
Air Plasma-Activated Medium Evokes a Death-Associated Perinuclear Mitochondrial Clustering

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
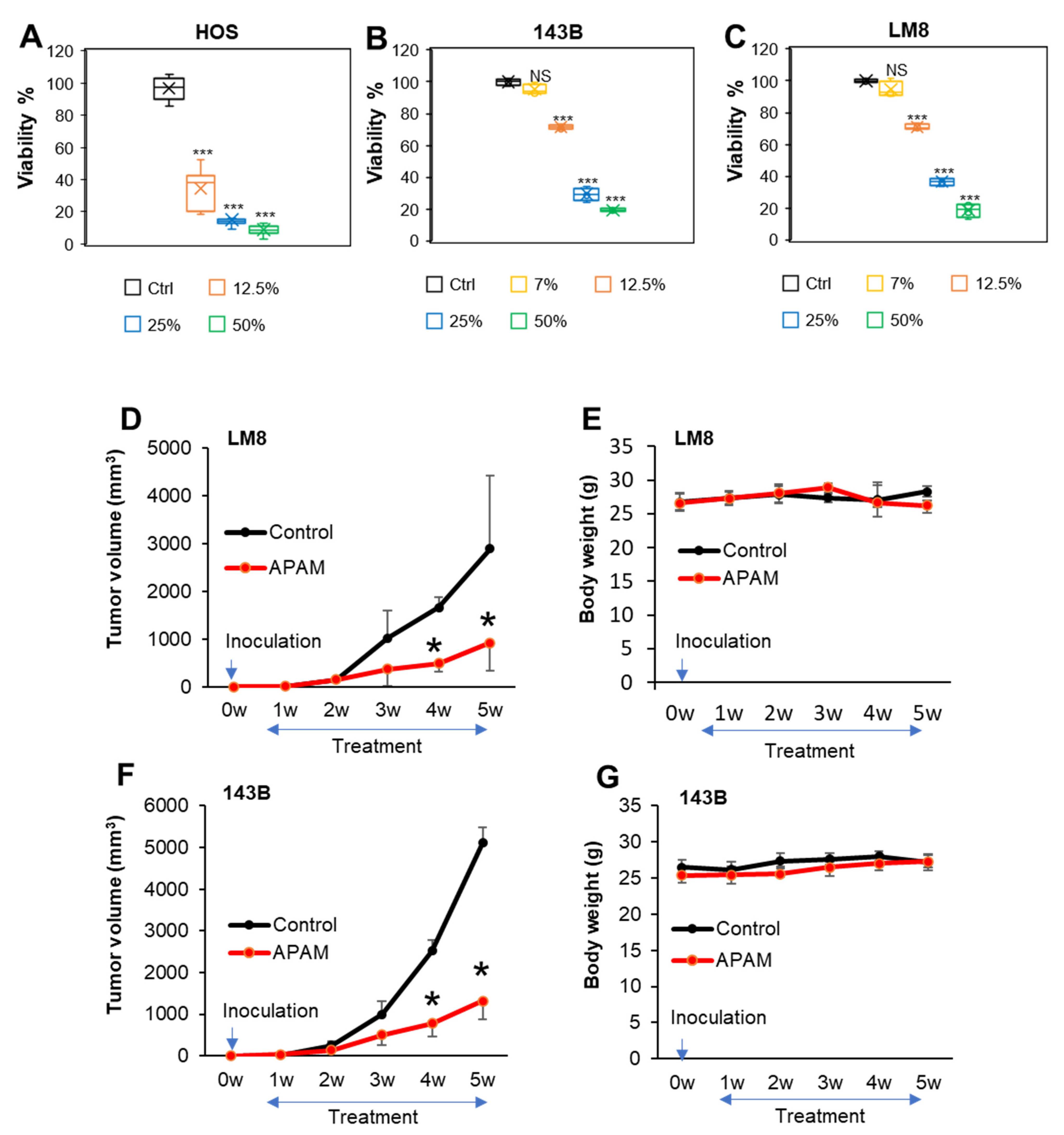
2.1. APAM Has Potent Antitumor Activity against OS In Vitro and In Vivo
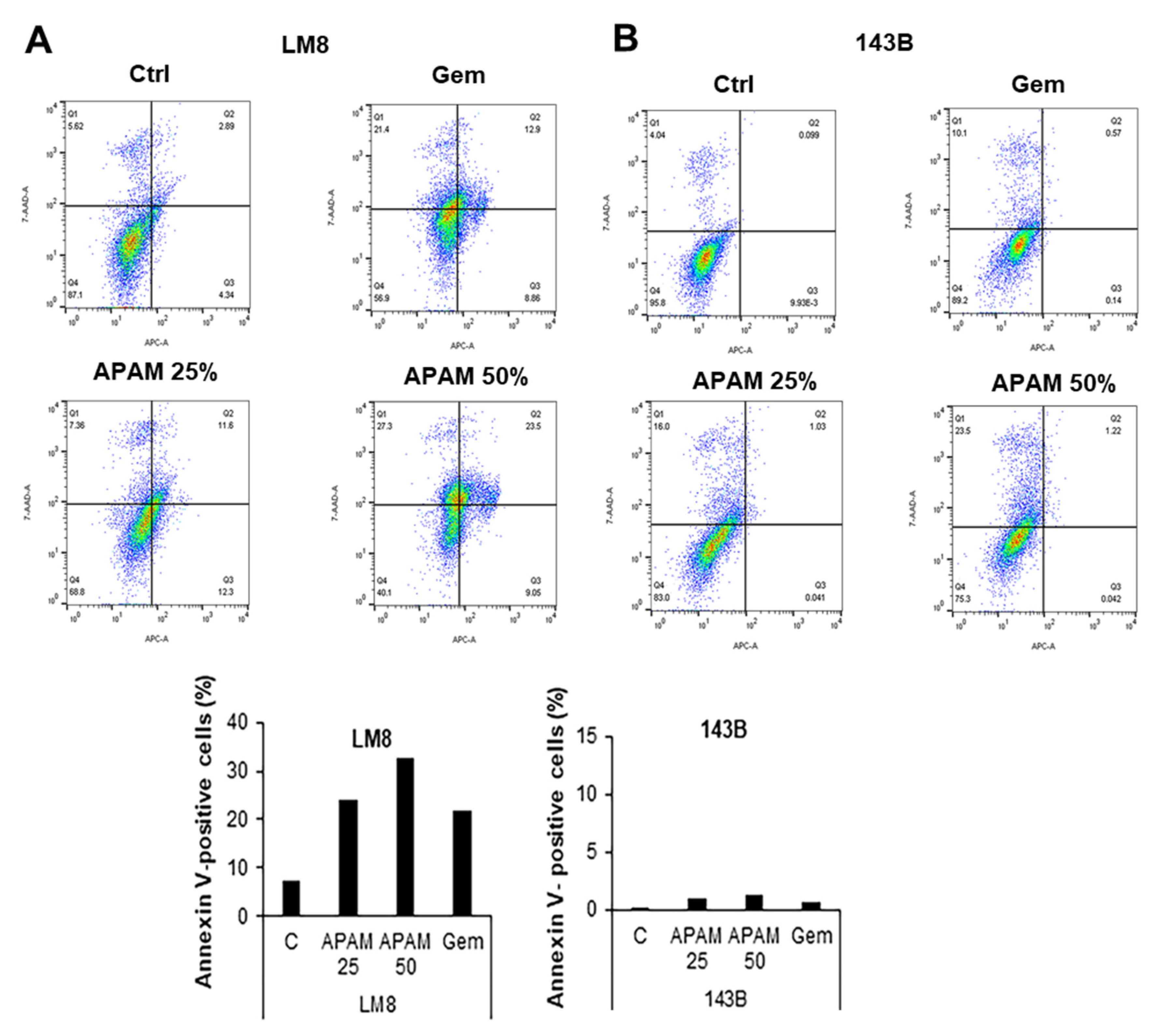
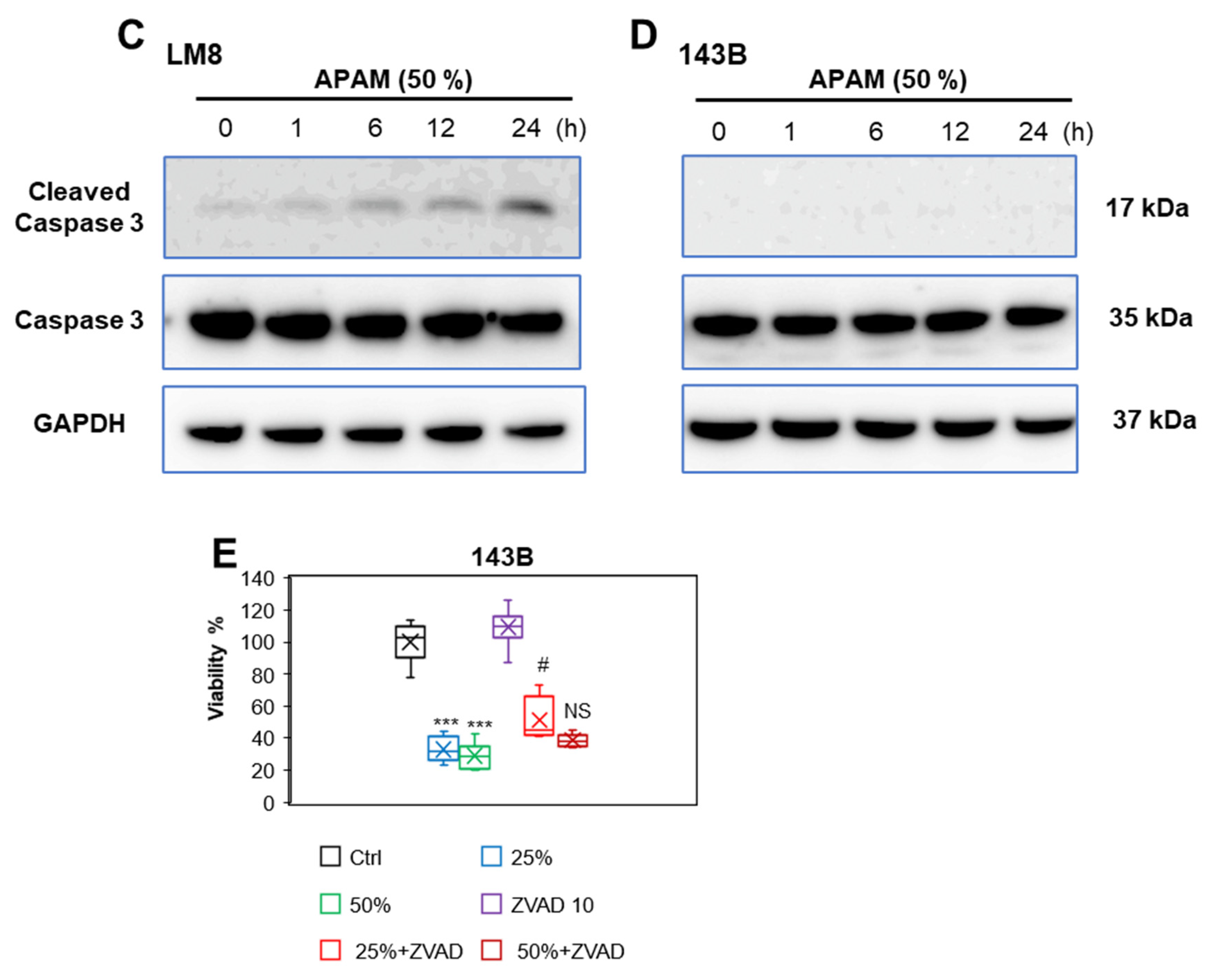
2.2. APAM Can Activate Apoptosis and Nonapoptotic Cell Death in OS Cells
2.3. ROS Plays a Vital Role in Cell Death
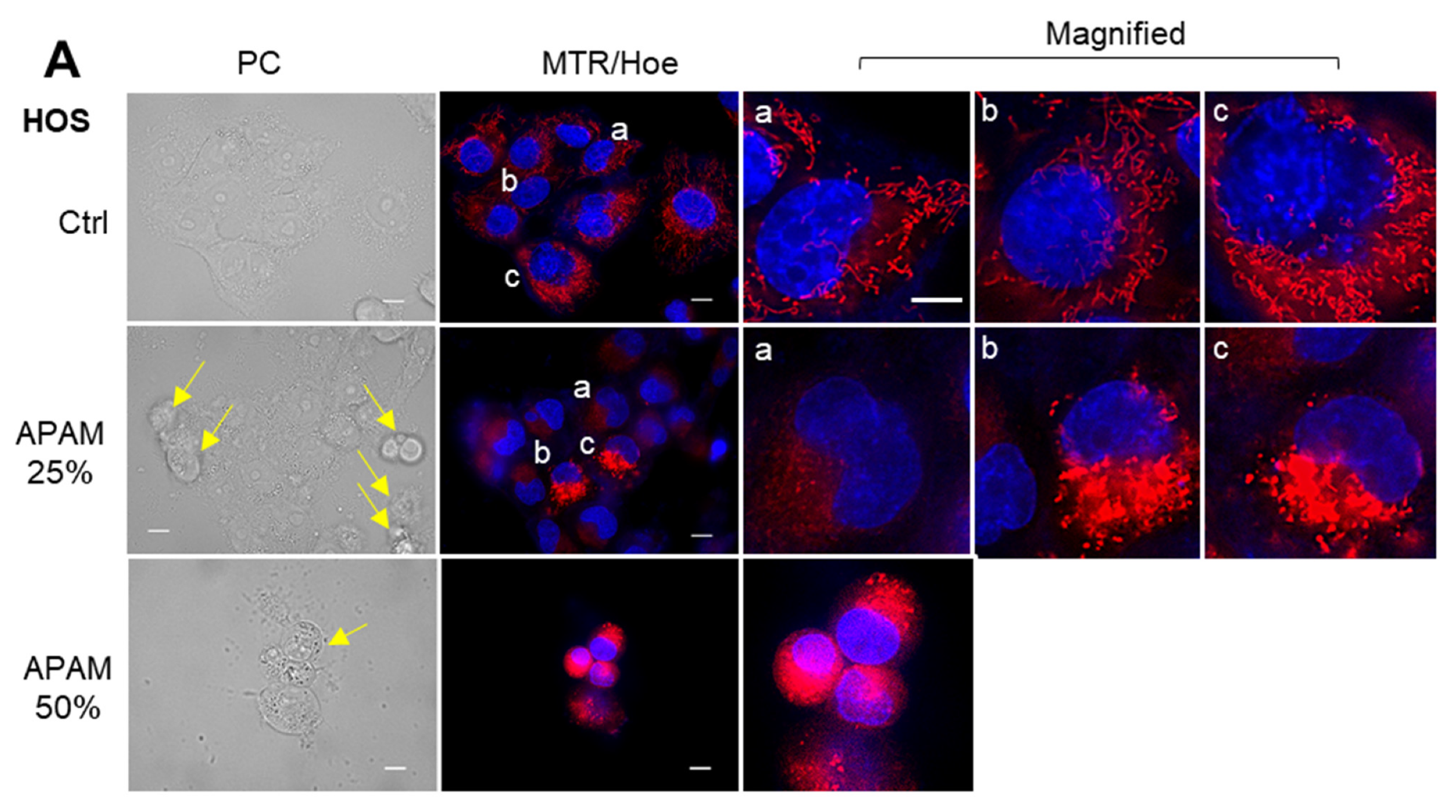
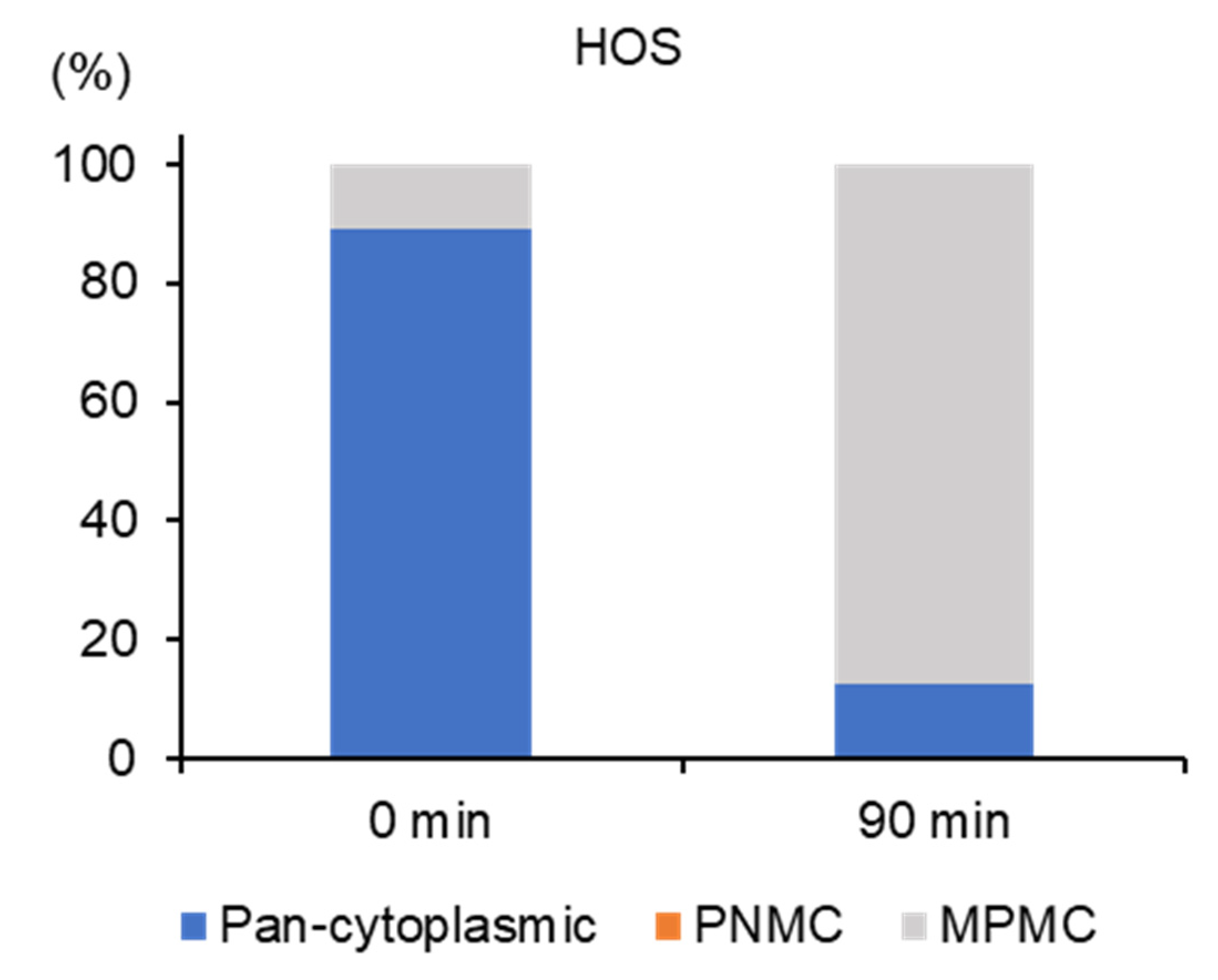
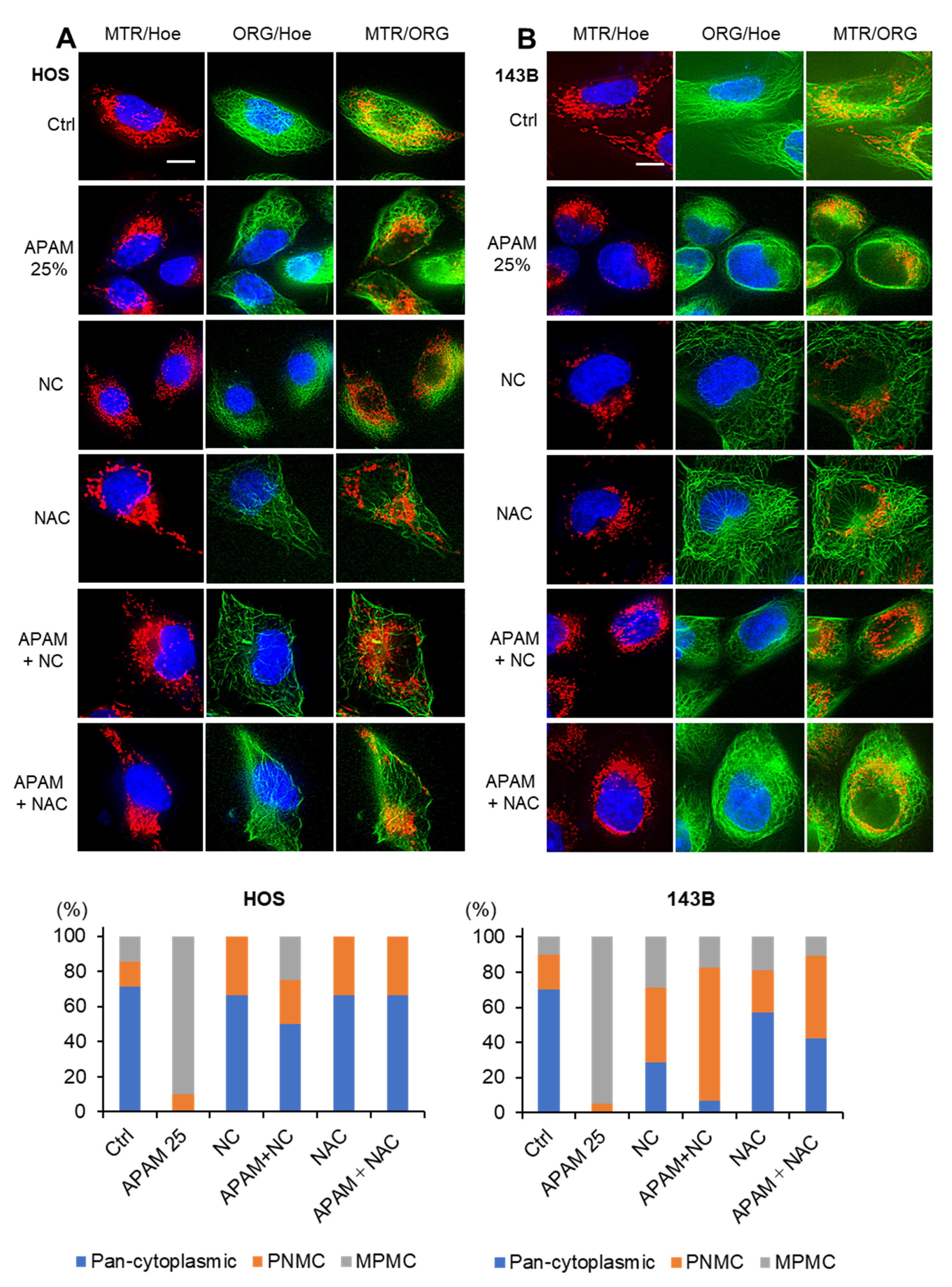
2.4. APAM Evokes MPMC in a Tumor-Specific Manner
2.5. MPMC Occurs in a Microtubule- and ROS-Dependent Manner
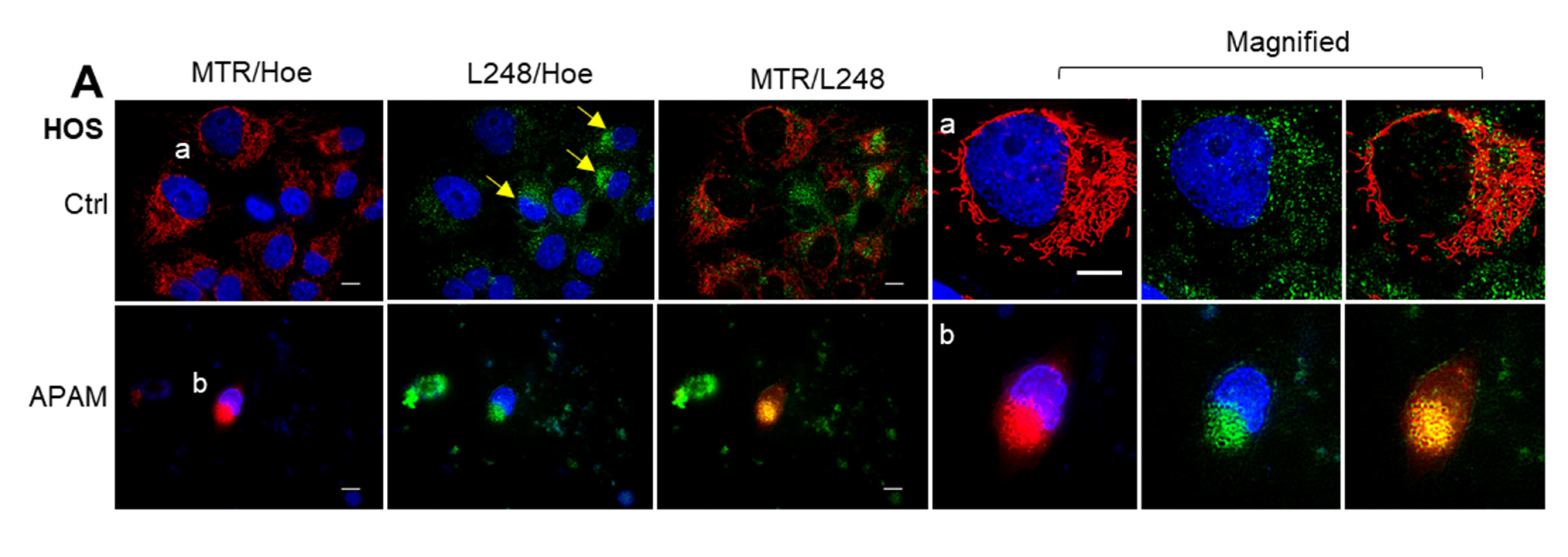
2.6. MPMC Involves LPO Accumulation and Cardiolipin (CL) Oxidation in the Mitochondrial Cluster
2.7. APAM Shows Similar Biological Effects in OC Cells
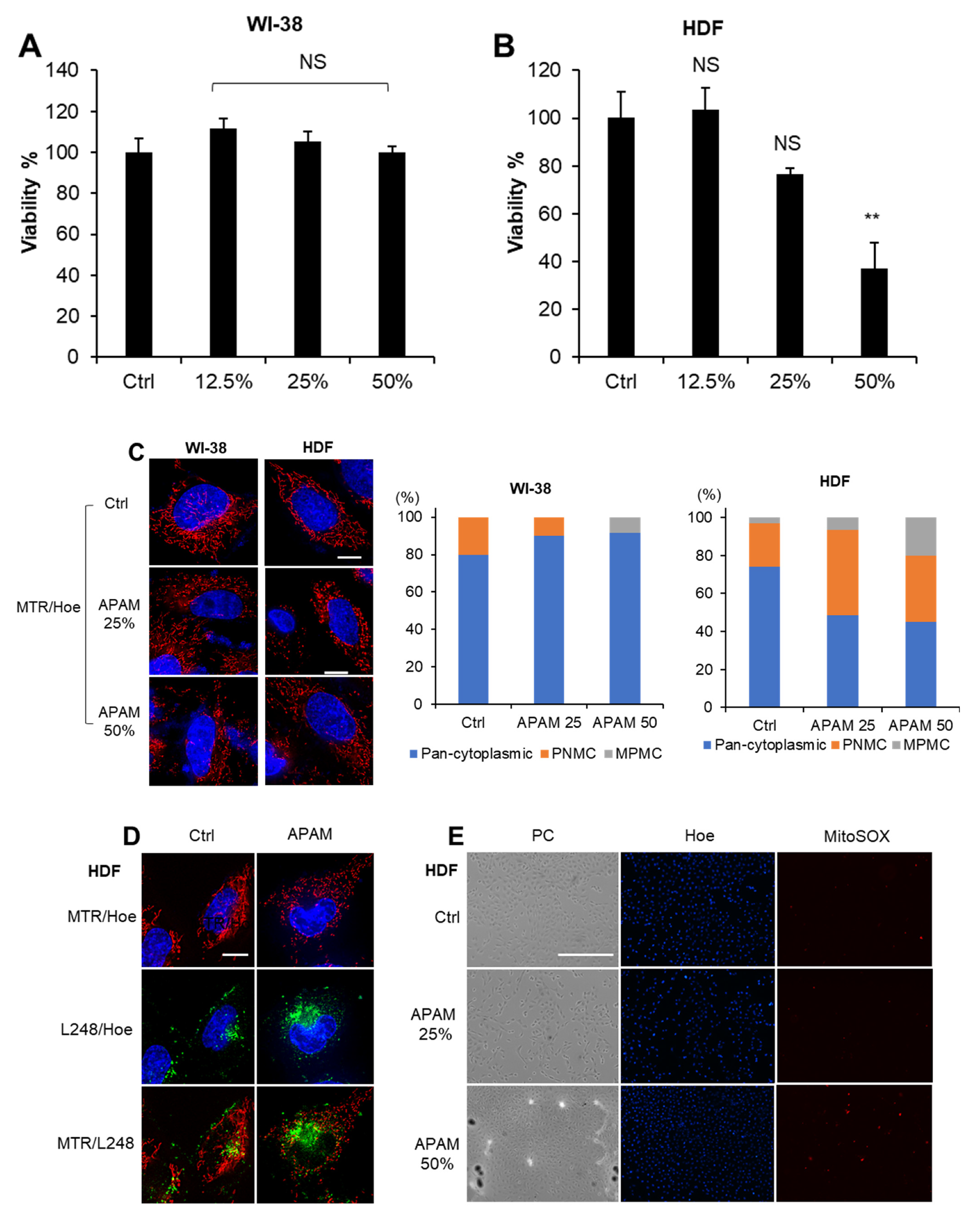
2.8. APAM Induces Minimal Cell Death, Mitochondrial Oxidative Stress, and MPMC in Noncancerous Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. APAM Preparation
4.4. Cell Viability Assay
4.5. Animals and In Vivo Antitumor Activity
4.6. Cell Death Assay
4.7. Western Blotting Analysis
4.8. Mitochondrial Network and Positioning Assays
4.9. Autophagy Assay
4.10. Quantitation of H2O2 in APAM
4.11. Measurements of Intracellular ROS Generation
4.12. LPO and CL Oxidation Analyses
