
Extracellular Events Involved in Cancer Cell–Cell Fusion



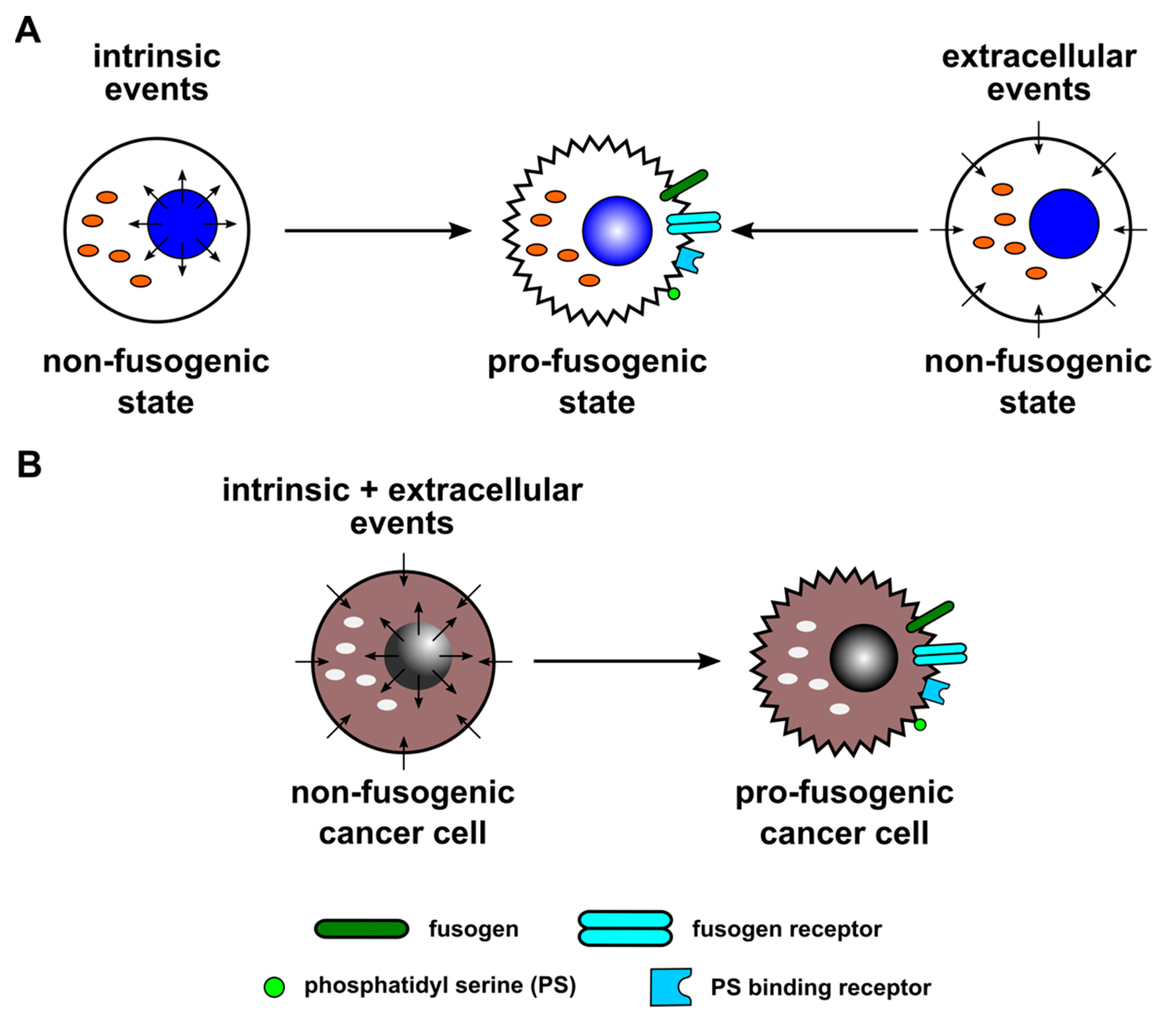
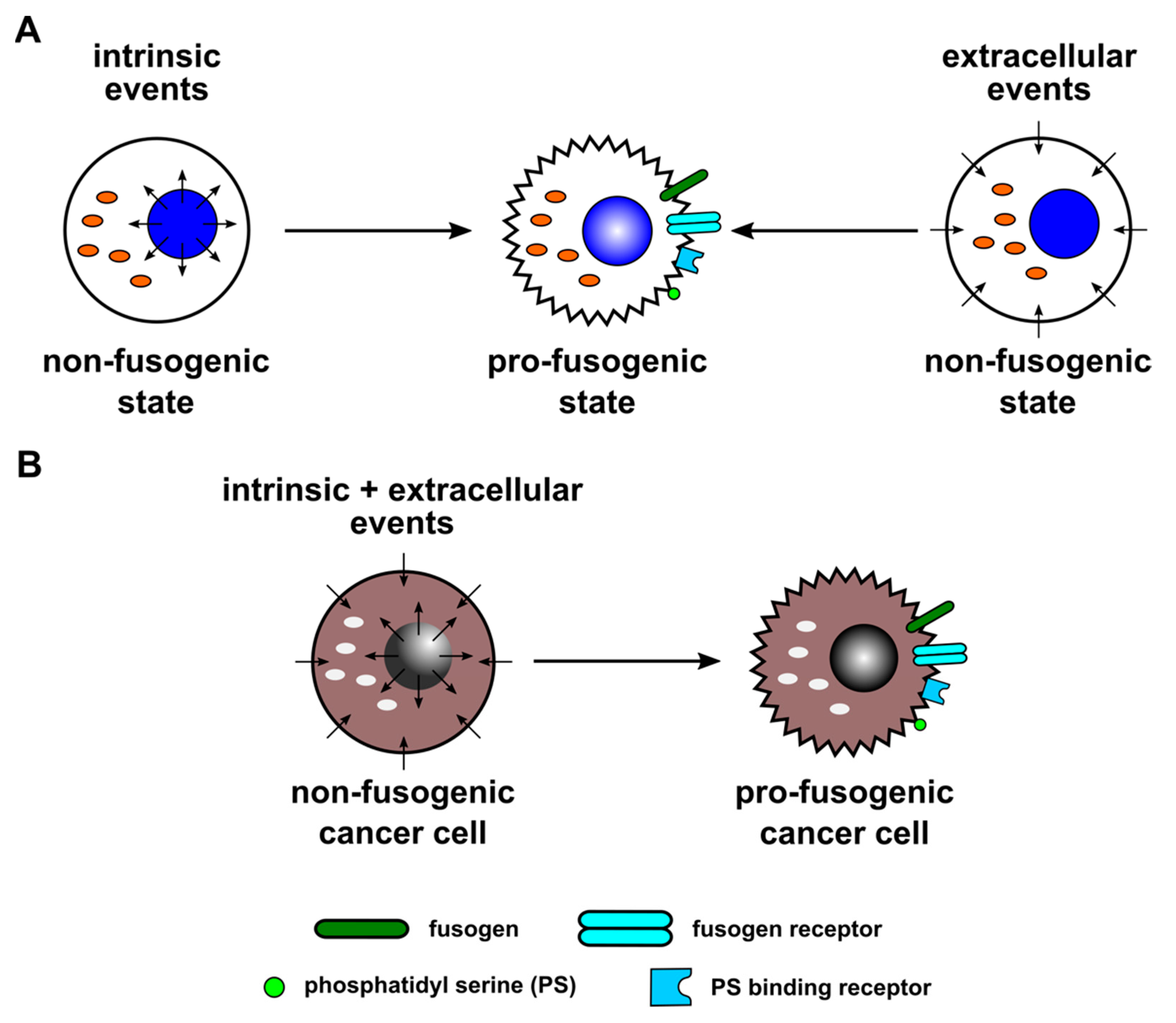{kind=link}
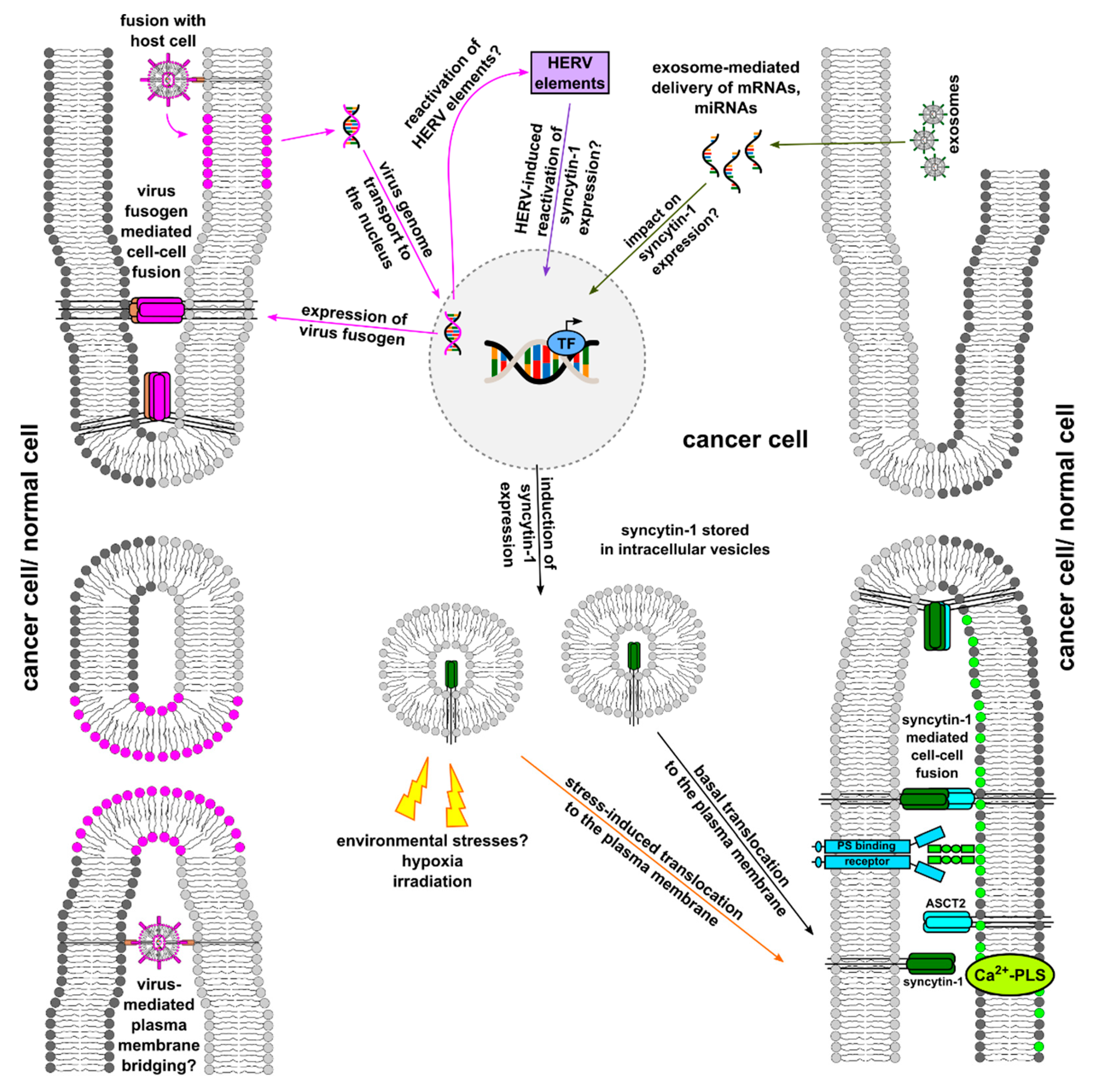
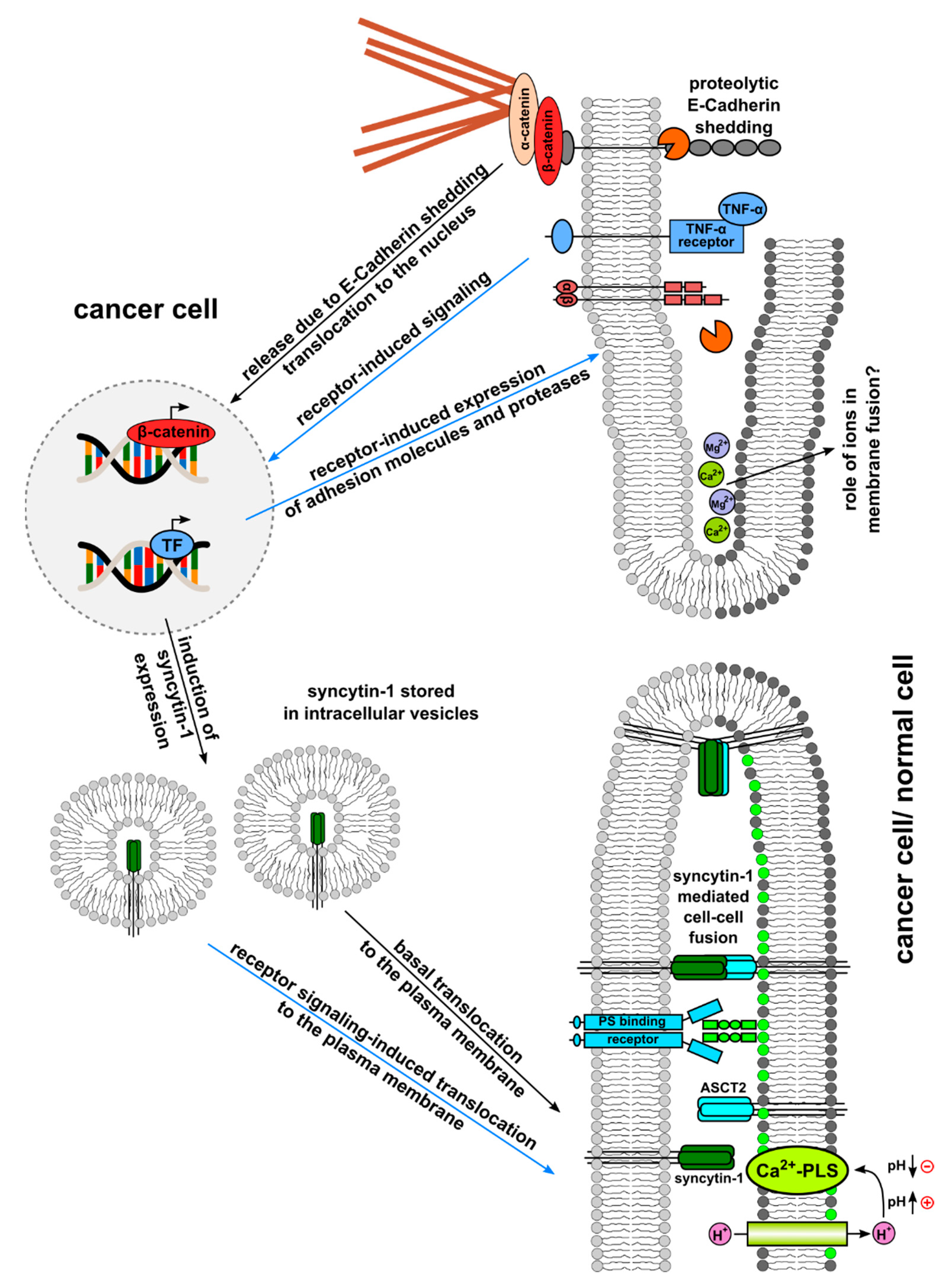{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Brief Facts of the Cell–Cell Fusion Machinery
1.2. Physiological and Non-Physiological Cell–Cell Fusion
1.3. Known Triggers for Cell–Cell Fusion
2. Inflammation/Inflammatory Cytokines in Cell Fusion
2.1. Role of TNF-α in Cell–Cell Fusion
2.2. Impact of Proteases and β-Catenin in Cell–Cell Fusion
3. Effects of pH and Ions in Cell Fusion
4. Virus-Mediated or -Associated Cancer Cell–Cell Fusion
4.1. Viruses as Bridgebuilders for Cell–Cell Merger
4.2. Viral Infection Induced Reactivation of HERV env Elements
4.2.1. HERV-K (HML-2) and Cancer
4.2.2. Reactivation of HERV Elements Due to Viral Infections
5. Role of Hypoxia in Cell Fusion
6. Exosomes/EVs in Cell Fusion
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ASCT2 | alanine, serine, cysteine transporter 2 (syncytin-1 receptor) |
| BMDCs | bone-marrow-derived cells |
| EMT | epithelial–mesenchymal transition |
| Ca2+-PLS | Ca2+-activated phospholipid scramblases |
| EBV | Epstein–Barr virus |
| EMT | epithelial-to-mesenchymal transition |
| EVs | extracellular vesicles |
| GCM1 | glial cell missing-2 |
| HBV | hepatitis B virus |
| HERV | human endogenous retroviral |
| HERV-FRD | human endogenous retrovirus-type FRD (syncytin-2) |
| HER2-W | human endogenous retrovirus-type W (syncytin-1) |
| HUVECs | human umbilical vein endothelial cells |
| HIF-1α | hypoxia-inducible factors-1α |
| HIV | human immunodeficiency virus |
| IL-6 | interleukin-6 |
| KSHV | Karposi’s sarcoma herpes virus |
| MMP-9 | matrix metalloproteinase-9 |
| MSC | mesenchymal stroma-/stem-like cells |
| OSCCs | oral squamous carcinoma cells |
| PHSP | post-hybrid selection process |
| PS | phosphatidylserine |
| PC | phosphatidylcholine |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus-2 |
| SCC-9 | squamous cell carcinoma cells 9 |
| SFV | Semliki Forest virus |
| TCF4 | T-cell factor 4 |
| TME | tumor microenvironment |
| TMEM16F | transmembrane member 16F |
| TNF-α | tumor necrosis factor-α |
| VCAM-1 | vascular cell adhesion molecule-1 |
| VLA-4 | very late antigen-4 |
| VSV | vesicular stomatitis virus |
References
- Dittmar, T.; Weiler, J.; Luo, T.; Hass, R. Cell-Cell Fusion Mediated by Viruses and HERV-Derived Fusogens in Cancer Initiation and Progression. Cancers 2021, 13, 5363. [Google Scholar] [CrossRef] [PubMed]
- Hass, R.; von der Ohe, J.; Dittmar, T. Hybrid Formation and Fusion of Cancer Cells In Vitro and In Vivo. Cancers 2021, 13, 4496. [Google Scholar] [CrossRef] [PubMed]
- Hass, R.; von der Ohe, J.; Dittmar, T. Cancer Cell Fusion and Post-Hybrid Selection Process (PHSP). Cancers 2021, 13, 4636. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, J.M.; Podbilewicz, B. The hallmarks of cell-cell fusion. Development 2017, 144, 4481–4495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podbilewicz, B. Virus and cell fusion mechanisms. Annu. Rev. Cell Dev. Biol. 2014, 30, 111–139. [Google Scholar] [CrossRef] [Green Version]
- Whitlock, J.M.; Chernomordik, L.V. Flagging fusion: Phosphatidylserine signaling in cell-cell fusion. J. Biol. Chem. 2021, 296, 100411. [Google Scholar] [CrossRef]
- Willkomm, L.; Bloch, W. State of the art in cell-cell fusion. In Cell Fusion; Methods in Molecular Biology; Springer: Cham, Switzerland, 2015; Volume 1313, pp. 1–19. [Google Scholar] [CrossRef]
- Aguilar, P.S.; Baylies, M.K.; Fleissner, A.; Helming, L.; Inoue, N.; Podbilewicz, B.; Wang, H.; Wong, M. Genetic basis of cell-cell fusion mechanisms. Trends Genet. 2013, 29, 427–437. [Google Scholar] [CrossRef] [Green Version]
- Helming, L.; Gordon, S. Molecular mediators of macrophage fusion. Trends Cell Biol. 2009, 19, 514–522. [Google Scholar] [CrossRef]
- Brukman, N.G.; Uygur, B.; Podbilewicz, B.; Chernomordik, L.V. How cells fuse. J. Cell Biol. 2019, 218, 1436–1451. [Google Scholar] [CrossRef]
- Petrany, M.J.; Millay, D.P. Cell Fusion: Merging Membranes and Making Muscle. Trends Cell Biol. 2019, 29, 964–973. [Google Scholar] [CrossRef]
- Gao, S.; Hu, J. Mitochondrial Fusion: The Machineries In and Out. Trends Cell Biol. 2021, 31, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Martens, S.; McMahon, H.T. Mechanisms of membrane fusion: Disparate players and common principles. Nat. Rev. Mol. Cell Biol. 2008, 9, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Margam, N.N. Structural Insights into Membrane Fusion Mediated by Convergent Small Fusogens. Cells 2021, 10, 160. [Google Scholar] [CrossRef]
- Ogle, B.M.; Cascalho, M.; Platt, J.L. Biological implications of cell fusion. Nat. Rev. Mol. Cell Biol. 2005, 6, 567–575. [Google Scholar] [CrossRef]
