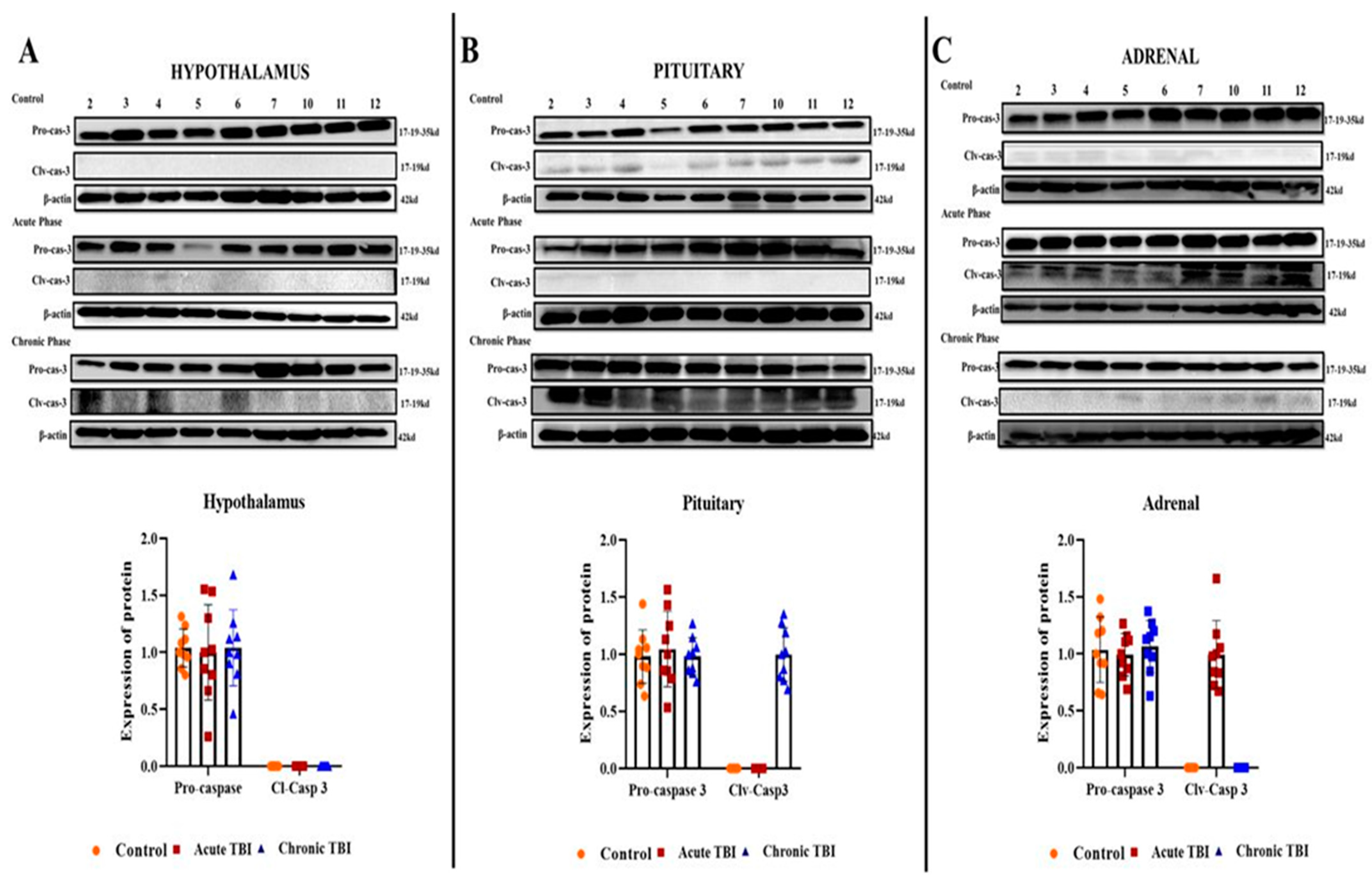
2.3. mTBI Increases Apoptosis First in the Acute Phase in the Adrenals and then in the Chronic Phase in the Pituitary
3. Discussion
4. Materials and Methods
4.1. Animals
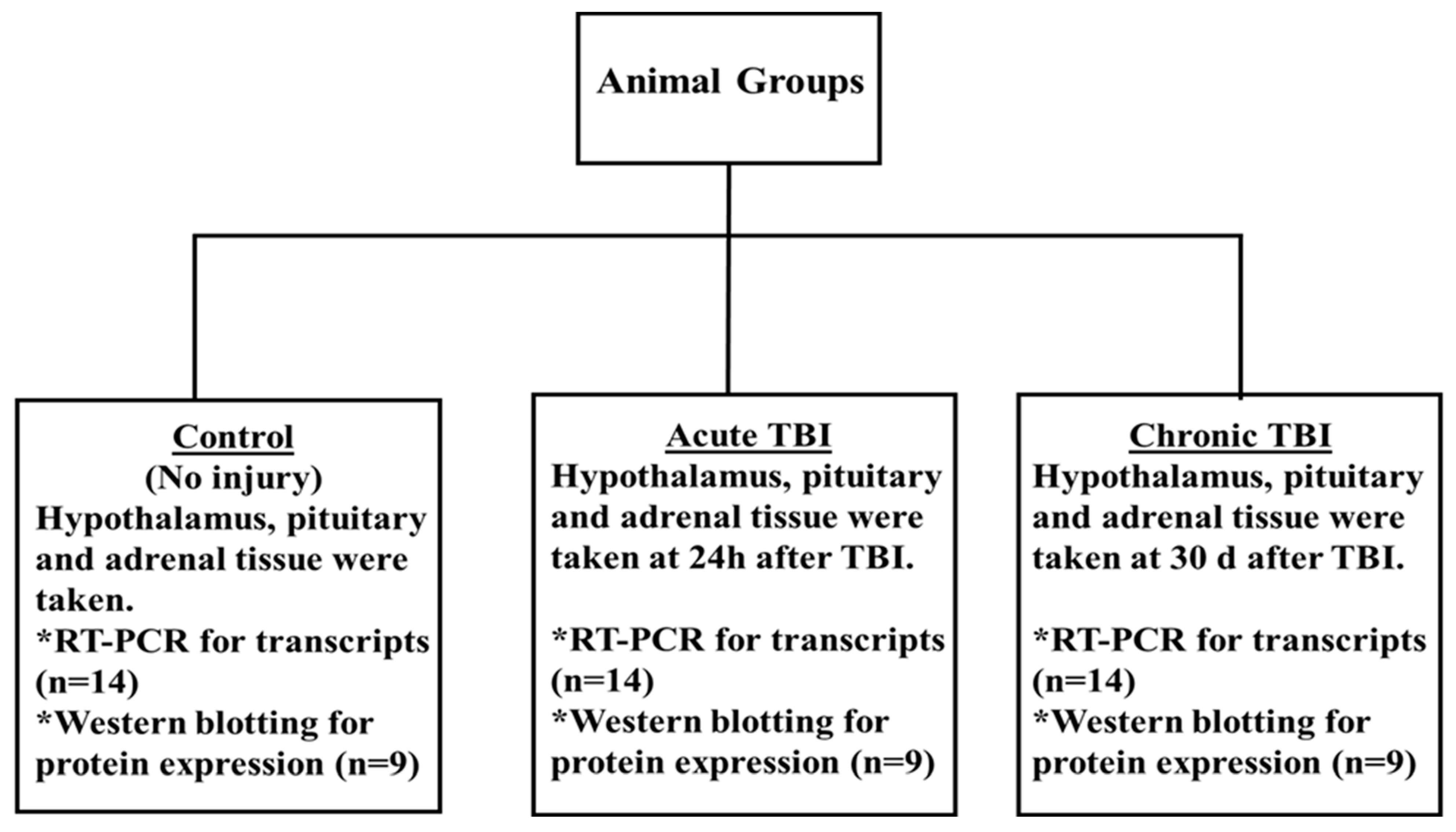
4.2. Experimental Design
4.3. Models of TBI
4.4. RNA Isolation and Real-Time PCR
4.5. Western Blot Analysis
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Özben, T. Pathophysiology of Cerebral Ischemia. In Free Radicals, Oxidative Stress, and Antioxidants; Springer: Boston, MA, USA, 1998; pp. 163–187. [Google Scholar]
- Mckee, A.C.; Daneshvar, D.H. The neuropathology of traumatic brain injury. Handb. Clin. Neurol. 2015, 127, 45–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiebert, J.B.; Shen, Q.; Thimmesch, A.R.; Pierce, J.D. Traumatic Brain Injury and Mitochondrial Dysfunction. Am. J. Med. Sci. 2015, 350, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.W.; McGeachy, M.; Bayir, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. Neuroinflammation in the Evolution of Secondary Injury, Repair, and Chronic Neurodegeneration after Traumatic Brain Injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef] [Green Version]
- Tanriverdi, F.; Taheri, S.; Ulutabanca, H.; Caglayan, A.O.; Ozkul, Y.; Dundar, M.; Selcuklu, A.; Unluhizarci, K.; Casanueva, F.F.; Kelestimur, F. Apolipoprotein E3/E3 genotype decreases the risk of pituitary dysfunction after traumatic brain injury due to various causes: Preliminary data. J. Neurotrauma 2008, 25, 1071–1077. [Google Scholar] [CrossRef]
- Karaca, Z.; Grossman, A.; Kelestimur, F. Investigation of the Hypothalamo-pituitary-adrenal (HPA) axis: A contemporary synthesis. Rev. Endocr. Metab. Disord. 2021, 22, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Tanriverdi, F.; Schneider, H.J.; Aimaretti, G.; Masel, B.E.; Casanueva, F.F.; Kelestimur, F. Pituitary dysfunction after traumatic brain injury: A clinical and pathophysiological approach. Endocr. Rev. 2015, 36, 305–342. [Google Scholar] [CrossRef] [Green Version]
- Bromberg, C.E.; Condon, A.M.; Ridgway, S.W.; Krishna, G.; Garcia-Filion, P.C.; Adelson, P.D.; Rowe, R.K.; Thomas, T.C. Sex-Dependent Pathology in the HPA Axis at a Sub-acute Period After Experimental Traumatic Brain Injury. Front. Neurol. 2020, 11, 946. [Google Scholar] [CrossRef]
- Russell, A.L.; Richardson, M.R.; Bauman, B.M.; Hernandez, I.M.; Saperstein, S.; Handa, R.J.; Wu, T.J. Differential Responses of the HPA Axis to Mild Blast Traumatic Brain Injury in Male and Female Mice. Endocrinology 2018, 159, 2363–2375. [Google Scholar] [CrossRef] [Green Version]