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Luetke, A.; Meyers, P.; Lewis, I.; Juergens, H. Osteosarcoma treatment—Where do we stand? A state of the art review. Cancer Treat. Rev. 2014, 40, 523–532. [Google Scholar] [CrossRef]
- Siegel, R.L.; Fedewa, S.A.; Miller, K.D.; Goding-Sauer, A.; Pinheiro, P.S.; Martinez-Tyson, D.; Jemal, A. Cancer statistics for Hispanics/Latinos, 2015. CA Cancer J. Clin. 2015, 65, 457–480. [Google Scholar] [CrossRef] [Green Version]
- Morrow, J.J.; Bayles, I.; Funnell, A.P.W.; Miller, T.E.; Saiakhova, A.; Lizardo, M.M.; Bartels, C.F.; Kapteijn, M.Y.; Hung, S.; Mendoza, A.; et al. Positively selected enhancer elements endow osteosarcoma cells with metastatic competence. Nat. Med. 2018, 24, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; MacLeod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Kornienko, A.; Mathieu, V.; Rastogi, S.K.; Lefranc, F.; Kiss, R. Therapeutic Agents Triggering Nonapoptotic Cancer Cell Death. J. Med. Chem. 2013, 56, 4823–4839. [Google Scholar] [CrossRef] [PubMed]
- Keidar, M.; Walk, R.M.; Shashurin, A.; Srinivasan, P.; Sandler, A.; Dasgupta, S.; Ravi, R.; Guerrero-Preston, R.; Trink, B. Cold plasma selectivity and the possibility of a paradigm shift in cancer therapy. Br. J. Cancer 2011, 105, 1295–1301. [Google Scholar] [CrossRef]
- Zucker, S.N.; Zirnheld, J.; Bagati, A.; Disanto, T.M.; Soye, B.D.; Wawrzyniak, J.A.; Etemadi, K.; Nikiforov, M.; Berezney, R. Preferential induction of apoptotic cell death in melanoma cells as compared with normal keratinocytes using