- Melzer, C.; von der Ohe, J.; Hass, R. In vitro fusion of normal and neoplastic breast epithelial cells with human mesenchymal stroma/stem cells (MSC) partially involves TNF receptor signaling. Stem Cells 2018, 36, 977–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melzer, C.; von der Ohe, J.; Hass, R. Enhanced metastatic capacity of breast cancer cells after interaction and hybrid formation with mesenchymal stroma/stem cells (MSC). Cell Commun. Signal. 2018, 16, 2. [Google Scholar] [CrossRef] [Green Version]
- Hass, R.; von der Ohe, J.; Ungefroren, H. Potential Role of MSC/Cancer Cell Fusion and EMT for Breast Cancer Stem Cell Formation. Cancers 2019, 11, 1432. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Otte, A.; Hass, R. Human mesenchymal stroma/stem cells exchange membrane proteins and alter functionality during interaction with different tumor cell lines. Stem Cells Dev. 2015, 24, 1205–1222. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Zhu, Y.; Sun, Z.; Ji, R.; Zhang, X.; Xu, W.; Yuan, X.; Zhang, B.; Yan, Y.; Yin, L.; et al. Tumorigenic hybrids between mesenchymal stem cells and gastric cancer cells enhanced cancer proliferation, migration and stemness. BMC Cancer 2015, 15, 793. [Google Scholar] [CrossRef]
- Wei, H.J.; Nickoloff, J.A.; Chen, W.H.; Liu, H.Y.; Lo, W.C.; Chang, Y.T.; Yang, P.C.; Wu, C.W.; Williams, D.F.; Gelovani, J.G.; et al. FOXF1 mediates mesenchymal stem cell fusion-induced reprogramming of lung cancer cells. Oncotarget 2014, 5, 9514–9529. [Google Scholar] [CrossRef] [Green Version]
- Melzer, C.; von der Ohe, J.; Hass, R. MSC stimulate ovarian tumor growth during intercellular communication but reduce tumorigenicity after fusion with ovarian cancer cells. Cell Commun. Signal. 2018, 16, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melzer, C.; von der Ohe, J.; Lehnert, H.; Ungefroren, H.; Hass, R. Cancer stem cell niche models and contribution by mesenchymal stroma/stem cells. Mol. Cancer 2017, 16, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hass, R.; Kasper, C.; Bohm, S.; Jacobs, R. Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell Commun. Signal. 2011, 9, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hass, R.; Otte, A. Mesenchymal stem cells as all-round supporters in a normal and neoplastic microenvironment. Cell Commun. Signal. 2012, 10, 26. [Google Scholar] [CrossRef] [Green Version]
- Melzer, C.; Yang, Y.; Hass, R. Interaction of MSC with tumor cells. Cell Commun. Signal. 2016, 14, 20. [Google Scholar] [CrossRef] [Green Version]
- Hass, R. Role of MSC in the Tumor Microenvironment. Cancers 2020, 12, 2107. [Google Scholar] [CrossRef]
- Melzer, C.; Ohe, J.V.; Luo, T.; Hass, R. Spontaneous Fusion of MSC with Breast Cancer Cells Can Generate Tumor Dormancy. Int. J. Mol. Sci. 2021, 22, 5930. [Google Scholar] [CrossRef]
- Melzer, C.; Ohe, J.V.; Hass, R. Altered Tumor Plasticity after Different Cancer Cell Fusions with MSC. Int. J. Mol. Sci. 2020, 21, 8347. [Google Scholar] [CrossRef]
- Hass, R.; von der Ohe, J.; Ungefroren, H. Impact of the Tumor Microenvironment on Tumor Heterogeneity and Consequences for Cancer Cell Plasticity and Stemness. Cancers 2020, 12, 3716. [Google Scholar] [CrossRef]
- Kreye, J.; Reincke, S.M.; Kornau, H.C.; Sanchez-Sendin, E.; Corman, V.M.; Liu, H.; Yuan, M.; Wu, N.C.; Zhu, X.; Lee, C.D.; et al. A Therapeutic Non-self-reactive SARS-CoV-2 Antibody Protects from Lung Pathology in a COVID-19 Hamster Model. Cell 2020, 183, 1058–1069. [Google Scholar] [CrossRef]
- Sugimoto, J.; Sugimoto, M.; Bernstein, H.; Jinno, Y.; Schust, D. A novel human endogenous retroviral protein inhibits cell-cell fusion. Sci. Rep. 2013, 3, 1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, P.; Ramirez-Martinez, A.; Li, H.; Cannavino, J.; McAnally, J.R.; Shelton, J.M.; Sanchez-Ortiz, E.; Bassel-Duby, R.; Olson, E.N. Control of muscle formation by the fusogenic micropeptide myomixer. Science 2017, 356, 323–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyado, K.; Yoshida, K.; Yamagata, K.; Sakakibara, K.; Okabe, M.; Wang, X.; Miyamoto, K.; Akutsu, H.; Kondo, T.; Takahashi, Y.; et al. The fusing ability of sperm is bestowed by CD9-containing vesicles released from eggs in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 12921–12926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, N.; Ikawa, M.; Isotani, A.; Okabe, M. The immunoglobulin superfamily protein Izumo is required for sperm to fuse with eggs. Nature 2005, 434, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Satouh, Y.; Nishimasu, H.; Kurabayashi, A.; Morita, J.; Fujihara, Y.; Oji, A.; Ishitani, R.; Ikawa, M.; Nureki, O. Structural and functional insights into IZUMO1 recognition by JUNO in mammalian fertilization. Nat. Commun. 2016, 7, 12198. [Google Scholar] [CrossRef] [Green Version]