- Kosari-Nasab, M.; Sadeghi, T.; Bashiri, H.; Shokouhi, G.; Salari, A.A. The blockade of corticotropin-releasing factor 1 receptor attenuates anxiety-related symptoms and hypothalamus-pituitary-adrenal axis reactivity in mice with mild traumatic brain injury. Behav. Pharmacol. 2019, 30, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Tapp, Z.M.; Godbout, J.P.; Kokiko-Cochran, O.N. A Tilted Axis: Maladaptive Inflammation and HPA Axis Dysfunction Contribute to Consequences of TBI. Front. Neurol. 2019, 10, 345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karaca, Z.; Tanriverdi, F.; Ünlühizarci, K.; Kelestimur, F. GH and Pituitary Hormone Alterations after Traumatic Brain Injury. Prog. Mol. Biol. Transl. Sci. 2016, 138, 167–191. [Google Scholar] [CrossRef] [PubMed]
- Tanriverdi, F.; Unluhizarci, K.; Kelestrimur, F. Persistent neuroinflammation may be involved in the pathogenesis of traumatic brain injury (TBI)-induced hypopituitarism: Potential genetic and autoimmune factors. J. Neurotrauma 2010, 27, 301–302. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lipinski, M.M. Autophagy in Neurotrauma: Good, Bad, or Dysregulated. Cells 2019, 8, 693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, M.S.; Bayır, H.; Kochanek, P.M.; Clark, R.S.B. The role of autophagy in acute brain injury: A state of flux? Neurobiol. Dis. 2019, 122, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Yang, W.; Wu, C.; Liu, B.; Lu, H.; Wang, H.; Yan, H. Assessment of the role of intracranial hypertension and stress on hippocampal cell apoptosis and hypothalamic-pituitary dysfunction after TBI. Sci. Rep. 2017, 7, 3805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Xu, R.; Zhu, X.; Li, Y.; Wang, Y.; Xu, W. MicroRNA-23a-3p improves traumatic brain injury through modulating the neurological apoptosis and inflammation response in mice. Cell Cycle 2019, 19, 24–38. [Google Scholar] [CrossRef]
- Ye, Y.; Zhang, P.; Qian, Y.; Yin, B.; Yan, M. The Effect of Pyrroloquinoline Quinone on the Expression of WISP1 in Traumatic Brain Injury. Stem Cells Int. 2017, 2017, 4782820. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Das, D.N.; Bhutia, S.K. Autophagy and apoptosis: Where do they meet? Apoptosis 2014, 19, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Cooper, K.F. Till death do us part: The marriage of autophagy and apoptosis. Oxid. Med. Cell. Longev. 2018, 2018, 4701275. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.N.; Rahman, S.U.; Sanders, N.C.; Tio, D.L.; Prolo, P.; Sutton, R.L. Injury severity differentially affects short- and long-term neuroendocrine outcomes of traumatic brain injury. J. Neurotrauma 2008, 25, 311–323. [Google Scholar] [CrossRef]
- Mele, C.; Pingue, V.; Caputo, M.; Zavattaro, M.; Pagano, L.; Prodam, F.; Nardone, A.; Aimaretti, G.; Marzullo, P. Neuroinflammation and hypothalamo-pituitary dysfunction: Focus of traumatic brain injury. Int. J. Mol. Sci. 2021, 22, 2686. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Kim, J.; Quan, W.; Lee, J.C.; Kim, M.S.; Kim, S.H.; Bae, J.W.; Hur, K.Y.; Lee, M.S. Autophagy deficiency in myeloid cells increases susceptibility to obesity-induced diabetes and experimental colitis. Autophagy 2016, 12, 1390–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueno, T.; Saji, S.; Sugimoto, M.; Masuda, N.; Kuroi, K.; Sato, N.; Takei, H.; Yamamoto, Y.; Ohno, S.; Yamashita, H.; et al. Clinical significance of the expression of autophagy-associated marker, beclin 1, in breast cancer patients who received neoadjuvant endocrine therapy. BMC Cancer 2016, 16, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasturi, B.S.; Stein, D.G. Traumatic brain injury causes long-term reduction in serum growth hormone and persistent astrocytosis in the cortico-hypothalamo-pituitary axis of adult male rats. J. Neurotrauma 2009, 26, 1315–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanriverdi, F.; De Bellis, A.; Bizzarro, A.; Sinisi, A.A.; Bellastella, G.; Pane, E.; Bellastella, A.; Unluhizarci, K.; Selcuklu, A.; Casanueva, F.F.; et al. Antipituitary antibodies after traumatic brain injury: Is head trauma-induced pituitary dysfunction associated with autoimmunity? Eur. J. Endocrinol. 2008, 159, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, H. Autophagy in traumatic brain injury: A new target for therapeutic intervention. Front. Mol. Neurosci. 2018, 11, 190. [Google Scholar] [CrossRef]
- Gao, Y.; Zhuang, Z.; Gao, S.; Li, X.; Zhang, Z.; Ye, Z.; Li, L.; Tang, C.; Zhou, M.; Han, X.; et al. Tetrahydrocurcumin reduces oxidative stress-induced apoptosis via the mitochondrial apoptotic pathway by modulating autophagy in rats after traumatic brain injury. Am. J. Transl. Res. 2017, 9, 887–899. [Google Scholar]
- Clark, R.S.B.; Bayir, H.; Chu, C.T.; Alber, S.M.; Kochanek, P.M.; Watkins, S.C. Autophagy is increased in mice after traumatic brain injury and is detectable in human brain after trauma and critical illness. Autophagy 2008, 4, 88–90. [Google Scholar] [CrossRef] [Green Version]
- Taheri, S.; Karaca, Z.; Rassoulzadegan, M.; Mehmetbeyoglu, E.; Zararsiz, G.; Sener, E.F.; Bayram, K.K.; Tufan, E.; Sahin, M.C.; Marasli, M.K.; et al. The Characterization of Sex Differences in Hypoglycemia-Induced Activation of HPA Axis on the Transcriptomic Level. Cell. Mol. Neurobiol. 2021, 42, 1523–1542. [Google Scholar] [CrossRef]
- Garrahy, A.; Agha, A. How should we interrogate the hypothalamic-pituitary-adrenal axis in patients with suspected hypopituitarism? BMC Endocr. Disord. 2016, 16, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, N.H.; Hadad, R.; Taylor, R.R.; Pilar, J.R.; Salazar, O.; Llompart-Pou, J.A.; Dietrich, W.D.; Keane, R.W.; Pérez-Bárcena, J.; de Rivero Vaccari, J.P. Inflammatory Biomarkers of Traumatic Brain Injury. Pharmaceuticals 2022, 15, 660. [Google Scholar] [CrossRef]
- Ozpolat, B.; Benbrook, D.M. Targeting autophagy in cancer management-strategies and developments. Cancer Manag. Res. 2015, 7, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Arozena, A.A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in health and disease: A comprehensive review. Biomed. Pharmacother. 2018, 104, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.-i.; Saiki, S.; Furuya, N.; Imamichi, Y.; Tsuboi, Y.; Hattori, N. P150 Glued Deficiency Impairs Effective Fusion Between Autophagosomes and Lysosomes Due to Their Redistribution to the Cell Periphery. Neurosci. Lett. 2019, 690, 181–187. [Google Scholar] [CrossRef]
- Li, G.; Qian, L.; Tang, X.; Chen, Y.; Zhao, Z.; Zhang, C. Long non-coding RNA growth arrest-specific 5 (GAS5) acts as a tumor suppressor by promoting autophagy in breast cancer. Mol. Med. Rep. 2020, 22, 2460–2468. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, B.; Chai, Y.; Dong, B.; Lei, P.; Jiang, R.; Zhang, J. Methylprednisolone exacerbates acute critical illness-related corticosteroid insufficiency associated with traumatic brain injury in rats. Brain Res. 2011, 1382, 298–307. [Google Scholar] [CrossRef]
- Scott, M.C.; Walters, A.J.; Olson, S.D.; Cox, C.S. Determining Sex-based Differences in Inflammatory Response in an Experimental Traumatic Brain Injury Model. Front. Immunol. 2022, 13, 753570. [Google Scholar] [CrossRef] [PubMed]
- Bilgen, M. A new device for experimental modeling of central nervous system injuries. Neurorehabil. Neural Repair 2005, 19, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Huang, X.; Li, S.; Yang, Z.; Zhang, F.; Chen, X.; Shan, H.; Tao, L.; Zhang, M. Surface-fill H 2 S-releasing Silk Fibroin Hydrogel for Brain Repair through the Repression of Neuronal Pyroptosis. Acta Biomater. 2022, 154, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Hamurcu, Z.; Deliba, N.; Nalbantoglu, U.; Sener, E.F.; Nurdinov, N. FOXM1 plays a role in autophagy by transcriptionally regulating Beclin-1 and LC3 genes in human triple-negative breast cancer cells. J. Mol. Med. 2019, 97, 491–508. [Google Scholar] [CrossRef] [PubMed]