a non-thermal plasma torch. Cancer Biol. Ther. 2012, 13, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, M.; Robert, E.; Lerondel, S.; Sarron, V.; Ries, D.; Dozias, S.; Sobilo, J.; Gosset, D.; Kieda, C.; Legrain, B.; et al. ROS implication in a new antitumor strategy based on non-thermal plasma. Int. J. Cancer 2011, 130, 2185–2194. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Preston, R.; Ogawa, T.; Uemura, M.; Shumulinsky, G.; Valle, B.L.; Pirini, F.; Ravi, R.; Sidransky, D.; Keidar, M.; Trink, B. Cold atmospheric plasma treatment selectively targets head and neck squamous cell carcinoma cells. Int. J. Mol. Med. 2014, 34, 941–946. [Google Scholar] [CrossRef] [Green Version]
- Hirst, A.M.; Simms, M.S.; Mann, V.M.; Maitland, N.; O’Connell, D.; Frame, F.M. Low-temperature plasma treatment induces DNA damage leading to necrotic cell death in primary prostate epithelial cells. Br. J. Cancer 2015, 112, 1536–1545. [Google Scholar] [CrossRef] [Green Version]
- Adachi, T.; Tanaka, H.; Nonomura, S.; Hara, H.; Kondo, S.-I.; Hori, M. Plasma-activated medium induces A549 cell injury via a spiral apoptotic cascade involving the mitochondrial–nuclear network. Free Radic. Biol. Med. 2015, 79, 28–44. [Google Scholar] [CrossRef]
- Saito, K.; Asai, T.; Fujiwara, K.; Sahara, J.; Koguchi, H.; Fukuda, N.; Suzuki-Karasaki, M.; Soma, M.; Suzuki-Karasaki, Y. Tumor-selective mitochondrial network collapse induced by atmospheric gas plasma-activated medium. Oncotarget 2016, 7, 19910–19927. [Google Scholar] [CrossRef] [Green Version]