- Abmayr, S.M.; Pavlath, G.K. Myoblast fusion: Lessons from flies and mice. Development 2012, 139, 641–656. [Google Scholar] [CrossRef] [Green Version]
- Melzer, C.; von der Ohe, J.; Hass, R. Involvement of Actin Cytoskeletal Components in Breast Cancer Cell Fusion with Human Mesenchymal Stroma/Stem-Like Cells. Int. J. Mol. Sci. 2019, 20, 876. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Platt, J.L. Molecular and cellular mechanisms of Mammalian cell fusion. Adv. Exp. Med. Biol. 2011, 713, 33–64. [Google Scholar] [CrossRef]
- Ferrand, J.; Noel, D.; Lehours, P.; Prochazkova-Carlotti, M.; Chambonnier, L.; Menard, A.; Megraud, F.; Varon, C. Human bone marrow-derived stem cells acquire epithelial characteristics through fusion with gastrointestinal epithelial cells. PLoS ONE 2011, 6, e19569. [Google Scholar] [CrossRef]
- Kemp, K.C.; Dey, R.; Verhagen, J.; Scolding, N.J.; Usowicz, M.M.; Wilkins, A. Aberrant cerebellar Purkinje cell function repaired in vivo by fusion with infiltrating bone marrow-derived cells. Acta Neuropathol. 2018, 135, 907–921. [Google Scholar] [CrossRef] [Green Version]
- Quintana-Bustamante, O.; Alvarez-Barrientos, A.; Kofman, A.V.; Fabregat, I.; Bueren, J.A.; Theise, N.D.; Segovia, J.C. Hematopoietic mobilization in mice increases the presence of bone marrow-derived hepatocytes via in vivo cell fusion. Hepatology 2006, 43, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Vassilopoulos, G.; Wang, P.R.; Russell, D.W. Transplanted bone marrow regenerates liver by cell fusion. Nature 2003, 422, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Fei, F.; Li, C.; Wang, X.; Du, J.; Liu, K.; Li, B.; Yao, P.; Li, Y.; Zhang, S. Syncytin 1, CD9, and CD47 regulating cell fusion to form PGCCs associated with cAMP/PKA and JNK signaling pathway. Cancer Med. 2019, 8, 3047–3058. [Google Scholar] [CrossRef] [PubMed]
- Strick, R.; Ackermann, S.; Langbein, M.; Swiatek, J.; Schubert, S.W.; Hashemolhosseini, S.; Koscheck, T.; Fasching, P.A.; Schild, R.L.; Beckmann, M.W.; et al. Proliferation and cell-cell fusion of endometrial carcinoma are induced by the human endogenous retroviral Syncytin-1 and regulated by TGF-beta. J. Mol. Med. 2007, 85, 23–38. [Google Scholar] [CrossRef]
- Yan, T.L.; Wang, M.; Xu, Z.; Huang, C.M.; Zhou, X.C.; Jiang, E.H.; Zhao, X.P.; Song, Y.; Song, K.; Shao, Z.; et al. Up-regulation of syncytin-1 contributes to TNF-alpha-enhanced fusion between OSCC and HUVECs partly via Wnt/beta-catenin-dependent pathway. Sci. Rep. 2017, 7, 40983. [Google Scholar] [CrossRef] [Green Version]
- Uygur, B.; Leikina, E.; Melikov, K.; Villasmil, R.; Verma, S.K.; Vary, C.P.H.; Chernomordik, L.V. Interactions with Muscle Cells Boost Fusion, Stemness, and Drug Resistance of Prostate Cancer Cells. Mol. Cancer Res. 2019, 17, 806–820. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Xu, J.; Wen, F.; Yang, F.; Li, X.; Geng, D.; Li, L.; Chen, J.; Zheng, J. Upregulation of syncytin-1 promotes invasion and metastasis by activating epithelial-mesenchymal transition-related pathway in endometrial carcinoma. OncoTargets Ther. 2019, 12, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Bjerregaard, B.; Holck, S.; Christensen, I.J.; Larsson, L.I. Syncytin is involved in breast cancer-endothelial cell fusions. Cell. Mol. Life Sci. 2006, 63, 1906–1911. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Zhuang, X.; Xia, X.; Li, X.; Xiao, K.; Liu, X. Correlation Between Promoter Hypomethylation and Increased Expression of Syncytin-1 in Non-Small Cell Lung Cancer. Int. J. Gen. Med. 2021, 14, 957–965. [Google Scholar] [CrossRef]
- Chignola, R.; Sega, M.; Molesini, B.; Baruzzi, A.; Stella, S.; Milotti, E. Collective radioresistance of T47D breast carcinoma cells is mediated by a Syncytin-1 homologous protein. PLoS ONE 2019, 14, e0206713. [Google Scholar] [CrossRef] [Green Version]
- Davies, P.S.; Powell, A.E.; Swain, J.R.; Wong, M.H. Inflammation and proliferation act together to mediate intestinal cell fusion. PLoS ONE 2009, 4, e6530. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.B.; Youssef, S.; Koleckar, K.; Holbrook, C.; Doyonnas, R.; Corbel, S.Y.; Steinman, L.; Rossi, F.M.; Blau, H.M. Extensive fusion of haematopoietic cells with Purkinje neurons in response to chronic inflammation. Nat. Cell Biol. 2008, 10, 575–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nygren, J.M.; Liuba, K.; Breitbach, M.; Stott, S.; Thoren, L.; Roell, W.; Geisen, C.; Sasse, P.; Kirik, D.; Bjorklund, A.; et al. Myeloid and lymphoid contribution to non-haematopoietic lineages through irradiation-induced heterotypic cell fusion. Nat. Cell Biol. 2008, 10, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Hotokezaka, H.; Sakai, E.; Ohara, N.; Hotokezaka, Y.; Gonzales, C.; Matsuo, K.; Fujimura, Y.; Yoshida, N.; Nakayama, K. Molecular analysis of RANKL-independent cell fusion of osteoclast-like cells induced by TNF-alpha, lipopolysaccharide, or peptidoglycan. J. Cell. Biochem. 