
Glioblastoma Molecular Classification Tool Based on mRNA Analysis: From Wet-Lab to Subtype

 , , ,
, , ,
Abstract
1. Introduction
2. Results
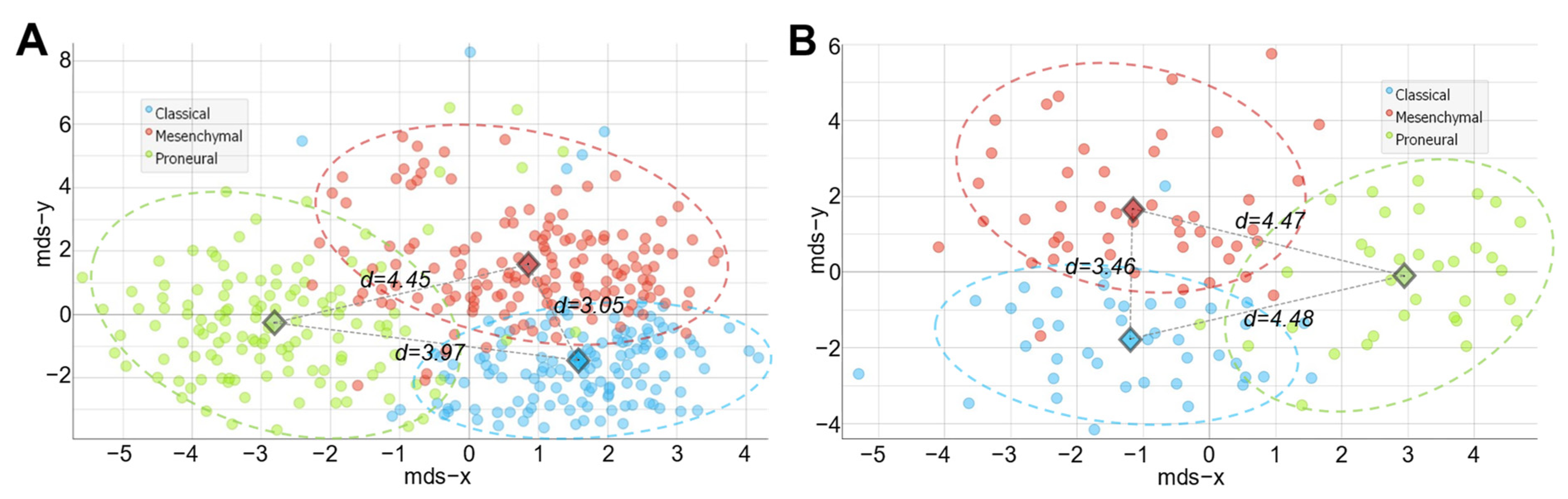
2.1. The Design of Classification-Relevant Markers Selection
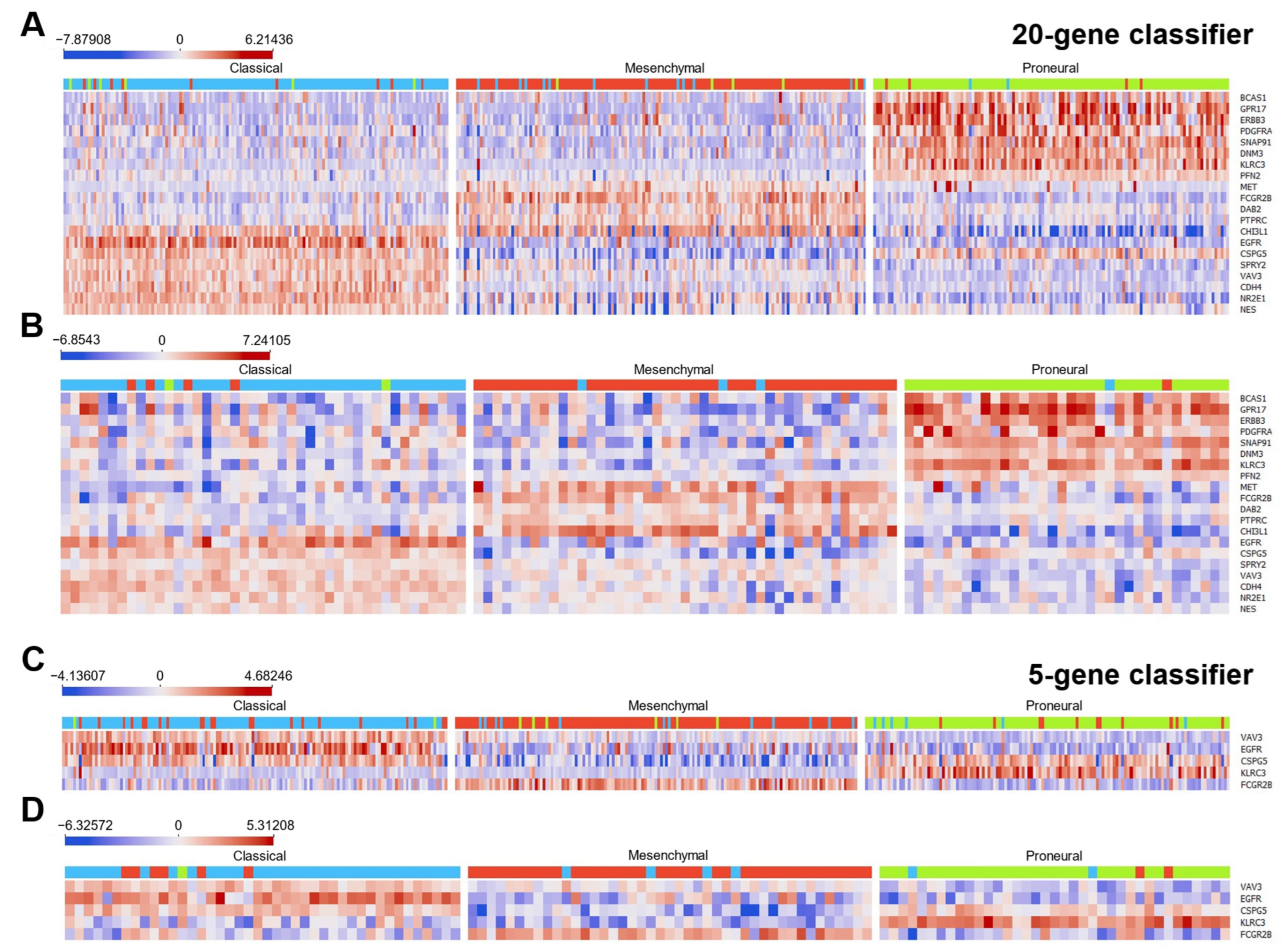
2.2. Classifier Development
2.3. Validation and Testing of Classification Models
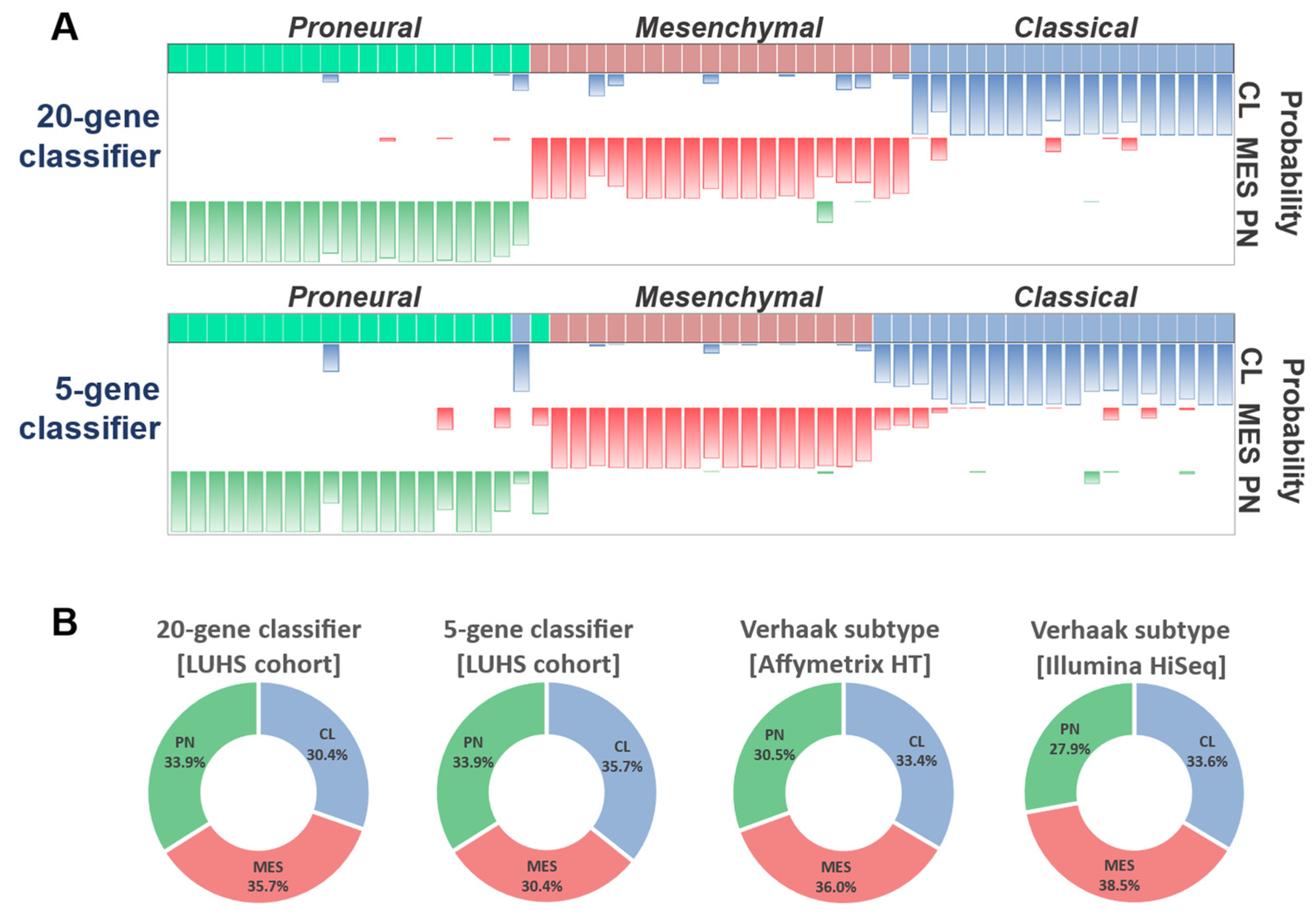
2.4. The Construction of the Gene Expression System Applying Ordinary qPCR for Glioblastoma Subtyping
2.5. Subtype Analysis in LUHS Cohort
3. Discussion
4. Materials and Methods
4.1. TCGA Gene Expression Data Processing
4.2. Patient Samples
4.3. RNA Isolation and qRT-PCR
4.4. Data Analysis
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, P.; Xia, Q.; Liu, L.; Li, S.; Dong, L. Current Opinion on Molecular Characterization for GBM Classification in Guiding Clinical Diagnosis, Prognosis, and Therapy. Front. Mol. Biosci. 2020, 7, 562798. [Google Scholar] [CrossRef]
- Lee, E.; Yong, R.L.; Paddison, P.; Zhu, J. Comparison of Glioblastoma (GBM) Molecular Classification Methods. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Paolillo, M.; Boselli, C.; Schinelli, S. Glioblastoma under Siege: An Overview of Current Therapeutic Strategies. Brain Sci. 2018, 8, 15. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular Subclasses of High-Grade Glioma Predict Prognosis, Delineate a Pattern of Disease Progression, and Resemble Stages in Neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG Island Methylator Phenotype That Defines a Distinct Subgroup of Glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef]
- Shen, R.; Mo, Q.; Schultz, N.; Seshan, V.E.; Olshen, A.B.; Huse, J.; Ladanyi, M.; Sander, C. Integrative Subtype Discovery in Glioblastoma Using ICluster. PLoS ONE 2012, 7, e35236. [Google Scholar] [CrossRef]