- Tokunaga, T.; Ando, T.; Suzuki-Karasaki, M.; Ito, T.; Onoe-Takahashi, A.; Ochiai, T.; Soma, M.; Suzuki-Karasaki, Y. Plasma-stimulated medium kills TRAIL-resistant human malignant cells by promoting caspase-independent cell death via membrane potential and calcium dynamics modulation. Int. J. Oncol. 2018, 52, 697–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Ando, T.; Suzuki-Karasaki, M.; Tokunaga, T.; Yoshida, Y.; Ochiai, T.; Tokuhashi, Y.; Suzuki-Karasaki, Y. Cold PSM, but not TRAIL, triggers autophagic cell death: A therapeutic advantage of PSM over TRAIL. Int. J. Oncol. 2018, 53, 503–514. [Google Scholar] [CrossRef]
- Tornin, J.; Sanz, M.M.; Rodríguez, A.; Labay, C.; Rodriguez, R.; Canal, C. Pyruvate Plays a Main Role in the Antitumoral Selectivity of Cold Atmospheric Plasma in Osteosarcoma. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Azzariti, A.; Iacobazzi, R.M.; Di Fonte, R.; Porcelli, L.; Gristina, R.; Favia, P.; Fracassi, F.; Trizio, I.; Silvestris, N.; Guida, G.; et al. Plasma-activated medium triggers cell death and the presentation of immune activating danger signals in melanoma and pancreatic cancer cells. Sci. Rep. 2019, 9, 4099. [Google Scholar] [CrossRef] [PubMed]
- Ando, T.; Suzuki-Karasaki, M.; Suzuki-Karasaki, M.; Ichikawa, J.; Ochiai, T.; Yoshida, Y.; Haro, H.; Suzuki-Karasaki, Y. Combined Anticancer Effect of Plasma-Activated Infusion and Salinomycin by Targeting Autophagy and Mitochondrial Morphology. Front. Oncol. 2021, 11, 3127. [Google Scholar] [CrossRef]