2007, 101, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Skokos, E.A.; Charokopos, A.; Khan, K.; Wanjala, J.; Kyriakides, T.R. Lack of TNF-alpha-induced MMP-9 production and abnormal E-cadherin redistribution associated with compromised fusion in MCP-1-null macrophages. Am. J. Pathol. 2011, 178, 2311–2321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiler, J.; Dittmar, T. Minocycline impairs TNF-alpha-induced cell fusion of M13SV1-Cre cells with MDA-MB-435-pFDR1 cells by suppressing NF-kappaB transcriptional activity and its induction of target-gene expression of fusion-relevant factors. Cell Commun. Signal. 2019, 17, 71. [Google Scholar] [CrossRef] [Green Version]
- Weiler, J.; Mohr, M.; Zanker, K.S.; Dittmar, T. Matrix metalloproteinase-9 (MMP9) is involved in the TNF-alpha-induced fusion of human M13SV1-Cre breast epithelial cells and human MDA-MB-435-pFDR1 cancer cells. Cell Commun. Signal. 2018, 16, 14. [Google Scholar] [CrossRef] [Green Version]
- Song, K.; Zhu, F.; Zhang, H.Z.; Shang, Z.J. Tumor necrosis factor-alpha enhanced fusions between oral squamous cell carcinoma cells and endothelial cells via VCAM-1/VLA-4 pathway. Exp. Cell Res. 2012, 318, 1707–1715. [Google Scholar] [CrossRef]
- Mohr, M.; Tosun, S.; Arnold, W.H.; Edenhofer, F.; Zanker, K.S.; Dittmar, T. Quantification of cell fusion events human breast cancer cells and breast epithelial cells using a Cre-LoxP-based double fluorescence reporter system. Cell. Mol. Life Sci. 2015, 72, 3769–3782. [Google Scholar] [CrossRef]
- Melzer, C.; von der Ohe, J.; Hass, R. Concise Review: Crosstalk of Mesenchymal Stroma/Stem-Like Cells with Cancer Cells Provides Therapeutic Potential. Stem Cells 2018, 36, 951–968. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.P.; Wang, K.G.; Chen, C.Y.; Yu, C.; Chuang, H.C.; Chen, H. Altered placental syncytin and its receptor ASCT2 expression in placental development and pre-eclampsia. BJOG 2006, 113, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Huppertz, B.; Bartz, C.; Kokozidou, M. Trophoblast fusion: Fusogenic proteins, syncytins and ADAMs, and other prerequisites for syncytial fusion. Micron 2006, 37, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; You, W.; Wang, Y.; Shan, T. The regulatory role of Myomaker and Myomixer-Myomerger-Minion in muscle development and regeneration. Cell. Mol. Life Sci. 2020, 77, 1551–1569. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Shpall, E.; Willerson, J.T.; Yeh, E.T. Fusion of human hematopoietic progenitor cells and murine cardiomyocytes is mediated by alpha 4 beta 1 integrin/vascular cell adhesion molecule-1 interaction. Circ. Res. 2007, 100, 693–702. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [Green Version]
- Takezawa, Y.; Yoshida, K.; Miyado, K.; Sato, M.; Nakamura, A.; Kawano, N.; Sakakibara, K.; Kondo, T.; Harada, Y.; Ohnami, N.; et al. beta-catenin is a molecular switch that regulates transition of cell-cell adhesion to fusion. Sci. Rep. 2011, 1, 68. [Google Scholar] [CrossRef] [Green Version]
- Maretzky, T.; Reiss, K.; Ludwig, A.; Buchholz, J.; Scholz, F.; Proksch, E.; de Strooper, B.; Hartmann, D.; Saftig, P. ADAM10 mediates E-cadherin shedding and regulates epithelial cell-cell adhesion, migration, and beta-catenin translocation. Proc. Natl. Acad. Sci. USA 2005, 102, 9182–9187. [Google Scholar] [CrossRef] [Green Version]
- Aghababaei, M.; Hogg, K.; Perdu, S.; Robinson, W.P.; Beristain, A.G. ADAM12-directed ectodomain shedding of E-cadherin potentiates trophoblast fusion. Cell Death Differ. 2015, 22, 1970–1984. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, K.; Jigami, T.; Taniue, K.; Morishita, Y.; Adachi, S.; Senda, T.; Nonaka, A.; Aburatani, H.; Nakamura, T.; Akiyama, T. Identification of a link between Wnt/β-catenin signalling and the cell fusion pathway. Nat. Commun. 2011, 2, 548. [Google Scholar] [CrossRef]
- Ghosh, S.; Paul, A.; Sen, E. Tumor necrosis factor alpha-induced hypoxia-inducible factor 1alpha-beta-catenin axis regulates major histocompatibility complex class I gene activation through chromatin remodeling. Mol. Cell. Biol. 2013, 33, 2718–2731. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Kim, D.H.; Na, H.K.; Surh, Y.J. TNF-alpha induces expression of urokinase-type plasminogen activator and beta-catenin activation through generation of ROS in human breast epithelial cells. Biochem. Pharmacol. 2010, 80, 2092–2100. [Google Scholar] [CrossRef] [PubMed]
- Brukman, N.G.; Li, X.; Podbilewicz, B. Fusexins, HAP2/GCS1 and Evolution of Gamete Fusion. Front. Cell Dev. Biol. 2021, 9, 824024. [Google Scholar] [CrossRef] [PubMed]