- Park, J.; Shim, J.K.; Yoon, S.J.; Kim, S.H.; Chang, J.H.; Kang, S.G. Transcriptome Profiling-Based Identification of Prognostic Subtypes and Multi-Omics Signatures of Glioblastoma. Sci. Rep. 2019, 9, 10555. [Google Scholar] [CrossRef]
- Teo, W.Y.; Sekar, K.; Seshachalam, P.; Shen, J.; Chow, W.Y.; Lau, C.C.; Yang, H.K.; Park, J.; Kang, S.G.; Li, X.; et al. Relevance of a TCGA-Derived Glioblastoma Subtype Gene-Classifier among Patient Populations. Sci. Rep. 2019, 9, 7442. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef]
- Madurga, R.; García-Romero, N.; Jiménez, B.; Collazo, A.; Pérez-Rodríguez, F.; Hernández-Laín, A.; Fernández-Carballal, C.; Prat-Acín, R.; Zanin, M.; Menasalvas, E.; et al. Normal Tissue Content Impact on the GBM Molecular Classification. Brief. Bioinform. 2020, 22, bbaa129. [Google Scholar] [CrossRef]
- Park, A.K.; Kim, P.; Ballester, L.Y.; Esquenazi, Y.; Zhao, Z. Subtype-Specific Signaling Pathways and Genomic Aberrations Associated with Prognosis of Glioblastoma. Neuro Oncol. 2019, 21, 59–70. [Google Scholar] [CrossRef]
- Erdem-Eraslan, L.; van den Bent, M.J.; Hoogstrate, Y.; Naz-Khan, H.; Stubbs, A.; van der Spek, P.; Bottcher, R.; Gao, Y.; de Wit, M.; Taal, W.; et al. Identification of Patients with Recurrent Glioblastoma Who May Benefit from Combined Bevacizumab and CCNU Therapy: A Report from the BELOB Trial. Cancer Res. 2016, 76, 525–534. [Google Scholar] [CrossRef]
- Chen, R.; Smith-Cohn, M.; Cohen, A.L.; Colman, H. Glioma Subclassifications and Their Clinical Significance. Neurotherapeutics 2017, 14, 284–297. [Google Scholar] [CrossRef]
- Zhao, R.; Pan, Z.; Li, B.; Zhao, S.; Zhang, S.; Qi, Y.; Qiu, J.; Gao, Z.; Fan, Y.; Guo, Q.; et al. Comprehensive Analysis of the Tumor Immune Microenvironment Landscape in Glioblastoma Reveals Tumor Heterogeneity and Implications for Prognosis and Immunotherapy. Front. Immunol. 2022, 13, 820673. [Google Scholar] [CrossRef]