- Yan, D.; Talbot, A.; Nourmohammadi, N.; Cheng, X.; Canady, J.; Sherman, J.H.; Keidar, M. Principles of using Cold Atmospheric Plasma Stimulated Media for Cancer Treatment. Sci. Rep. 2015, 5, 18339. [Google Scholar] [CrossRef] [Green Version]
- Girard, P.-M.; Arbabian, A.; Fleury, M.; Bauville, G.; Puech, V.; Dutreix, M.; Sousa, J.S. Synergistic Effect of H2O2 and NO2 in Cell Death Induced by Cold Atmospheric He Plasma. Sci. Rep. 2016, 6, 29098. [Google Scholar] [CrossRef] [Green Version]
- Kurake, N.; Tanaka, H.; Ishikawa, K.; Kondo, T.; Sekine, M.; Nakamura, K.; Kajiyama, H.; Kikkawa, F.; Mizuno, M.; Hori, M. Cell survival of glioblastoma grown in medium containing hydrogen peroxide and/or nitrite, or in plasma-activated medium. Arch. Biochem. Biophys. 2016, 605, 102–108. [Google Scholar] [CrossRef]
- Bauer, G. The synergistic effect between hydrogen peroxide and nitrite, two long-lived molecular species from cold atmospheric plasma, triggers tumor cells to induce their own cell death. Redox Biol. 2019, 26, 101291. [Google Scholar] [CrossRef]
- Landes, T.; Martinou, J.-C. Mitochondrial outer membrane permeabilization during apoptosis: The role of mitochondrial fission. Biochim. Et Biophys. Acta (BBA)-Bioenerg. 2011, 1813, 540–545. [Google Scholar] [CrossRef]