- Vance, T.D.R.; Lee, J.E. Virus and eukaryote fusogen superfamilies. Curr. Biol. 2020, 30, R750–R754. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, G.; Heidmann, O.; Degrelle, S.A.; Vernochet, C.; Lavialle, C.; Letzelter, C.; Bernard-Stoecklin, S.; Hassanin, A.; Mulot, B.; Guillomot, M.; et al. Captured retroviral envelope syncytin gene associated with the unique placental structure of higher ruminants. Proc. Natl. Acad. Sci. USA 2013, 110, E828–E837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornelis, G.; Funk, M.; Vernochet, C.; Leal, F.; Tarazona, O.A.; Meurice, G.; Heidmann, O.; Dupressoir, A.; Miralles, A.; Ramirez-Pinilla, M.P.; et al. An endogenous retroviral envelope syncytin and its cognate receptor identified in the viviparous placental Mabuya lizard. Proc. Natl. Acad. Sci. USA 2017, 114, E10991–E11000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, P.; Yang, H. Molecular underpinning of intracellular pH regulation on TMEM16F. J. Gen. Physiol. 2021, 153, e202012704. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef]
- Boedtkjer, E.; Pedersen, S.F. The Acidic Tumor Microenvironment as a Driver of Cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef] [Green Version]
- Boedtkjer, E. Na+,HCO3− cotransporter NBCn1 accelerates breast carcinogenesis. Cancer Metastasis Rev. 2019, 38, 165–178. [Google Scholar] [CrossRef]
- Mondal Roy, S.; Sarkar, M. Membrane fusion induced by small molecules and ions. J. Lipids 2011, 2011, 528784. [Google Scholar] [CrossRef] [Green Version]
- Compton, A.A.; Schwartz, O. They Might Be Giants: Does Syncytium Formation Sink or Spread HIV Infection? PLoS Pathog. 2017, 13, e1006099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Symeonides, M.; Murooka, T.T.; Bellfy, L.N.; Roy, N.H.; Mempel, T.R.; Thali, M. HIV-1-Induced Small T Cell Syncytia Can Transfer Virus Particles to Target Cells through Transient Contacts. Viruses 2015, 7, 6590–6603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornich, B.F.; Grosskopf, A.K.; Schlagowski, S.; Tenbusch, M.; Kleine-Weber, H.; Neipel, F.; Stahl-Hennig, C.; Hahn, A.S. SARS-CoV-2 and SARS-CoV spike-mediated cell-cell fusion differ in the requirements for receptor expression and proteolytic activation. J. Virol. 2021, 95, e00002-21. [Google Scholar] [CrossRef]
- Moss, B. Membrane fusion during poxvirus entry. Semin. Cell Dev. Biol. 2016, 60, 89–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diller, J.R.; Parrington, H.M.; Patton, J.T.; Ogden, K.M. Rotavirus Species B Encodes a Functional Fusion-Associated Small Transmembrane Protein. J. Virol. 2019, 93, e00813-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Duelli, D.; Lazebnik, Y. Cell-to-cell fusion as a link between viruses and cancer. Nat. Rev. Cancer 2007, 7, 968–976. [Google Scholar] [CrossRef]
- Burton, C.; Bartee, E. Syncytia Formation in Oncolytic Virotherapy. Mol. Ther. Oncolytics 2019, 15, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Krabbe, T.; Altomonte, J. Fusogenic Viruses in Oncolytic Immunotherapy. Cancers 2018, 10, 216. [Google Scholar] [CrossRef]
- Matveeva, O.V.; Shabalina, S.A. Prospects for Using Expression Patterns of Paramyxovirus Receptors as Biomarkers for Oncolytic Virotherapy. Cancers 2020, 12, 3659. [Google Scholar] [CrossRef]
- Chen, J.; Foroozesh, M.; Qin, Z. Transactivation of human endogenous retroviruses by tumor viruses and their functions in virus-associated malignancies. Oncogenesis 2019, 8, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Liu, L.; Wang, X.; Liu, Y.; Wang, M.; Zhu, F. HBV X Protein induces overexpression of HERV-W env through NF-kappaB in HepG2 cells. Virus Genes 2017, 53, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Del Valle, L.; Miley, W.; Whitby, D.; Ochoa, A.C.; Flemington, E.K.; Qin, Z. Transactivation of human endogenous retrovirus K (HERV-K) by KSHV promotes Kaposi’s sarcoma development. Oncogene 2018, 37, 4534–4545. [Google Scholar] [CrossRef] [PubMed]
- Uleri, E.; Mei, A.; Mameli, G.; Poddighe, L.; Serra, C.; Dolei, A. HIV Tat acts on endogenous retroviruses of the W family and this occurs via Toll-like receptor 4: Inference for neuroAIDS. AIDS 2014, 28, 2659–2670. [Google Scholar] [CrossRef]
- Mameli, G.; Poddighe, L.; Mei, A.; Uleri, E.; Sotgiu, S.; Serra, C.; Manetti, R.; Dolei, A. Expression and activation by Epstein Barr virus of human endogenous retroviruses-W in blood cells and astrocytes: Inference for multiple sclerosis. PLoS ONE 2012, 7, e44991. [Google Scholar] [CrossRef]
- Marston, J.L.; Greenig, M.; Singh, M.; Bendall, M.L.; Duarte, R.R.R.; Feschotte, C.; Iniguez, L.P.; Nixon, D.F. SARS-CoV-2 infection mediates differential expression of human endogenous retroviruses and long interspersed nuclear elements. JCI Insight 2021, 6, e147170. [Google Scholar] [CrossRef]
- Dervan, E.; Bhattacharyya, D.D.; McAuliffe, J.D.; Khan, F.H.; Glynn, S.A. Ancient Adversary—HERV-K (HML-2) in Cancer. Front. Oncol. 2021, 11, 658489. [Google Scholar] [CrossRef]
- Curty, G.; Marston, J.L.; de Mulder Rougvie, M.; Leal, F.E.; Nixon, D.F.; Soares, M.A. Human Endogenous Retrovirus K in Cancer: A Potential Biomarker and Immunotherapeutic Target. Viruses 2020, 12, 726. [Google Scholar] [CrossRef]
- de Parseval, N.; Heidmann, T. Human endogenous retroviruses: From infectious elements to human genes. Cytogenet. Genome Res. 2005, 110, 318–332. [Google Scholar] [CrossRef]
- Gao, Y.; Yu, X.F.; Chen, T. Human endogenous retroviruses in cancer: Expression, regulation and function. Oncol. Lett. 2021, 21, 121. [Google Scholar] [CrossRef]
- Garcia-Montojo, M.; Doucet-O’Hare, T.; Henderson, L.; Nath, A. Human endogenous retrovirus-K (HML-2): A comprehensive review. Crit. Rev. Microbiol. 2018, 44, 715–738. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Sechi, L.A.; Kelvin, D.J. Human Endogenous Retrovirus K (HML-2) in Health and Disease. Front. Microbiol. 2020, 11, 1690. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.J.; Rosenkrantz, J.L.; Carbone, L.; Chavez, S.L. Endogenous Retroviruses: With Us and against Us. Front. Chem. 2017, 5, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johanning, G.L.; Malouf, G.G.; Zheng, X.; Esteva, F.J.; Weinstein, J.N.; Wang-Johanning, F.; Su, X. Expression of human endogenous retrovirus-K is strongly associated with the basal-like breast cancer phenotype. Sci. Rep. 2017, 7, 41960. [Google Scholar] [CrossRef] [Green Version]
- Cardelli, M.; Doorn, R.V.; Larcher, L.; Donato, M.D.; Piacenza, F.; Pierpaoli, E.; Giacconi, R.; Malavolta, M.; Rachakonda, S.; Gruis, N.A.; et al. Association of HERV-K and LINE-1 hypomethylation with reduced disease-free survival in melanoma patients. Epigenomics 2020, 12, 1689–1706. [Google Scholar] [CrossRef]
- Ma, W.; Hong, Z.; Liu, H.; Chen, X.; Ding, L.; Liu, Z.; Zhou, F.; Yuan, Y. Human Endogenous Retroviruses-K (HML-2) Expression Is Correlated with Prognosis and Progress of Hepatocellular Carcinoma. Biomed. Res. Int. 2016, 2016, 8201642. [Google Scholar] [CrossRef]
- Zhou, F.; Li, M.; Wei, Y.; Lin, K.; Lu, Y.; Shen, J.; Johanning, G.L.; Wang-Johanning, F. Activation of HERV-K Env protein is essential for tumorigenesis and metastasis of breast cancer cells. Oncotarget 2016, 7, 84093–84117. [Google Scholar] [CrossRef] [Green Version]
- Lemaitre, C.; Tsang, J.; Bireau, C.; Heidmann, T.; Dewannieux, M. A human endogenous retrovirus-derived gene that can contribute to oncogenesis by activating the ERK pathway and inducing migration and invasion. PLoS Pathog. 2017, 13, e1006451. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Radvanyi, L.; Yin, B.; Rycaj, K.; Li, J.; Chivukula, R.; Lin, K.; Lu, Y.; Shen, J.; Chang, D.Z.; et al. Downregulation of Human Endogenous Retrovirus Type K (HERV-K) Viral env RNA in Pancreatic Cancer Cells Decreases Cell Proliferation and Tumor Growth. Clin. Cancer Res. 2017, 23, 5892–5911. [Google Scholar] [CrossRef]
- Huang, G.; Li, Z.; Wan, X.; Wang, Y.; Dong, J. Human endogenous retroviral K element encodes fusogenic activity in melanoma cells. J. Carcinog. 2013, 12, 5. [Google Scholar] [CrossRef]
- Durnaoglu, S.; Lee, S.K.; Ahnn, J. Syncytin, envelope protein of human endogenous retrovirus (HERV): No longer ‘fossil’ in human genome. Anim. Cells Syst. 2021, 25, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liu, L.; Liu, Y.; Zhou, P.; Yan, Q.; Yu, H.; Chen, X.; Zhu, F. Implication of human endogenous retrovirus W family envelope in hepatocellular carcinoma promotes MEK/ERK-mediated metastatic invasiveness and doxorubicin resistance. Cell Death Discov. 2021, 7, 177. [Google Scholar] [CrossRef] [PubMed]
- Uygur, B.; Melikov, K.; Arakelyan, A.; Margolis, L.B.; Chernomordik, L.V. Syncytin 1 dependent horizontal transfer of marker genes from retrovirally transduced cells. Sci. Rep. 2019, 9, 17637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolosa, J.M.; Schjenken, J.E.; Clifton, V.L.; Vargas, A.; Barbeau, B.; Lowry, P.; Maiti, K.; Smith, R. The endogenous retroviral envelope protein syncytin-1 inhibits LPS/PHA-stimulated cytokine responses in human blood and is sorted into placental exosomes. Placenta 2012, 33, 933–941. [Google Scholar] [CrossRef]
- Vargas, A.; Zhou, S.; Ethier-Chiasson, M.; Flipo, D.; Lafond, J.; Gilbert, C.; Barbeau, B. Syncytin proteins incorporated in placenta exosomes are important for cell uptake and show variation in abundance in serum exosomes from patients with preeclampsia. FASEB J. 2014, 28, 3703–3719. [Google Scholar] [CrossRef]
- Yu, H.; Liu, T.; Zhao, Z.; Chen, Y.; Zeng, J.; Liu, S.; Zhu, F. Mutations in 3′-long terminal repeat of HERV-W family in chromosome 7 upregulate syncytin-1 expression in urothelial cell carcinoma of the bladder through interacting with c-Myb. Oncogene 2014, 33, 3947–3958. [Google Scholar] [CrossRef]