- Hamurcu, Z.; Delibaşı, N.; Geçene, S.; Şener, E.F.; Dönmez-Altuntaş, H.; Özkul, Y.; Canatan, H.; Ozpolat, B. Targeting LC3 and Beclin-1 autophagy genes suppresses proliferation, survival, migration and invasion by inhibition of Cyclin-D1 and uPAR/Integrin β1/ Src signaling in triple negative breast cancer cells. J. Cancer Res. Clin. Oncol. 2018, 144, 415–430. [Google Scholar] [CrossRef] [PubMed]
- Bordeaux, J.; Welsh, A.W.; Agarwal, S.; Killiam, E.; Baquero, M.T.; Hanna, J.A.; Anagnostou, V.K.; Rimm, D.L. Antibody validation. Biotechniques 2010, 48, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Pillai-Kastoori, L.; Heaton, S.; Shiflett, S.D.; Roberts, A.C.; Solache, A.; Schutz-Geschwender, A.R. Antibody validation for Western blot: By the user, for the user. J. Biol. Chem. 2020, 295, 926–939. [Google Scholar] [CrossRef]
- Uhlen, M.; Bandrowski, A.; Carr, S.; Edwards, A.; Ellenberg, J.; Lundberg, E.; Rimm, D.L.; Rodriguez, H.; Hiltke, T.; Snyder, M.; et al. A proposal for validation of antibodies. Nat. Methods 2016, 13, 823–827. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taheri, S.; Karaca, Z.; Mehmetbeyoglu, E.; Hamurcu, Z.; Yilmaz, Z.; Dal, F.; Çınar, V.; Ulutabanca, H.; Tanriverdi, F.; Unluhizarci, K.; et al. The Role of Apoptosis and Autophagy in the Hypothalamic-Pituitary-Adrenal (HPA) Axis after Traumatic Brain Injury (TBI). Int. J. Mol. Sci. 2022, 23, 15699. https://doi.org/10.3390/ijms232415699
Taheri S, Karaca Z, Mehmetbeyoglu E, Hamurcu Z, Yilmaz Z, Dal F, Çınar V, Ulutabanca H, Tanriverdi F, Unluhizarci K, et al. The Role of Apoptosis and Autophagy in the Hypothalamic-Pituitary-Adrenal (HPA) Axis after Traumatic Brain Injury (TBI). International Journal of Molecular Sciences. 2022; 23(24):15699. https://doi.org/10.3390/ijms232415699
Chicago/Turabian StyleTaheri, Serpil, Züleyha Karaca, Ecmel Mehmetbeyoglu, Zuhal Hamurcu, Zeynep Yilmaz, Fatma Dal, Venhar Çınar, Halil Ulutabanca, Fatih Tanriverdi, Kursad Unluhizarci, and et al. 2022. "The Role of Apoptosis and Autophagy in the Hypothalamic-Pituitary-Adrenal (HPA) Axis after Traumatic Brain Injury (TBI)" International Journal of Molecular Sciences 23, no. 24: 15699. https://doi.org/10.3390/ijms232415699