


Identification of the Third Case of PSEN1 Tyr389His Variant in Early-Onset Alzheimer’s Disease in Korea


{kind=link}
Abstract
1. Introduction
2. Results
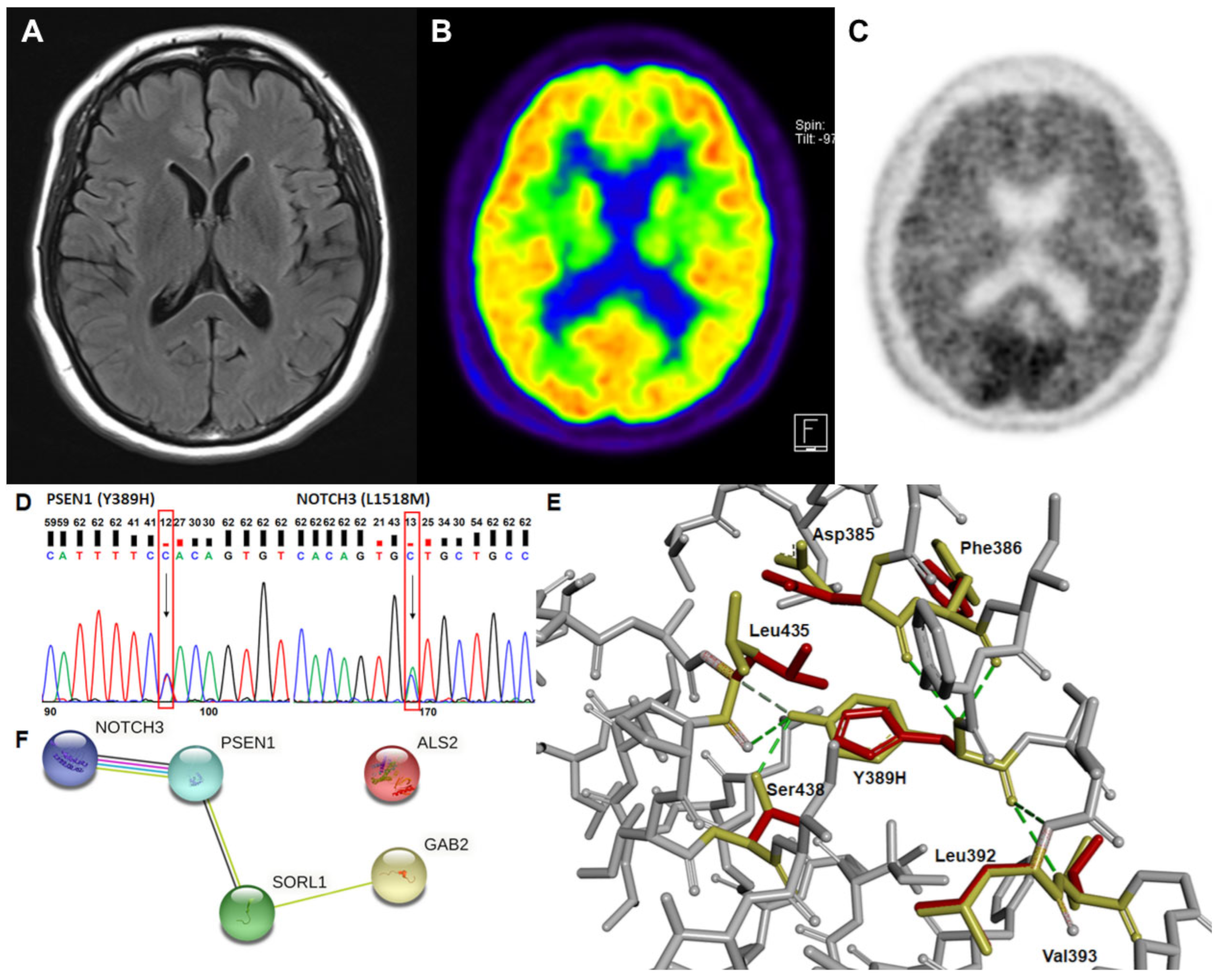
2.1. Clinical Characterization
2.2. Genetic Analysis and In Silico Prediction
3. Discussion
4. Method
4.1. Subject
4.2. DNA Purification and Genetic Screening
4.3. In Silico Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Jonghe, C.; Esselens, C.; Kumar-Singh, S.; Craessaerts, K.; Serneels, S.; Checler, F.; Annaert, W.; Van Broeckhoven, C.; De Strooper, B. Pathogenic APP mutations near the gamma-secretase cleavage site differentially affect Abeta secretion and APP C-terminal fragment stability. Hum. Mol. Genet. 2001, 10, 1665–1671. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhou, R.; Yang, G.; Shi, Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Abeta42 and Abeta40 peptides by gamma-secretase. Proc. Natl. Acad. Sci. USA 2017, 114, E476–E485. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Gutiérrez, L.; Bammens, L.; Benilova, I.; Vandersteen, A.; Benurwar, M.; Borgers, M.; Lismont, S.; Zhou, L.; Van Cleynenbreugel, S.; Esselmann, H.; et al. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012, 31, 2261–2274. [Google Scholar] [CrossRef] [PubMed]
- Giau, V.V.; Bagyinszky, E.; Youn, Y.C.; An, S.S.A.; Kim, S. APP, PSEN1, and PSEN2 Mutations in Asian Patients with Early-Onset Alzheimer Disease. Int. J. Mol. Sci. 2019, 20, 4757. [Google Scholar] [CrossRef] [PubMed]
- Takada, L.T.; Aláez-Verson, C.; Burgute, B.D.; Nitrini, R.; Sosa, A.L.; Castilhos, R.M.; Chaves, M.F.; Longoria, E.-M.; Carrillo-Sánchez, K.; Brucki, S.M.D.; et al. Discovery and validation of dominantly inherited Alzheimer’s disease mutations in populations from Latin America. Alzheimer’s Res. Ther. 2022, 14, 108. [Google Scholar] [CrossRef] [PubMed]
- Kimberly, W.T.; Xia, W.; Rahmati, T.; Wolfe, M.S.; Selkoe, D.J. The transmembrane aspartates in presenilin 1 and 2 are obligatory for gamma-secretase activity and amyloid beta-protein generation. J. Biol. Chem. 2000, 275, 3173–3178. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Cho, H.; Kim, H.J.; Na, D.L.; Seo, S.W.; Ki, C.S. PSEN1 variants in Korean patients with clinically suspicious early-onset familial Alzheimer’s disease. Sci. Rep. 2020, 10, 3480. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Kim, H.J.; Kim, Y.E.; Jang, H.; Cho, S.H.; Kim, S.J.; Na, D.L.; Won, H.H.; Ki, C.S.; Seo, S.W. Analysis of dementia-related gene variants in APOE ε4 noncarrying Korean patients with early-onset Alzheimer’s disease. Neurobiol. Aging 2020, 85, 155.e5–155.e8. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Liu, H.; Liu, X.; Zhang, W.; Zhang, S.; Jiao, B. APP, PSEN1, and PSEN2 Variants in Alzheimer’s Disease: Systematic Re-evaluation According to ACMG Guidelines. Front. Aging Neurosci. 2021, 13, 695808. [Google Scholar] [CrossRef] [PubMed]
- Park, D.G.; Min, J.H.; Sohn, S.H.; Sohn, Y.B.; Yoon, J.H. Ataxia Associated with CADASIL: A Pathology-Confirmed Case Report and Literature Review. Cerebellum 2020, 19, 907–910. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.; Dilliott, A.A.; Binns, M.A.; Breen, D.P.; Evans, E.C.; Beaton, D.; McLaughlin, P.M.; Kwan, D.; Holmes, M.F.; Ozzoude, M.; et al. Parkinson’s Disease, NOTCH3 Genetic Variants, and White Matter Hyperintensities. Mov. Disord. 2020, 35, 2090–2095. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shim, K.H.; Kang, S.; An, S.S.A.; Kang, M.J. Identification of the Third Case of PSEN1 Tyr389His Variant in Early-Onset Alzheimer’s Disease in Korea. Int. J. Mol. Sci. 2022, 23, 16192. https://doi.org/10.3390/ijms232416192
Shim KH, Kang S, An SSA, Kang MJ. Identification of the Third Case of PSEN1 Tyr389His Variant in Early-Onset Alzheimer’s Disease in Korea. International Journal of Molecular Sciences. 2022; 23(24):16192. https://doi.org/10.3390/ijms232416192
Chicago/Turabian StyleShim, Kyu Hwan, Sangjoon Kang, Seong Soo A. An, and Min Ju Kang. 2022. "Identification of the Third Case of PSEN1 Tyr389His Variant in Early-Onset Alzheimer’s Disease in Korea" International Journal of Molecular Sciences 23, no. 24: 16192. https://doi.org/10.3390/ijms232416192
APA StyleShim, K. H., Kang, S., An, S. S. A., & Kang, M. J. (2022). Identification of the Third Case of PSEN1 Tyr389His Variant in Early-Onset Alzheimer’s Disease in Korea. International Journal of Molecular Sciences, 23(24), 16192. https://doi.org/10.3390/ijms232416192