- Larsen, J.M.; Christensen, I.J.; Nielsen, H.J.; Hansen, U.; Bjerregaard, B.; Talts, J.F.; Larsson, L.I. Syncytin immunoreactivity in colorectal cancer: Potential prognostic impact. Cancer Lett. 2009, 280, 44–49. [Google Scholar] [CrossRef]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Emami Nejad, A.; Najafgholian, S.; Rostami, A.; Sistani, A.; Shojaeifar, S.; Esparvarinha, M.; Nedaeinia, R.; Haghjooy Javanmard, S.; Taherian, M.; Ahmadlou, M.; et al. The role of hypoxia in the tumor microenvironment and development of cancer stem cell: A novel approach to developing treatment. Cancer Cell Int. 2021, 21, 62. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, L.; Li, X.F. Hypoxia and the Tumor Microenvironment. Technol. Cancer Res. Treat. 2021, 20, 15330338211036304. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archacka, K.; Grabowska, I.; Mierzejewski, B.; Graffstein, J.; Gorzynska, A.; Krawczyk, M.; Rozycka, A.M.; Kalaszczynska, I.; Muras, G.; Streminska, W.; et al. Hypoxia preconditioned bone marrow-derived mesenchymal stromal/stem cells enhance myoblast fusion and skeletal muscle regeneration. Stem Cell Res. Ther. 2021, 12, 448. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; He, Y.; Liu, D.; Zhao, L.; Fang, S.; Tan, B.; Dong, S.; Wang, Y.; He, T.; Bi, Y. Hypoxia Preconditioning Promotes the Proliferation and Migration of Human Urine-Derived Stem Cells in Chronically Injured Liver of Mice by Upregulating CXCR4. Stem Cells Dev. 2021, 30, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Yart, L.; Bastida-Ruiz, D.; Allard, M.; Dietrich, P.Y.; Petignat, P.; Cohen, M. Linking unfolded protein response to ovarian cancer cell fusion. BMC Cancer 2022, 22, 622. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.M.; Yan, T.L.; Xu, Z.; Wang, M.; Zhou, X.C.; Jiang, E.H.; Liu, K.; Shao, Z.; Shang, Z.J. Hypoxia Enhances Fusion of Oral Squamous Carcinoma Cells and Epithelial Cells Partly via the Epithelial-Mesenchymal Transition of Epithelial Cells. Biomed. Res. Int. 2018, 2018, 5015203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsat, E.; Wyplosz, P.; Malassine, A.; Guibourdenche, J.; Porquet, D.; Nessmann, C.; Evain-Brion, D. Hypoxia impairs cell fusion and differentiation process in human cytotrophoblast, in vitro. J. Cell. Physiol. 1996, 168, 346–353. [Google Scholar] [CrossRef]
- Kudo, Y.; Boyd, C.A.; Sargent, I.L.; Redman, C.W. Hypoxia alters expression and function of syncytin and its receptor during trophoblast cell fusion of human placental BeWo cells: Implications for impaired trophoblast syncytialisation in pre-eclampsia. Biochim. Biophys. Acta 2003, 1638, 63–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Luo, T.; von der Ohe, J.; Hass, R. MSC-Derived Extracellular Vesicles in Tumors and Therapy. Cancers 2021, 13, 5212. [Google Scholar] [CrossRef]
- Sharma, R.; Huang, X.; Brekken, R.A.; Schroit, A.J. Detection of phosphatidylserine-positive exosomes for the diagnosis of early-stage malignancies. Br. J. Cancer 2017, 117, 545–552. [Google Scholar] [CrossRef] [Green Version]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Mathivanan, S.; Ji, H.; Simpson, R.J. Exosomes: Extracellular organelles important in intercellular communication. J. Proteom. 2010, 73, 1907–1920. [Google Scholar] [CrossRef] [PubMed]
- Baglio, S.R.; Rooijers, K.; Koppers-Lalic, D.; Verweij, F.J.; Perez Lanzon, M.; Zini, N.; Naaijkens, B.; Perut, F.; Niessen, H.W.; Baldini, N.; et al. Human bone marrow- and adipose-mesenchymal stem cells secrete exosomes enriched in distinctive miRNA and tRNA species. Stem Cell Res. Ther. 2015, 6, 127. [Google Scholar] [CrossRef] [Green Version]
- Keller, S.; Sanderson, M.P.; Stoeck, A.; Altevogt, P. Exosomes: From biogenesis and secretion to biological function. Immunol. Lett. 2006, 107, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Tkach, M.; Thery, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [Green Version]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Bucan, V.; Baehre, H.; von der Ohe, J.; Otte, A.; Hass, R. Acquisition of new tumor cell properties by MSC-derived exosomes. Int J Oncol 2015, 47, 244–252. [Google Scholar] [CrossRef] [Green Version]
- Mandel, K.; Yang, Y.; Schambach, A.; Glage, S.; Otte, A.; Hass, R. Mesenchymal stem cells directly interact with breast cancer cells and promote tumor cell growth in vitro and in vivo. Stem Cells Dev. 2013, 22, 3114–3127. [Google Scholar] [CrossRef]
- Liao, C.M.; Luo, T.; von der Ohe, J.; de Juan Mora, B.; Schmitt, R.; Hass, R. Human MSC-Derived Exosomes Reduce Cellular Senescence in Renal Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 13562. [Google Scholar] [CrossRef]