- Elgass, K.; Pakay, J.; Ryan, M.; Palmer, C. Recent advances into the understanding of mitochondrial fission. Biochim. Biophys. Acta 2013, 1833, 150–161. [Google Scholar] [CrossRef] [Green Version]
- Twig, G.; Shirihai, O.S. The Interplay Between Mitochondrial Dynamics and Mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef] [Green Version]
- Hoppins, S.; Lackner, L.; Nunnari, J. The Machines that Divide and Fuse Mitochondria. Annu. Rev. Biochem. 2007, 76, 751–780. [Google Scholar] [CrossRef] [PubMed]
- Soubannier, V.; McBride, H.M. Positioning mitochondrial plasticity within cellular signaling cascades. Biochim. Biophys. Acta (BBA)-Bioenerg. 2009, 1793, 154–170. [Google Scholar] [CrossRef] [Green Version]
- Pendin, D.; Filadi, R.; Pizzo, P. The Concerted Action of Mitochondrial Dynamics and Positioning: New Characters in Cancer Onset and Progression. Front. Oncol. 2017, 7, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Schumacker, P.T.; Chandel, N.S. Mitochondrial Reactive Oxygen Species Trigger Hypoxia-Inducible Factor-Dependent Extension of the Replicative Life Span during Hypoxia. Mol. Cell. Biol. 2007, 27, 5737–5745. [Google Scholar] [CrossRef] [Green Version]
- Desireddi, J.R.; Farrow, K.N.; Marks, J.D.; Waypa, G.B.; Schumacker, P.T. Hypoxia Increases ROS Signaling and Cytosolic Ca2+ in Pulmonary Artery Smooth Muscle Cells of Mouse Lungs Slices. Antioxid. Redox Signal. 2010, 12, 595–602. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Regulation of Metabolism by Hypoxia-Inducible Factor 1. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 347–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Mehdi, A.-B.; Pastukh, V.M.; Swiger, B.M.; Reed, D.J.; Patel, M.R.; Bardwell, G.C.; Pastukh, V.V.; Alexeyev, M.F.; Gillespie, M.N. Perinuclear Mitochondrial Clustering Creates an Oxidant-Rich Nuclear Domain Required for Hypoxia-Induced Transcription. Sci. Signal. 2012, 5, ra47. [Google Scholar] [CrossRef] [Green Version]
- Akita, M.; Suzuki-Karasaki, M.; Fujiwara, K.; Nakagawa, C.; Soma, M.; Yoshida, Y.; Ochiai, T.; Tokuhashi, Y.; Suzuki-Karasaki, Y. Mitochondrial division inhibitor-1 induces mitochondrial hyperfusion and sensitizes human cancer cells to TRAIL-induced apoptosis. Int. J. Oncol. 2014, 45, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Suzuki-Karasaki, Y.; Fujiwara, K.; Saito, K.; Suzuki-Karasaki, M.; Ochiai, T.; Soma, M. Distinct effects of TRAIL on the mitochondrial network in human cancer cells and normal cells: Role of plasma membrane depolarization. Oncotarget 2015, 6, 21572–21588. [Google Scholar] [CrossRef] [Green Version]
- Hollenbeck, P.J.; Saxton, W.M. The axonal transport of mitochondria. J. Cell Sci. 2005, 118, 5411–5419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunniff, B.; McKenzie, A.J.; Heintz, N.H.; Howe, A.K. AMPK activity regulates trafficking of mitochondria to the leading edge during cell migration and matrix invasion. Mol. Biol. Cell 2016, 27, 2662–2674. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kanai, Y.; Okada, Y.; Nonaka, S.; Takeda, S.; Harada, A.; Hirokawa, N. Targeted Disruption of Mouse Conventional Kinesin Heavy Chain kif5B, Results in Abnormal Perinuclear Clustering of Mitochondria. Cell 1998, 93, 1147–1158. [Google Scholar] [CrossRef] [Green Version]
- Pilling, A.D.; Horiuchi, D.; Lively, C.M.; Saxton, W.M. Kinesin-1 and Dynein Are the Primary Motors for Fast Transport of Mitochondria inDrosophilaMotor Axons. Mol. Biol. Cell 2006, 17, 2057–2068. [Google Scholar] [CrossRef] [Green Version]
- Hasler, S.L.-A. Cholesterol, Cardiolipin, and Mitochondria Permeabilisation. Anti-Cancer Agents Med. Chem. 2012, 12, 329–339. [Google Scholar] [CrossRef]
- Kaminskyy, V.; Zhivotovsky, B. Free Radicals in Cross Talk Between Autophagy and Apoptosis. Antioxid. Redox Signal. 2014, 21, 86–102. [Google Scholar] [CrossRef]
- Ahmadpour, S.; Mahéo, K.; Servais, S.; Brisson, L.; Dumas, J.-F. Cardiolipin, the Mitochondrial Signature Lipid: Implication in Cancer. Int. J. Mol. Sci. 2020, 21, 8031. [Google Scholar] [CrossRef] [PubMed]
- Petit, J.-M.; Maftah, A.; Ratinaud, M.-H.; Julien, R. 10N-Nonyl acridine orange interacts with cardiolipin and allows the quantification of this phospholipid in isolated mitochondria. JBIC J. Biol. Inorg. Chem. 1992, 209, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vučković, A.-M.; Travain, V.B.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020, 28, 101328. [Google Scholar] [CrossRef]
- Tochigi, M.; Inoue, T.; Suzuki-Karasaki, M.; Ochiai, T.; Ra, C.; