

The Role of Inflammation and Oxidative Stress in Rheumatic Heart Disease



Abstract
1. Introduction
2. Rheumatic Heart Disease (RHD)
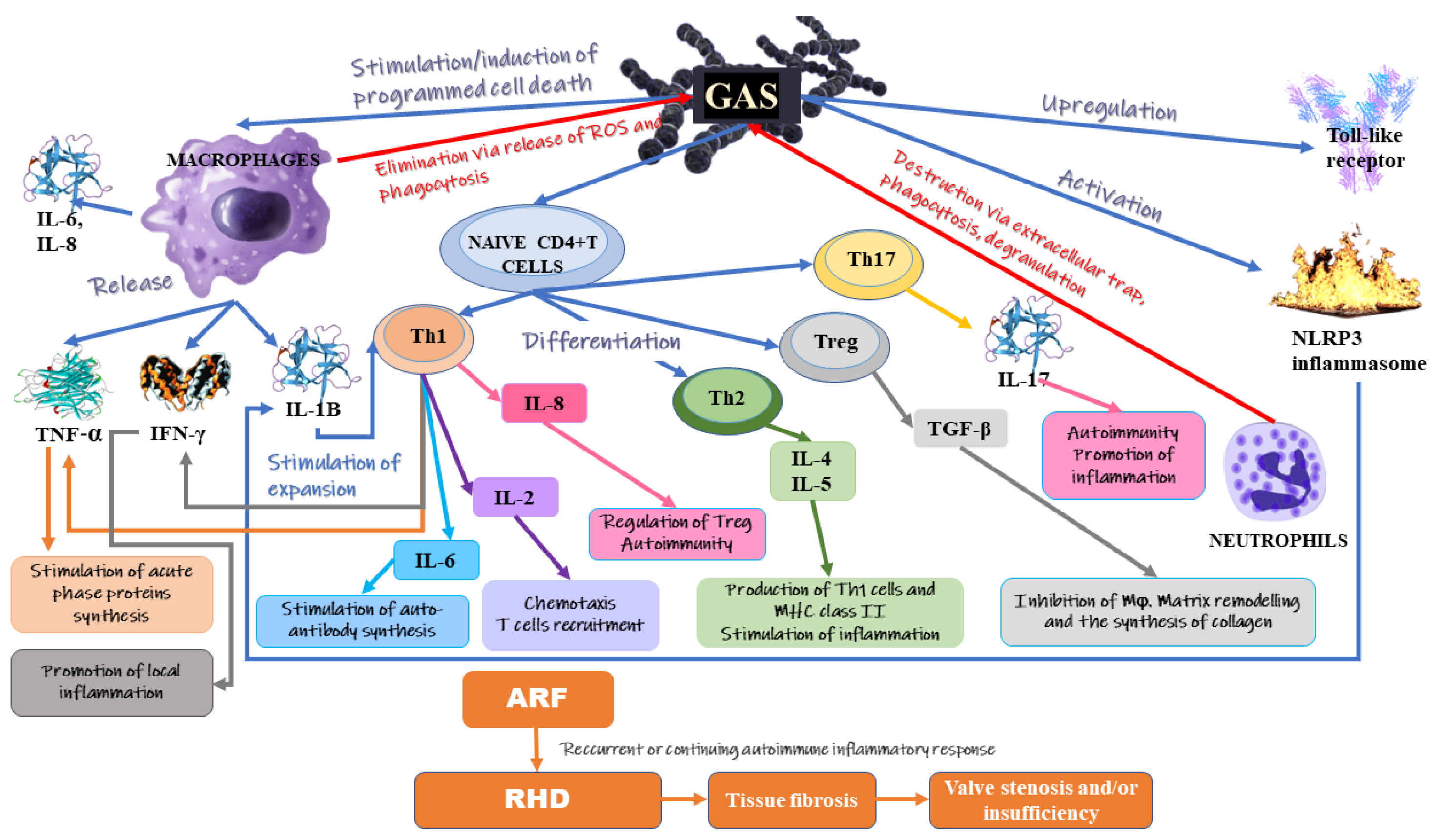
3. Inflammation
4. Oxidative Stress
5. Treatment
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lu, Q.; Sun, Y.; Duan, Y.; Li, B.; Xia, J.; Yu, S.; Zhang, G. Comprehensive microRNA profiling reveals potential augmentation of the IL1 pathway in rheumatic heart valve disease. BMC Cardiovasc. Disord. 2018, 18, 53. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, M.; Sarkar, S.; Makol, A.; Sandip Singh, R.; Saikia, U.N.; Banerjee, D.; Chopra, S.; Chakraborti, A. Anti-endothelial cell antibody rich sera from rheumatic heart disease patients induces proinflammatory phenotype and methylation alteration in endothelial cells. Genes Dis. 2018, 5, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Carapetis, J.R. Rheumatic heart disease in developing countries. N. Engl. J. Med. 2007, 357, 439–441. [Google Scholar] [CrossRef] [PubMed]
- Butt, H.I.; Shahbaz, A.; Nawaz, H.; Butt, K. Comparative Clinical Characteristics of Rheumatic Heart Disease Patients Undergoing Surgical Valve Replacement. Cureus 2019, 11, e4889. [Google Scholar] [CrossRef]
- Marijon, E.; Mirabel, M.; Celermajer, D.S.; Jouven, X. Rheumatic heart disease. Lancet 2012, 379, 953–964. [Google Scholar] [CrossRef]
- Passos, L.S.A.; Nunes, M.C.P.; Aikawa, E. Rheumatic Heart Valve Disease Pathophysiology and Underlying Mechanisms. Front. Cardiovasc. Med. 2020, 7, 612716. [Google Scholar] [CrossRef]
- Attar, A.; Marzban, P.; Moaref, A.; Aghasadeghi, K. The association of plasma high-sensitivity C-reactive protein level with rheumatic heart disease: The possible role of inflammation. Indian Heart J. 2018, 70, 346–349. [Google Scholar] [CrossRef]
- Van de Rijn, I.; Zabriskie, J.B.; McCarty, M. Group A streptococcal antigens cross-reactive with myocardium. Purification of heart-reactive antibody and isolation and characterization of the streptococcal antigen. J. Exp. Med. 1977, 146, 579–599. [Google Scholar] [CrossRef]
- Ayoub, E.M.; Taranta, A.; BARTLEY, T.D. Effect of valvular surgery on antibody to the group A streptococcal carbohydrate. Circulation 1974, 50, 144–150. [Google Scholar] [CrossRef][Green Version]
- Remenyi, B.; Carapetis, J.; Wyber, R.; Taubert, K.; Mayosi, B.M. Position statement of the World Heart Federation on the prevention and control of rheumatic heart disease. Nat. Rev. Cardiol. 2013, 10, 284–292. [Google Scholar] [CrossRef]
- Watkins, D.A.; Johnson, C.O.; Colquhoun, S.M.; Karthikeyan, G.; Beaton, A.; Bukhman, G.; Forouzanfar, M.H.; Longenecker, C.T.; Mayosi, B.M.; Mensah, G.A. Global, regional, and national burden of rheumatic heart disease, 1990–2015. N. Engl. J. Med. 2017, 377, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Gewitz, M.H.; Baltimore, R.S.; Tani, L.Y.; Sable, C.A.; Shulman, S.T.; Carapetis, J.; Remenyi, B.; Taubert, K.A.; Bolger, A.F.; Beerman, L. Revision of the Jones Criteria for the diagnosis of acute rheumatic fever in the era of Doppler echocardiography: A scientific statement from the American Heart Association. Circulation 2015, 131, 1806–1818. [Google Scholar] [CrossRef] [PubMed]
- Albert, D.A.; Harel, L.; Karrison, T. The treatment of rheumatic carditis: A review and meta-analysis. Medicine 1995, 74, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Buleu, F.; Sirbu, E.; Caraba, A.; Dragan, S. Heart Involvement in Inflammatory Rheumatic Diseases: A Systematic Literature Review. Medicina 2019, 55, 249. [Google Scholar] [CrossRef] [PubMed]
- Amaya-Amaya, J.; Montoya-Sánchez, L.; Rojas-Villarraga, A. Cardiovascular involvement in autoimmune diseases. BioMed Res. Int. 2014, 2014, 367359. [Google Scholar] [CrossRef]
- Rajamannan, N.M.; Nealis, T.B.; Subramaniam, M.; Pandya, S.; Stock, S.R.; Ignatiev, C.I.; Sebo, T.J.; Rosengart, T.K.; Edwards, W.D.; McCarthy, P.M.; et al. Calcified rheumatic valve neoangiogenesis is associated with vascular endothelial growth factor expression and osteoblast-like bone formation. Circulation 2005, 111, 3296–3301. [Google Scholar] [CrossRef]
- Hutcheson, J.D.; Goettsch, C.; Bertazzo, S.; Maldonado, N.; Ruiz, J.L.; Goh, W.; Yabusaki, K.; Faits, T.; Bouten, C.; Franck, G.; et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat. Mater. 2016, 15, 335–343. [Google Scholar] [CrossRef]
- New, S.E.; Goettsch, C.; Aikawa, M.; Marchini, J.F.; Shibasaki, M.; Yabusaki, K.; Libby, P.; Shanahan, C.M.; Croce, K.; Aikawa, E. Macrophage-derived matrix vesicles: An alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ. Res. 2013, 113, 72–77. [Google Scholar] [CrossRef]
- Krohn, J.B.; Hutcheson, J.D.; Martínez-Martínez, E.; Aikawa, E. Extracellular vesicles in cardiovascular calcification: Expanding current paradigms. J. Physiol. 2016, 594, 2895–2903. [Google Scholar] [CrossRef]
- Cunningham, M.W. Rheumatic fever, autoimmunity, and molecular mimicry: The streptococcal connection. Int. Rev. Immunol. 2014, 33, 314–329. [Google Scholar] [CrossRef]
- Martin, W.J.; Steer, A.C.; Smeesters, P.R.; Keeble, J.; Inouye, M.; Carapetis, J.; Wicks, I.P. Post-infectious group A streptococcal autoimmune syndromes and the heart. Autoimmun. Rev. 2015, 14, 710–725. [Google Scholar] [CrossRef] [PubMed]
- Kirvan, C.A.; Galvin, J.E.; Hilt, S.; Kosanke, S.; Cunningham, M.W. Identification of streptococcal m-protein cardiopathogenic epitopes in experimental autoimmune valvulitis. J. Cardiovasc. Transl. Res. 2014, 7, 172–181. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kanagasingam, A.; Francis, G.R.; Komagarajah, B.; Ladchumanan, D.; Sivapramyan, A.; Packiyarajah, P.; Megatheepan, R.; Madura, J. Pattern of Rheumatic valvular involvement and its contribution towards valvular malfunction in young adults. Ceylon Med. J. 2019, 64, 91–97. [Google Scholar] [CrossRef]
- Guedes, C.; Bianchi-Fior, P.; Cormier, B.; Barthelemy, B.; Rat, A.C.; Boissier, M.C. Cardiac manifestations of rheumatoid arthritis: A case-control transesophageal echocardiography study in 30 patients. Arthritis Rheum. 2001, 45, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.H.; Sareli, P.; Pocock, W.A.; Barlow, J.B. The spectrum of severe rheumatic mitral valve disease in a developing country. Correlations among clinical presentation, surgical pathologic findings, and hemodynamic sequelae. Ann. Intern. Med. 1994, 120, 177–183. [Google Scholar] [CrossRef]
- Lambova, S. Cardiac manifestations in systemic sclerosis. World J. Cardiol. 2014, 6, 993. [Google Scholar] [CrossRef]
- Luo, T.; Han, J.; Meng, X. Features of rheumatic mitral valves and a grading system to identify suitable repair cases in China. J. Thorac. Dis. 2017, 9, 3138. [Google Scholar] [CrossRef]
- Veinot, J.P. Pathology of inflammatory native valvular heart disease. Cardiovascular Pathology 2006, 15, 243–251. [Google Scholar] [CrossRef]
- Shiba, M.; Sugano, Y.; Ikeda, Y.; Okada, H.; Nagai, T.; Ishibashi-Ueda, H.; Yasuda, S.; Ogawa, H.; Anzai, T. Presence of increased inflammatory infiltrates accompanied by activated dendritic cells in the left atrium in rheumatic heart disease. PLoS ONE 2018, 13, e0203756. [Google Scholar] [CrossRef]
- Marcus, G.M.; Smith, L.M.; Ordovas, K.; Scheinman, M.M.; Kim, A.M.; Badhwar, N.; Lee, R.J.; Tseng, Z.H.; Lee, B.K.; Olgin, J.E. Intracardiac and extracardiac markers of inflammation during atrial fibrillation. Heart Rhythm. 2010, 7, 149–154. [Google Scholar] [CrossRef]
- Pandit, B.N.; Aggarwal, P.; Subramaniyan, S.; Gujral, J.S.; Nath, R.K. Largest giant left atrium in rheumatic heart disease. J. Cardiol. Cases 2021, 24, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.L.; Martin, W.J.; Minigo, G.; Keeble, J.L.; Garnham, A.L.; Pacini, G.; Smyth, G.K.; Speed, T.P.; Carapetis, J.; Wicks, I.P. Dysregulated IL-1β-GM-CSF Axis in Acute Rheumatic Fever That Is Limited by Hydroxychloroquine. Circulation 2018, 138, 2648–2661. [Google Scholar] [CrossRef] [PubMed]
- Ralston, S.H.; Penman, I.D.; Strachan, M.W.; Hobson, R. Davidson’s Principles and Practice of Medicine E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Ambari, A.M.; Setianto, B.; Santoso, A.; Radi, B.; Dwiputra, B.; Susilowati, E.; Tulrahmi, F.; Doevendans, P.A.; Cramer, M.J. Angiotensin Converting Enzyme Inhibitors (ACEIs) Decrease the Progression of Cardiac Fibrosis in Rheumatic Heart Disease Through the Inhibition of IL-33/sST2. Front. Cardiovasc. Med. 2020, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Ghati, N.; Sharique, M.; Sharma, S.; Shetkar, S.; Karmakar, S.; Naik, N.; Lakshmy, R.; Thakur, B.; Agarwal, A.; et al. Role of inflammation in initiation and maintenance of atrial fibrillation in rheumatic mitral stenosis-An analytical cross-sectional study. J. Arrhythm. 2020, 36, 1007–1015. [Google Scholar] [CrossRef] [PubMed]
- Rifaie, O.; Badr, M.; Salam, A.A.; Galal, H. Colchicine ameliorates the chronic inflammatory state in patients with chronic rheumatic valvular heart disease: A pilot study. Egypt Heart J. 2020, 72, 42. [Google Scholar] [CrossRef]
- Guilherme, L.; Ramasawmy, R.; Kalil, J. Rheumatic fever and rheumatic heart disease: Genetics and pathogenesis. Scand. J. Immunol. 2007, 66, 199–207. [Google Scholar] [CrossRef]
- Beltrame, M.H.; Catarino, S.J.; Goeldner, I.; Boldt, A.B.; de Messias-Reason, I.J. The lectin pathway of complement and rheumatic heart disease. Front. Pediatr. 2014, 2, 148. [Google Scholar] [CrossRef]
- Gomaa, M.H.; Ali, S.S.; Fattouh, A.M.; Hamza, H.S.; Badr, M.M. MBL2 gene polymorphism rs1800450 and rheumatic fever with and without rheumatic heart disease: An Egyptian pilot study. Pediatr. Rheumatol. Online J. 2018, 16, 24. [Google Scholar] [CrossRef]
- Gui, T.; Sun, Y.; Shimokado, A.; Muragaki, Y. The Roles of Mitogen-Activated Protein Kinase Pathways in TGF-β-Induced Epithelial-Mesenchymal Transition. J. Signal Transduct. 2012, 2012, 289243. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Mavrogeni, S.I.; Markousis-Mavrogenis, G.; Koutsogeorgopoulou, L.; Dimitroulas, T.; Vartela, V.; Rigopoulos, A.; Noutsias, M.; Kolovou, G. Pathophysiology and imaging of heart failure in women with autoimmune rheumatic diseases. Heart Fail Rev. 2019, 24, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Tormin, J.; Nascimento, B.R.; Sable, C.A.; da Silva, J.L.P.; Brandao-de-Resende, C.; Rocha, L.P.C.; Pinto, C.H.R.; Neves, E.G.A.; Macedo, F.V.B.; Fraga, C.L.; et al. Cytokine gene functional polymorphisms and phenotypic expression as predictors of evolution from latent to clinical rheumatic heart disease. Cytokine 2021, 138, 155370. [Google Scholar] [CrossRef]
- Chopra, P.; Gulwani, H. Pathology and pathogenesis of rheumatic heart disease. Indian J. Pathol. Microbiol. 2007, 50, 685–697. [Google Scholar] [PubMed]
- Korkmaz, A.; Doğanay, B.; Basyigit, F.; Çöteli, C.; Yildiz, A.; Gursoy, T.; Guray, U.; Elalmis, O.U. Serum Thiol Levels and Thiol/Disulfide Homeostasis in Patients with Rheumatic Mitral Valve Disease and Healthy Subjects. Arq. Bras. Cardiol. 2021, 117, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Carapetis, J.R.; McDonald, M.; Wilson, N.J. Acute rheumatic fever. Lancet 2005, 366, 155–168. [Google Scholar] [CrossRef]
- Sharma, S.; Sharma, G.; Hote, M.; Devagourou, V.; Kesari, V.; Arava, S.; Airan, B.; Ray, R. Light and electron microscopic features of surgically excised left atrial appendage in rheumatic heart disease patients with atrial fibrillation and sinus rhythm. Cardiovasc. Pathol. 2014, 23, 319–326. [Google Scholar] [CrossRef]
- Eigenbrod, T.; Pelka, K.; Latz, E.; Kreikemeyer, B.; Dalpke, A.H. TLR8 senses bacterial RNA in human monocytes and plays a nonredundant role for recognition of Streptococcus pyogenes. J. Immunol. 2015, 195, 1092–1099. [Google Scholar] [CrossRef]
- Valderrama, J.A.; Riestra, A.M.; Gao, N.J.; LaRock, C.N.; Gupta, N.; Ali, S.R.; Hoffman, H.M.; Ghosh, P.; Nizet, V. Group A streptococcal M protein activates the NLRP3 inflammasome. Nat. Microbiol. 2017, 2, 1425–1434. [Google Scholar] [CrossRef]
- Movert, E.; Lienard, J.; Valfridsson, C.; Nordström, T.; Johansson-Lindbom, B.; Carlsson, F. Streptococcal M protein promotes IL-10 production by cGAS-independent activation of the STING signaling pathway. PLoS Pathog. 2018, 14, e1006969. [Google Scholar] [CrossRef]
- DuPage, M.; Bluestone, J.A. Harnessing the plasticity of CD4+ T cells to treat immune-mediated disease. Nat. Rev. Immunol. 2016, 16, 149–163. [Google Scholar] [CrossRef]
- Soderholm, A.T.; Barnett, T.C.; Sweet, M.J.; Walker, M.J. Group A streptococcal pharyngitis: Immune responses involved in bacterial clearance and GAS-associated immunopathologies. J. Leukoc. Biol. 2018, 103, 193–213. [Google Scholar] [CrossRef] [PubMed]
- Fieber, C.; Kovarik, P. Responses of innate immune cells to group A Streptococcus. Front. Cell. Infect. Microbiol. 2014, 4, 140. [Google Scholar] [CrossRef] [PubMed]
- Döhrmann, S.; Cole, J.N.; Nizet, V. Conquering neutrophils. PLoS Pathog. 2016, 12, e1005682. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, N.; Bi, S.; Wang, X.; Wang, B. Co-activation of Th17 and antibody responses provides efficient protection against mucosal infection by group A Streptococcus. PLoS ONE 2016, 11, e0168861. [Google Scholar] [CrossRef][Green Version]
- Bozinovski, S.; Seow, H.J.; Chan, S.P.J.; Anthony, D.; McQualter, J.; Hansen, M.; Jenkins, B.J.; Anderson, G.P.; Vlahos, R. Innate cellular sources of interleukin-17A regulate macrophage accumulation in cigarette-smoke-induced lung inflammation in mice. Clin. Sci. 2015, 129, 785–796. [Google Scholar] [CrossRef]
- Moser, M.; Leo, O. Key concepts in immunology. Vaccine 2010, 28, C2–C13. [Google Scholar] [CrossRef]
- Guilherme, L.; Kalil, J. Rheumatic fever and rheumatic heart disease: Cellular mechanisms leading autoimmune reactivity and disease. J. Clin. Immunol. 2010, 30, 17–23. [Google Scholar] [CrossRef]
- Becher, B.; Tugues, S.; Greter, M. GM-CSF: From growth factor to central mediator of tissue inflammation. Immunity 2016, 45, 963–973. [Google Scholar] [CrossRef]
- Cooper, K.A.; Donovan, J.L.; Waterhouse, A.L.; Williamson, G. Cocoa and health: A decade of research. Br. J. Nutr. 2008, 99, 1–11. [Google Scholar] [CrossRef]
- Sonderegger, I.; Iezzi, G.; Maier, R.; Schmitz, N.; Kurrer, M.; Kopf, M. GM-CSF mediates autoimmunity by enhancing IL-6–dependent Th17 cell development and survival. J. Exp. Med. 2008, 205, 2281–2294. [Google Scholar] [CrossRef]
- Kemeny, E.; Grieve, T.; Marcus, R.; Sareli, P.; Zabriskie, J.B. Identification of mononuclear cells and T cell subsets in rheumatic valvulitis. Clin. Immunol. Immunopathol. 1989, 52, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Guilherme, L.; Cury, P.; Demarchi, L.M.; Coelho, V.; Abel, L.; Lopez, A.P.; Oshiro, S.E.; Aliotti, S.; Cunha-Neto, E.; Pomerantzeff, P.M. Rheumatic heart disease: Proinflammatory cytokines play a role in the progression and maintenance of valvular lesions. Am. J. Pathol. 2004, 165, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Walker, G.A.; Masters, K.S.; Shah, D.N.; Anseth, K.S.; Leinwand, L.A. Valvular myofibroblast activation by transforming growth factor-β: Implications for pathological extracellular matrix remodeling in heart valve disease. Circ. Res. 2004, 95, 253–260. [Google Scholar] [CrossRef]
- Morris, K.; Mohan, C.; Wahi, P.; Anand, I.; Ganguly, N. Increase in activated T cells and reduction in suppressor/cytotoxic T cells in acute rheumatic fever and active rheumatic heart disease: A longitudinal study. J. Infect. Dis. 1993, 167, 979–983. [Google Scholar] [CrossRef] [PubMed]
- Faé, K.C.; da Silva, D.D.; Oshiro, S.E.; Tanaka, A.C.; Pomerantzeff, P.M.; Douay, C.; Charron, D.; Toubert, A.; Cunningham, M.W.; Kalil, J. Mimicry in recognition of cardiac myosin peptides by heart-intralesional T cell clones from rheumatic heart disease. J. Immunol. 2006, 176, 5662–5670. [Google Scholar] [CrossRef]
- Bhatia, R.; Narula, J.; Reddy, K.; Koicha, M.; Malaviya, A.; Pothineni, R.; Tandon, R.; Bhatia, M. Lymphocyte subsets in acute rheumatic fever and rheumatic heart disease. Clin. Cardiol. 1989, 12, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, A.; Grover, A.; Ganguly, N. Superantigen-induced T cell responses in acute rheumatic fever and chronic rheumatic heart disease patients. Clin. Exp. Immunol. 1999, 116, 100–106. [Google Scholar] [CrossRef]
- Yi, Y.-S. Role of inflammasomes in inflammatory autoimmune rheumatic diseases. Korean J. Physiol. Pharmacol. 2018, 22, 1–15. [Google Scholar] [CrossRef]
- LaRock, C.N.; Nizet, V. Inflammasome/IL-1β responses to streptococcal pathogens. Front. Immunol. 2015, 6, 518. [Google Scholar] [CrossRef]
- Gasse, P.; Riteau, N.; Vacher, R.; Michel, M.-L.; Fautrel, A.; Di Padova, F.; Fick, L.; Charron, S.; Lagente, V.; Eberl, G. IL-1 and IL-23 mediate early IL-17A production in pulmonary inflammation leading to late fibrosis. PLoS ONE 2011, 6, e23185. [Google Scholar] [CrossRef]
- Kim, L.; Yang, W.I.; Shin, D.H.; Jung, I.M.; Park, H.K.; Chang, B.C. Overexpression of transforming growth factor-β1 in the valvular fibrosis of chronic rheumatic heart disease. J. Korean Med. Sci. 2008, 23, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, T.; Mukherjee, S.; Ghosh, S.; Biswas, M.; Dutta, S.; Pattari, S.; Chatterjee, S.; Bandyopadhyay, A. Clinical significance of markers of collagen metabolism in rheumatic mitral valve disease. PLoS ONE 2014, 9, e90527. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Ye, Y.; Yin, Y.; Sang, P.; Li, L.; Wang, J.; Wan, W.; Li, R.; Bai, X.; Xie, Y.; et al. Association of matrix metalloprotease 1, 3, and 12 polymorphisms with rheumatic heart disease in a Chinese Han population. BMC Med. Genet. 2018, 19, 27. [Google Scholar] [CrossRef] [PubMed]
- Soares, A.C.D.; Passos, L.S.A.; Sable, C.; Beaton, A.; Ribeiro, V.T.; Gollob, K.J.; Dutra, W.O.; Nunes, M.C.P. Circulating cytokines predict severity of rheumatic heart disease. Int. J. Cardiol. 2019, 289, 107–109. [Google Scholar] [CrossRef] [PubMed]
- Faé, K.C.; Palacios, S.A.; Nogueira, L.G.; Oshiro, S.E.; Demarchi, L.M.; Bilate, A.M.; Pomerantzeff, P.M.; Brandão, C.; Thomaz, P.G.; dos Reis, M. CXCL9/Mig mediates T cells recruitment to valvular tissue lesions of chronic rheumatic heart disease patients. Inflammation 2013, 36, 800–811. [Google Scholar] [CrossRef]
- Sharma, N.; Toor, D. Interleukin-10: Role in increasing susceptibility and pathogenesis of rheumatic fever/rheumatic heart disease. Cytokine 2017, 90, 169–176. [Google Scholar] [CrossRef]
- Toor, D.; Vohra, H. Immune responsiveness during disease progression from acute rheumatic fever to chronic rheumatic heart disease. Microbes Infect. 2012, 14, 1111–1117. [Google Scholar] [CrossRef]
- Yeğin, O.; Coşkun, M.; Ertuğ, H. Cytokines in acute rheumatic fever. Eur. J. Pediatr. 1997, 156, 25–29. [Google Scholar] [CrossRef]
- Dinarello, C.A. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood J. Am. Soc. Hematol. 2011, 117, 3720–3732. [Google Scholar] [CrossRef]
- Azevedo, P.M.; Bauer, R.; de Falco Caparbo, V.; Silva, C.A.A.; Bonfá, E.; Pereira, R.M.R. Interleukin-1 receptor antagonist gene (IL1RN) polymorphism possibly associated to severity of rheumatic carditis in a Brazilian cohort. Cytokine 2010, 49, 109–113. [Google Scholar] [CrossRef]
- Dienz, O.; Eaton, S.M.; Bond, J.P.; Neveu, W.; Moquin, D.; Noubade, R.; Briso, E.M.; Charland, C.; Leonard, W.J.; Ciliberto, G. The induction of antibody production by IL-6 is indirectly mediated by IL-21 produced by CD4+ T cells. J. Exp. Med. 2009, 206, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Moon, B.-I.; Kim, T.H.; Seoh, J.-Y. Functional modulation of regulatory T cells by IL-2. PLoS ONE 2015, 10, e0141864. [Google Scholar] [CrossRef] [PubMed]
- Bas, H.D.; Baser, K.; Yavuz, E.; Bolayir, H.A.; Yaman, B.; Unlu, S.; Cengel, A.; Bagriacik, E.U.; Yalcin, R. A shift in the balance of regulatory T and T helper 17 cells in rheumatic heart disease. J. Investig. Med. 2014, 62, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Varma, S.; Kumar, H.M.; Yusaf, J.; Goyal, M.; Mehta, V.; Tyagi, S. Circulating level of regulatory T cells in rheumatic heart disease: An observational study. Indian Heart J. 2016, 68, 342–348. [Google Scholar] [CrossRef][Green Version]
- Chen, A.; Wen, J.; Lu, C.; Lin, B.; Xian, S.; Huang, F.; Wu, Y.; Zeng, Z. Inhibition of miR-155-5p attenuates the valvular damage induced by rheumatic heart disease. Int. J. Mol. Med. 2020, 45, 429–440. [Google Scholar] [CrossRef]
- Dong, H.; Sun, Y.; Shan, F.; Sun, Q.; Yang, B. Down-regulation of miR-101 contributes to rheumatic heart disease through up-regulating TLR2. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2015, 21, 1500. [Google Scholar]
- Li, N.; Lian, J.; Zhao, S.; Zheng, D.; Yang, X.; Huang, X.; Shi, X.; Sun, L.; Zhou, Q.; Shi, H. Detection of differentially expressed MicroRNAs in rheumatic heart disease: miR-1183 and miR-1299 as potential diagnostic biomarkers. BioMed Res. Int. 2015, 2015, 524519. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, C.; Liu, L.; Xi, A.; Chen, B.; Li, Y.; Du, J. Macrophage-derived mir-155-containing exosomes suppress fibroblast proliferation and promote fibroblast inflammation during cardiac injury. Mol. Ther. 2017, 25, 192–204. [Google Scholar] [CrossRef]
- O’Connell, R.M.; Kahn, D.; Gibson, W.S.; Round, J.L.; Scholz, R.L.; Chaudhuri, A.A.; Kahn, M.E.; Rao, D.S.; Baltimore, D. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity 2010, 33, 607–619. [Google Scholar] [CrossRef]
- Thai, T.-H.; Calado, D.P.; Casola, S.; Ansel, K.M.; Xiao, C.; Xue, Y.; Murphy, A.; Frendewey, D.; Valenzuela, D.; Kutok, J.L. Regulation of the germinal center response by microRNA-155. Science 2007, 316, 604–608. [Google Scholar] [CrossRef]
- Zhang, Z.; Liang, K.; Zou, G.; Chen, X.; Shi, S.; Wang, G.; Zhang, K.; Li, K.; Zhai, S. Inhibition of miR-155 attenuates abdominal aortic aneurysm in mice by regulating macrophage-mediated inflammation. Biosci. Rep. 2018, 38, BSR20171432. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Huang, C.-X.; Rao, P.-P.; Cao, G.-Q.; Zhang, Y.; Zhou, J.-P.; Zhu, L.-Y.; Liu, M.-X.; Zhang, G.-G. MicroRNA-155 inhibition attenuates endoplasmic reticulum stress-induced cardiomyocyte apoptosis following myocardial infarction via reducing macrophage inflammation. Eur. J. Pharmacol. 2019, 857, 172449. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Huang, H.; Xie, Q.; Wang, Z.; Fan, Y.; Kong, B.; Huang, D.; Xiao, Y. MiR-155 knockout in fibroblasts improves cardiac remodeling by targeting tumor protein p53-inducible nuclear protein 1. J. Cardiovasc. Pharmacol. Ther. 2016, 21, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Dayer, J.M.; Manger, B. Interleukin-1 function and role in rheumatic disease. Nat. Rev. Rheumatol. 2016, 12, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Borthwick, L. The IL-1 Cytokine Family and its Role in Inflammation and Fibrosis in the Lung. In Seminars in Immunopathology; Springer: Berlin/Heidelberg, Germany, 2016; pp. 517–534. [Google Scholar]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The interleukin-1 family: Back to the future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef]
- Abbas, A.K.; Lichtman, A.; Pillai, S. Basic Immunology: Functions and Disorders of the Immune System, 6e: Sae-E-Book; Elsevier: Chennai, India, 2019. [Google Scholar]
- Bilik, M.Z.; Kaplan, İ.; Polat, N.; Akil, M.A.; Akyüz, A.; Acet, H.; Yüksel, M.; İnci, Ü.; Kayan, F.; Toprak, N. Serum Levels of IL-17 and IL-23 in Patients With Rheumatic Mitral Stenosis. Medicine 2016, 95, e3562. [Google Scholar] [CrossRef]
- Wen, Y.; Zeng, Z.; Gui, C.; Li, L.; Li, W. Changes in the expression of Th17 cell-associated cytokines in the development of rheumatic heart disease. Cardiovasc. Pathol. 2015, 24, 382–387. [Google Scholar] [CrossRef]
- Ali, S.K.; Eldaim, I.N.; Osman, S.H.; Bakhite, S.M. Clinical and echocardiographic features of children with rheumatic heart disease and their serum cytokine profile. Pan Afr. Med. J. 2012, 13, 36. [Google Scholar]
- Nakashima, H.; Miyake, K.; Inoue, Y.; Shimizu, S.; Akahoshi, M.; Tanaka, Y.; Otsuka, T.; Harada, M. Association between IL-4 genotype and IL-4 production in the Japanese population. Genes Immun. 2002, 3, 107–109. [Google Scholar] [CrossRef]
- Gölbasi, Z.; Uçar, O.; Keles, T.; Sahin, A.; Cagli, K.; Camsari, A.; Diker, E.; Aydogdu, S. Increased levels of high sensitive C-reactive protein in patients with chronic rheumatic valve disease: Evidence of ongoing inflammation. Eur. J. Heart Fail 2002, 4, 593–595. [Google Scholar] [CrossRef]
- Güngör, N.K. Overweight and obesity in children and adolescents. J. Clin. Res. Pediatr. Endocrinol. 2014, 6, 129. [Google Scholar] [CrossRef]
- Pulimamidi, V.K.; Murugesan, V.; Rajappa, M.; Satheesh, S.; Harichandrakumar, K.T. Increased levels of markers of oxidative stress and inflammation in patients with rheumatic mitral stenosis predispose to left atrial thrombus formation. J. Clin. Diagn. Res. 2013, 7, 2445. [Google Scholar] [PubMed]
- Hadi, H.A.; Alsheikh-Ali, A.A.; Mahmeed, W.A.; Al Suwaidi, J.M. Inflammatory cytokines and atrial fibrillation: Current and prospective views. J. Inflamm. Res. 2010, 3, 75. [Google Scholar] [CrossRef] [PubMed]
- Polat, N.; Yildiz, A.; Yuksel, M.; Bilik, M.Z.; Aydin, M.; Acet, H.; Akil, M.A.; Oylumlu, M.; Kaya, H.; Ertas, F. Association of neutrophil–lymphocyte ratio with the presence and severity of rheumatic mitral valve stenosis. Clin. Appl. Thromb. Hemost. 2014, 20, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Akboğa, M.K.; Akyel, A.; Şahinarslan, A.; Yayla, Ç.; Alsancak, Y.; Gökalp, G.; Nurkoç, S.; Abacı, A. Neutrophil-to-lymphocyte ratio is increased in patients with rheumatic mitral valve stenosis? Anatol. J. Cardiol. 2015, 15, 380. [Google Scholar] [CrossRef] [PubMed]
- Elemary, M.; Elmeligy, N.; Tabl, M.; Abd Elhaleem, A.E. Usefulness of novel hematologic inflammatory parameter: Neutrophil to lymphocyte ratio in patients with rheumatic valve diseases. Am. J. Res. Commun. 2016, 4, 43–62. [Google Scholar]
- Singal, A.K.; Devagourou, V.; Hote, M.P.; Choudhary, S.K.; Parakh, N.; Ray, R.; Lakshmy, R.; Karthikeyan, G. Detecting sub-clinical disease activity in patients with chronic rheumatic valvular heart disease. Indian Heart J. 2021, 73, 313–318. [Google Scholar] [CrossRef]
- Habeeb, N.M.; Al Hadidi, I.S. Ongoing inflammation in children with rheumatic heart disease. Cardiol. Young 2011, 21, 334–339. [Google Scholar] [CrossRef]
- Polat, N.; Yildiz, A.; Alan, S.; Toprak, N. Association of pentraxin-3 with the severity of rheumatic mitral valve stenosis. Acta Cardiol. 2015, 70, 409–413. [Google Scholar] [CrossRef]
- Hafez, M.; Yahia, S.; Eldars, W.; Eldegla, H.; Matter, M.; Attia, G.; Hawas, S. Prediction of residual valvular lesions in rheumatic heart disease: Role of adhesion molecules. Pediatr. Cardiol. 2013, 34, 583–590. [Google Scholar] [CrossRef]
- Galvin, J.E.; Hemric, M.E.; Ward, K.; Cunningham, M.W. Cytotoxic mAb from rheumatic carditis recognizes heart valves and laminin. J. Clin. Invest. 2000, 106, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Januska, R.; Meyer, A.; Fleck, M.; Luchner, A. Soluble ST2-A Potential Biomarker of Rheu-matic Heart Disease. Clin. Med. Rev. Case Rep. 2019, 6, 255. [Google Scholar]
- Howell, E.J.; Butcher, J.T. Valvular heart diseases in the developing world: Developmental biology takes center stage. J. Heart Valve Dis. 2012, 21, 234. [Google Scholar]
- Hudson, B.G.; Tryggvason, K.; Sundaramoorthy, M.; Neilson, E.G. Alport’s syndrome, Goodpasture’s syndrome, and type IV collagen. N. Engl. J. Med. 2003, 348, 2543–2556. [Google Scholar] [CrossRef]
- Dinkla, K.; Rohde, M.; Jansen, W.T.; Kaplan, E.L.; Chhatwal, G.S.; Talay, S.R. Rheumatic fever–associated Streptococcus pyogenes isolates aggregate collagen. J. Clin. Investig. 2003, 111, 1905–1912. [Google Scholar] [CrossRef] [PubMed]
- Dinkla, K.; Talay, S.R.; Mörgelin, M.; Graham, R.M.; Rohde, M.; Nitsche-Schmitz, D.P.; Chhatwal, G.S. Crucial role of the CB3-region of collagen IV in PARF-induced acute rheumatic fever. PLoS ONE 2009, 4, e4666. [Google Scholar] [CrossRef]
- Sari, A.; Davutoglu, V.; Bozkurt, E.; Tarakcioglu, M.; Erciyas, K. Effect of periodontitis on oxidative stress parameters in patients with rheumatic heart valve disease. Arch. Oral Biol. 2021, 121, 104961. [Google Scholar] [CrossRef]
- Thanan, R.; Oikawa, S.; Hiraku, Y.; Ohnishi, S.; Ma, N.; Pinlaor, S.; Yongvanit, P.; Kawanishi, S.; Murata, M. Oxidative stress and its significant roles in neurodegenerative diseases and cancer. Int. J. Mol. Sci. 2014, 16, 193–217. [Google Scholar] [CrossRef] [PubMed]
- Rabus, M.; Demirbağ, R.; Sezen, Y.; Konukoğlu, O.; Yildiz, A.; Erel, O.; Zeybek, R.; Yakut, C. Plasma and tissue oxidative stress index in patients with rheumatic and degenerative heart valve disease. Turk. Kardiyol. Dern. Ars. 2008, 36, 536–540. [Google Scholar]
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Taniyama, Y.; Griendling, K.K. Reactive oxygen species in the vasculature: Molecular and cellular mechanisms. Hypertension 2003, 42, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, S.; Jikimoto, T.; Saegusa, J. [Pathological roles of oxidative stress in autoimmune diseases]. Rinsho Byori 2003, 51, 126–132. [Google Scholar]
- Karataş, Z.; Baysal, T.; Sap, F.; Altın, H.; Çiçekler, H. The role of tenascin-C and oxidative stress in rheumatic and congenital heart valve diseases: An observational study. Anadolu Kardiyol. Derg. 2013, 13, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Chiu-Braga, Y.Y.; Hayashi, S.Y.; Schafranski, M.; Messias-Reason, I.J. Further evidence of inflammation in chronic rheumatic valve disease (CRVD): High levels of advanced oxidation protein products (AOPP) and high sensitive C-reactive protein (hs-CRP). Int. J. Cardiol. 2006, 109, 275–276. [Google Scholar] [CrossRef]
- Łuczak, A.; Madej, M.; Kasprzyk, A.; Doroszko, A. Role of the eNOS Uncoupling and the Nitric Oxide Metabolic Pathway in the Pathogenesis of Autoimmune Rheumatic Diseases. Oxid. Med. Cell. Longev. 2020, 2020, 1417981. [Google Scholar] [CrossRef]
- Buie, J.J.; Renaud, L.L.; Muise-Helmericks, R.; Oates, J.C. IFN-α Negatively Regulates the Expression of Endothelial Nitric Oxide Synthase and Nitric Oxide Production: Implications for Systemic Lupus Erythematosus. J. Immunol. 2017, 199, 1979–1988. [Google Scholar] [CrossRef]
- Felger, J.C.; Li, L.; Marvar, P.J.; Woolwine, B.J.; Harrison, D.G.; Raison, C.L.; Miller, A.H. Tyrosine metabolism during interferon-alpha administration: Association with fatigue and CSF dopamine concentrations. Brain Behav. Immun. 2013, 31, 153–160. [Google Scholar] [CrossRef]
- Kitagami, T.; Yamada, K.; Miura, H.; Hashimoto, R.; Nabeshima, T.; Ohta, T. Mechanism of systemically injected interferon-alpha impeding monoamine biosynthesis in rats: Role of nitric oxide as a signal crossing the blood-brain barrier. Brain Res. 2003, 978, 104–114. [Google Scholar] [CrossRef]
- Ursini, F.; Leporini, C.; Bene, F.; D’Angelo, S.; Mauro, D.; Russo, E.; de Sarro, G.; Olivieri, I.; Pitzalis, C.; Lewis, M.; et al. Anti-TNF-alpha agents and endothelial function in rheumatoid arthritis: A systematic review and meta-analysis. Sci. Rep. 2017, 7, 5346. [Google Scholar] [CrossRef]
- Kumar, V.; Ganguly, N.K.; Anand, I.S.; Wahi, P.L. Release of oxygen free radicals by macrophages and neutrophils in patients with rheumatic fever. Eur. Heart J. 1991, 12 (Suppl. D), 163–165. [Google Scholar] [CrossRef] [PubMed]
- RHDAustralia (ARF/RHD Writing Group). The 2020 Australian Guideline for Prevention, Diagnosis and Management of Acute Rheumatic Fever and Rheumatic Heart Disease. Version 3.2. Available online: https://www.rhdaustralia.org.au/system/files/fileuploads/arf_rhd_guidelines_3.2_edition_march_2022.pdf (accessed on 30 November 2022).
- Ralph, A.P.; Currie, B.J. Therapeutics for rheumatic fever and rheumatic heart disease. Aust. Prescr. 2022, 45, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Cilliers, A.; Adler, A.J.; Saloojee, H. Anti-inflammatory treatment for carditis in acute rheumatic fever. Cochrane Database Syst. Rev. 2015, 5, Cd003176. [Google Scholar] [CrossRef] [PubMed]
- Beaton, A.; Okello, E.; Rwebembera, J.; Grobler, A.; Engelman, D.; Alepere, J.; Canales, L.; Carapetis, J.; DeWyer, A.; Lwabi, P.; et al. Secondary Antibiotic Prophylaxis for Latent Rheumatic Heart Disease. N. Engl. J. Med. 2022, 386, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.J.; Concannon, A.; Malcolm, J.; Davidakova, S.; Martin, W.J.; Webb, R.; Moreland, N.J. The Treatment of Acute Rheumatic Fever: Novel Use of Hydroxychloroquine. Pediatr. Infect. Dis. J. 2020, 39, e120–e122. [Google Scholar] [CrossRef]
- WHO. Rheumatic Fever and Rheumatic Heart Disease; Technical Report Series; No. 923; World Health Organization: Geneva, Switzerland, 2004; pp. 1–122. [Google Scholar]
- Li, M.; Xu, S.; Geng, Y.; Sun, L.; Wang, R.; Yan, Y.; Wang, H.; Li, Y.; Yi, Q.; Zhang, Y.; et al. The protective effects of L-carnitine on myocardial ischaemia-reperfusion injury in patients with rheumatic valvular heart disease undergoing CPB surgery are associated with the suppression of NF-κB pathway and the activation of Nrf2 pathway. Clin. Exp. Pharmacol. Physiol. 2019, 46, 1001–1012. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, M.; Tai, Y.; Tao, J.; Zhou, W.; Han, Y.; Wei, W.; Wang, Q. Triggers of Cardiovascular Diseases in Rheumatoid Arthritis. Curr. Probl. Cardiol. 2022, 47, 100853. [Google Scholar] [CrossRef]


{kind=link}
| Population/Type of Material | Main Results | Ref. |
|---|---|---|
| Inflammation | ||
| Mitral valve tissue (n = 28) from chronic RHD patients undergoing valve replacement surgery |
| [2] |
| Peripheral blood mononuclear cells from an Australian Aboriginal ARF |
| [32] |
| Formalin-fixed autopsy specimens from consecutive RHD patients |
| [29] |
| 30 patients with rheumatic heart disease undergoing mitral valve replacement |
| [47] |
| Human PBMCs isolated from heparinized blood of healthy donors |
| [48] |
| Model of autoimmune heart inflammatory disease (myocarditis) |
| [61] |
| 13 valve specimens from nine patients with rheumatic carditis |
| [62] |
| 20 heart tissue infiltrates from 14 RHD patients |
| [63] |
| Patients with ARF and ARHD |
| [65] |
| Surgical fragments obtained during valve correction surgery from 6 severe RHD patients |
| [66] |
| 53 patients with ARF, 78 patients with chronic RHD vs. 20 normal control subjects and 39 patients with USP |
| [67] |
| 30 rheumatic mitral valves and in 15 control valves. |
| [72] |
| 89 with RHD |
| [75] |
| Cardiac tissue biopsies obtained from chronic RHD patients |
| [76] |
| 27 patients with acute rheumatic fever (RF), 12 with only arthritis (RFA) and 15 with rheumatic heart disease (RHD) |
| [79] |
| Forty patients with rheumatic MVD and 23 controls |
| [84] |
| 70 adults of RHD and 35 controls |
| [85] |
| Cardiac tissues of 11 RHD patients and 11 controls |
| [87] |
| Rheumatic heart disease (RHD) patients and healthy controls |
| [88] |
| 80 patients with rheumatic MS: group 1—35 patients with rheumatic mitral stenosis and left atrium; group 2—45 patients with rheumatic mitral stenosis without left atrium |
| [105] |
| 314 patients with RMVS, 57 healthy persons in control group |
| [108] |
| Oxidative stress | ||
| 56 patients who underwent valve replacement due to rheumatic (n = 32) and degenerative (n = 24) heart valve disease. |
| [122] |
| 25 rheumatic HVD paediatric patients and 25 paediatric congenital HVD patients and 20 healthy age-matched control subjects |
| [127] |
| 90 patients with CRVD |
| [128] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franczyk, B.; Gluba-Brzózka, A.; Rysz-Górzyńska, M.; Rysz, J. The Role of Inflammation and Oxidative Stress in Rheumatic Heart Disease. Int. J. Mol. Sci. 2022, 23, 15812. https://doi.org/10.3390/ijms232415812
Franczyk B, Gluba-Brzózka A, Rysz-Górzyńska M, Rysz J. The Role of Inflammation and Oxidative Stress in Rheumatic Heart Disease. International Journal of Molecular Sciences. 2022; 23(24):15812. https://doi.org/10.3390/ijms232415812
Chicago/Turabian StyleFranczyk, Beata, Anna Gluba-Brzózka, Magdalena Rysz-Górzyńska, and Jacek Rysz. 2022. "The Role of Inflammation and Oxidative Stress in Rheumatic Heart Disease" International Journal of Molecular Sciences 23, no. 24: 15812. https://doi.org/10.3390/ijms232415812
APA StyleFranczyk, B., Gluba-Brzózka, A., Rysz-Górzyńska, M., & Rysz, J. (2022). The Role of Inflammation and Oxidative Stress in Rheumatic Heart Disease. International Journal of Molecular Sciences, 23(24), 15812. https://doi.org/10.3390/ijms232415812