- Melzer, C.; Rehn, V.; Yang, Y.; Bahre, H.; von der Ohe, J.; Hass, R. Taxol-Loaded MSC-Derived Exosomes Provide a Therapeutic Vehicle to Target Metastatic Breast Cancer and Other Carcinoma Cells. Cancers 2019, 11, 798. [Google Scholar] [CrossRef] [Green Version]
- Melzer, C.; Ohe, J.V.; Hass, R. Anti-Tumor Effects of Exosomes Derived from Drug-Incubated Permanently Growing Human MSC. Int. J. Mol. Sci. 2020, 21, 7311. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Thery, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Record, M. Intercellular communication by exosomes in placenta: A possible role in cell fusion? Placenta 2014, 35, 297–302. [Google Scholar] [CrossRef]
- Howcroft, T.K.; Zhang, H.G.; Dhodapkar, M.; Mohla, S. Vesicle transfer and cell fusion: Emerging concepts of cell-cell communication in the tumor microenvironment. Cancer Biol. Ther. 2011, 12, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar]
- Chakraborty, A.; Lazova, R.; Davies, S.; Backvall, H.; Ponten, F.; Brash, D.; Pawelek, J. Donor DNA in a renal cell carcinoma metastasis from a bone marrow transplant recipient. Bone Marrow Transplant. 2004, 34, 183–186. [Google Scholar] [CrossRef]
- LaBerge, G.; Duvall, E.; Grasmick, Z.; Haedicke, K.; Galan, A.; Pawelek, J. A melanoma patient with macrophage-cancer cell hybrids in the primary tumor, a lymph node metastasis and a brain metastasis. Cancer Genet. 2021, 256–257, 162–164. [Google Scholar] [CrossRef]
- LaBerge, G.S.; Duvall, E.; Grasmick, Z.; Haedicke, K.; Pawelek, J. A Melanoma Lymph Node Metastasis with a Donor-Patient Hybrid Genome following Bone Marrow Transplantation: A Second Case of Leucocyte-Tumor Cell Hybridization in Cancer Metastasis. PLoS ONE 2017, 12, e0168581. [Google Scholar] [CrossRef]
- Lazova, R.; Laberge, G.S.; Duvall, E.; Spoelstra, N.; Klump, V.; Sznol, M.; Cooper, D.; Spritz, R.A.; Chang, J.T.; Pawelek, J.M. A Melanoma Brain Metastasis with a Donor-Patient Hybrid Genome following Bone Marrow Transplantation: First Evidence for Fusion in Human Cancer. PLoS ONE 2013, 8, e66731. [Google Scholar] [CrossRef]
- Yilmaz, Y.; Lazova, R.; Qumsiyeh, M.; Cooper, D.; Pawelek, J. Donor Y chromosome in renal carcinoma cells of a female BMT recipient: Visualization of putative BMT-tumor hybrids by FISH. Bone Marrow Transplant. 2005, 35, 1021–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manjunath, Y.; Mitchem, J.B.; Suvilesh, K.N.; Avella, D.M.; Kimchi, E.T.; Staveley-O’Carroll, K.F.; Deroche, C.B.; Pantel, K.; Li, G.; Kaifi, J.T. Circulating giant tumor-macrophage fusion cells are independent prognosticators in non-small cell lung cancer patients. J. Thorac. Oncol. 2020, 15, 1460–1471. [Google Scholar] [CrossRef] [PubMed]
- Dietz, M.S.; Sutton, T.L.; Walker, B.S.; Gast, C.E.; Zarour, L.; Sengupta, S.K.; Swain, J.R.; Eng, J.; Parappilly, M.; Limbach, K.; et al. Relevance of circulating hybrid cells as a non-invasive biomarker for myriad solid tumors. Sci. Rep. 2021, 11, 13630. [Google Scholar] [CrossRef] [PubMed]
- Gast, C.E.; Silk, A.D.; Zarour, L.; Riegler, L.; Burkhart, J.G.; Gustafson, K.T.; Parappilly, M.S.; Roh-Johnson, M.; Goodman, J.R.; Olson, B.; et al. Cell fusion potentiates tumor heterogeneity and reveals circulating hybrid cells that correlate with stage and survival. Sci. Adv. 2018, 4, eaat7828. [Google Scholar] [CrossRef] [Green Version]
- Clawson, G.A.; Kimchi, E.; Patrick, S.D.; Xin, P.; Harouaka, R.; Zheng, S.; Berg, A.; Schell, T.; Staveley-O’Carroll, K.F.; Neves, R.I.; et al. Circulating tumor cells in melanoma patients. PLoS ONE 2012, 7, e41052. [Google Scholar] [CrossRef] [Green Version]
- Clawson, G.A.; Matters, G.L.; Xin, P.; Imamura-Kawasawa, Y.; Du, Z.; Thiboutot, D.M.; Helm, K.F.; Neves, R.I.; Abraham, T. Macrophage-Tumor Cell Fusions from Peripheral Blood of Melanoma Patients. PLoS ONE 2015, 10, e0134320. [Google Scholar] [CrossRef] [Green Version]
- Muthu, S.; Bapat, A.; Jain, R.; Jeyaraman, N.; Jeyaraman, M. Exosomal therapy—A new frontier in regenerative medicine. Stem Cell Investig. 2021, 8, 7. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dittmar, T.; Hass, R. Extracellular Events Involved in Cancer Cell–Cell Fusion. Int. J. Mol. Sci. 2022, 23, 16071. https://doi.org/10.3390/ijms232416071
Dittmar T, Hass R. Extracellular Events Involved in Cancer Cell–Cell Fusion. International Journal of Molecular Sciences. 2022; 23(24):16071. https://doi.org/10.3390/ijms232416071
Chicago/Turabian StyleDittmar, Thomas, and Ralf Hass. 2022. "Extracellular Events Involved in Cancer Cell–Cell Fusion" International Journal of Molecular Sciences 23, no. 24: 16071. https://doi.org/10.3390/ijms232416071
APA StyleDittmar, T., & Hass, R. (2022). Extracellular Events Involved in Cancer Cell–Cell Fusion. International Journal of Molecular Sciences, 23(24), 16071. https://doi.org/10.3390/ijms232416071