- Bianconi, A.; Aruta, G.; Rizzo, F.; Salvati, L.F.; Zeppa, P.; Garbossa, D.; Cofano, F. Systematic Review on Tumor Microenvironment in Glial Neoplasm: From Understanding Pathogenesis to Future Therapeutic Perspectives. Int. J. Mol. Sci. 2022, 23, 4166. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, N.; Regev, A.; He, S.; Hemann, M.T. Integrated Regulatory Models for Inference of Subtype-specific Susceptibilities in Glioblastoma. Mol. Syst. Biol. 2020, 16, e9506. [Google Scholar] [CrossRef]
- Vessières, A.; Quissac, E.; Lemaire, N.; Alentorn, A.; Domeracka, P.; Pigeon, P.; Sanson, M.; Idbaih, A.; Verreault, M. Heterogeneity of Response to Iron-Based Metallodrugs in Glioblastoma Is Associated with Differences in Chemical Structures and Driven by FAS Expression Dynamics and Transcriptomic Subtypes. Int. J. Mol. Sci. 2021, 22, 10404. [Google Scholar] [CrossRef]
- Pal, S.; Bi, Y.; MacYszyn, L.; Showe, L.C.; O’Rourke, D.M.; Davuluri, R.V. Isoform-Level Gene Signature Improves Prognostic Stratification and Accurately Classifies Glioblastoma Subtypes. Nucleic Acids Res. 2014, 42, e64. [Google Scholar] [CrossRef][Green Version]
- Crisman, T.J.; Zelaya, I.; Laks, D.R.; Zhao, Y.; Kawaguchi, R.; Gao, F.; Kornblum, H.I.; Coppola, G. Identification of an Efficient Gene Expression Panel for Glioblastoma Classification. PLoS ONE 2016, 11, e0164649. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.; Guan, G.; Zhao, W.; Zhuang, M. A Risk Classification System With Five-Gene for Survival Prediction of Glioblastoma Patients. Front. Neurol. 2019, 10, 745. [Google Scholar] [CrossRef]
- Brennan, C.; Momota, H.; Hambardzumyan, D.; Ozawa, T.; Tandon, A.; Pedraza, A.; Holland, E. Glioblastoma Subclasses Can Be Defined by Activity among Signal Transduction Pathways and Associated Genomic Alterations. PLoS ONE 2009, 4, e7752. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Gill, B.J.; Pisapia, D.J.; Malone, H.R.; Goldstein, H.; Lei, L.; Sonabend, A.; Yun, J.; Samanamud, J.; Sims, J.S.; Banu, M.; et al. MRI-Localized Biopsies Reveal Subtype-Specific Differences in Molecular and Cellular Composition at the Margins of Glioblastoma. Proc. Natl. Acad. Sci. USA 2014, 111, 12550–12555. [Google Scholar] [CrossRef]
- Sandmann, T.; Bourgon, R.; Garcia, J.; Li, C.; Cloughesy, T.; Chinot, O.L.; Wick, W.; Nishikawa, R.; Mason, W.; Henriksson, R.; et al. Patients With Proneural Glioblastoma May Derive Overall Survival Benefit From the Addition of Bevacizumab to First-Line Radiotherapy and Temozolomide: Retrospective Analysis of the AVAglio Trial. J. Clin. Oncol. 2015, 33, 2735–2744. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Sander, C.; Stuart, J.M.; Chang, K.; Creighton, C.J.; et al. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and Interpreting Cancer Genomics Data via the Xena Platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.W.; Squatrito, M. GlioVis Data Portal for Visualization and Analysis of Brain Tumor Expression Datasets. Neuro Oncol. 2017, 19, 139–141. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classifier Development | ||||||
| Dataset: Agilent G4502 | Logistic regression | SVM | ||||
| 20-gene model | 5 top ranked genes model | 5 selected genes model | 20-gene model | 5 top ranked genes model | 5 selected genes model | |
| Classification accuracy | 0.948 | 0.877 | 0.937 | 0.948 | 0.877 | 0.932 |
| Area under ROC curve (AUC) | 0.995 | 0.888 | 0.995 | 0.994 | 0.888 | 0.994 |
| Classifier testing | ||||||
| Dataset: Affymetrix HT | ||||||
| Classification accuracy | 0.907 | 0.833 | 0.859 | 0.914 | 0.835 | 0.859 |
| Area under ROC curve (AUC) | 0.967 | 0.933 | 0.946 | 0.986 | 0.946 | 0.961 |
| Dataset: Illumina HiSeq 2000 | ||||||
| Classification accuracy | 0.91 | 0.885 | 0.877 | 0.893 | 0.869 | 0.868 |
| Area under ROC curve (AUC) | 0.998 | 0.991 | 0.992 | 0.987 | 0.977 | 0.985 |
| Gene Name | Forward Primer 5′ --> 3′ | Revers Primer 5′ --> 3′ | Amplicon Lenght, bp | Primer Amount µM | Annealing Temp., °C | ||
|---|---|---|---|---|---|---|---|
| 20-gene classifier | 5-gene class. | CSPG5 | CTCTACCTGCTCAAGACGGA | GCACTAGGATCATCATTTGGGT | 133 | 0.33 | 62 |
| EGFR | GGACCAGACAACTGTATCCA | AAGATTTATTAGGACCCGTAGGTG | 172 | 0.67 | 60 | ||
| FCGR2B | CTGTGCTTTCTGAGTGGCTG | TGACTGTGGTTTGCTTGTGG | 189 | 0.29 | 62 | ||
| KLRC3 | ATATGACTGCCAAGGTTTACTG | CTCTTCCCAAGTTCTTCTTTCC | 246 | 0.29 | 60 | ||
| VAV3 | CTCAAACTACCAGAGAAACGGAC | ATCTCCTTTCAGAAGTTCAACGG | 176 | 0.33 | 60 | ||
| BCAS1 | AGACAAATGACATCAGACTCCA | CTTCTGCTTGTTCATCTCGG | 131 | 0.42 | 60 | ||
| CDH4 | CCGTCCCAGAATATGTTCAC | GCCATAGTTGAGATTTCCTTCC | 137 | 0.42 | 58 | ||
| CHI3L1 | GTCTCAAACAGGCTTTGTGG | GTAGATGATGTGGGTACAGAGG | 153 | 0.42 | 60 | ||
| DAB2 | CAGTTGAGAATGGGAGTGAGG | GTGGGAAAGAAGTTGAGATTGG | 240 | 0.33 | 54 | ||
| DNM3 | TCCTCAAGGTCTGAGAACCA | GTCCTTCTTCCCATCTATGTCC | 159 | 0.42 | 60 | ||
| ERBB3 | ATGCTGAGAACCAATACCAGAC | CAAACTTCCCATCGTAGACCT | 255 | 0.42 | 60 | ||
| GPR17 | AGCAGCTAGAGGATGTCCA | TGGAGTCAGAGCCTGAGAG | 87 | 0.29 | 60 | ||
| MET | CACTGCTTTAATAGGACACTTCTG | AGGTGGATATAGATGTTAAGAGGAC | 147 | 0.42 | 60 | ||
| NES | GTTGGAACAGAGGTTGGAGG | AAAGCTGAGGGAAGTCTTGG | 173 | 0.42 | 60 | ||
| NR2E1 | TCAAGTGGGCTAAGAGTGTG | ACCGTTCATGCCAGATACAG | 160 | 0.29 | 60 | ||
| PDGFRA | ACAACCTCTACACCACACTG | ATGATCTCGTAGACTTCACTGG | 180 | 0.29 | 60 | ||
| PFN2 | GTTTCTTTACCAACGGTTTGAC | CATGACTATAACCAATGCTCTACC | 169 | 0.42 | 60 | ||
| PTPRC | TAAGACAACAGTGGAGAAAGGAC | CAAATGCCAAGAGTTTAAGCCA | 96 | 0.42 | 60 | ||
| SNAP91 | CCCAGTCAGCACTTCTAAACC | CAGCCAAAGAATCCTCTCCC | 154 | 0.42 | 60 | ||
| SPRY2 | GGAAGTTGGTCTAAAGCAGAGG | CACATCTGAACTCCGTGATCG | 137 | 0.29 | 60 | ||
| Endogenous contr. | ACTB | AGAGCTACGAGCTGCCTGAC | AGCACTGTGTTGGCGTACAG | 184 | 0.083 | 60 | |
| GAPDH | TCAAGATCATCAGCAATGCCT | CATGAGTCCTTCCACGATACC | 94 | 0.42 | 60 | ||
| YWHAZ | CCGTTACTTGGCTGAGGTTG | TGCTTGTTGTGACTGATCGAC | 67 | 0.42 | 62 | ||
| Features | LUHS Cohort n = 56 | Affymetrix HG-U133a n = 419 | Illumina HiSeq 2000 n = 122 |
|---|---|---|---|
| Gender | |||
| Female | 29 (51.8%) | 166 (39.6%) | 47 (38.5%) |
| Male | 27 (48.2%) | 253 (60.4%) | 75 (61.5%) |
| Age (years) | mean 58.66 | mean 58.2 | mean 60 |
| ≤60 | 30 (53.6%) | 216 (51.6%) | 53 (53.4%) |
| >60 | 26 (46.4%) | 203 (48.4%) | 69 (56.6%) |
| Survival (months) | mean 17.78 | mean 14.51 | mean 11.3 |
| ≤12 | 20 (35.7%) | 242 (57.7%) | 72 (59%) |
| >12 | 36 (64.3%) | 177 (42.3%) | 50 (41%) |
| IDH1 mutation | Unexplored n = 107 | Unexplored n = 13 | |
| Wild-type | 50 (89.3%) | 286 (91.6%) | 101 (92.7%) |
| Mutant | 6 (10.7%) | 26 (8.4%) | 8 (7.3%) |
| MGMT methylation | Unexplored n = 146 | Unexplored n = 34 | |
| Unmeth | 28 (50%) | 137 (50.2%) | 46 (52.3%) |
| Meth | 28 (50%) | 136 (49.8%) | 42 (47.7%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steponaitis, G.; Kucinskas, V.; Golubickaite, I.; Skauminas, K.; Saudargiene, A. Glioblastoma Molecular Classification Tool Based on mRNA Analysis: From Wet-Lab to Subtype. Int. J. Mol. Sci. 2022, 23, 15875. https://doi.org/10.3390/ijms232415875
Steponaitis G, Kucinskas V, Golubickaite I, Skauminas K, Saudargiene A. Glioblastoma Molecular Classification Tool Based on mRNA Analysis: From Wet-Lab to Subtype. International Journal of Molecular Sciences. 2022; 23(24):15875. https://doi.org/10.3390/ijms232415875
Chicago/Turabian StyleSteponaitis, Giedrius, Vytautas Kucinskas, Ieva Golubickaite, Kestutis Skauminas, and Ausra Saudargiene. 2022. "Glioblastoma Molecular Classification Tool Based on mRNA Analysis: From Wet-Lab to Subtype" International Journal of Molecular Sciences 23, no. 24: 15875. https://doi.org/10.3390/ijms232415875
APA StyleSteponaitis, G., Kucinskas, V., Golubickaite, I., Skauminas, K., & Saudargiene, A. (2022). Glioblastoma Molecular Classification Tool Based on mRNA Analysis: From Wet-Lab to Subtype. International Journal of Molecular Sciences, 23(24), 15875. https://doi.org/10.3390/ijms232415875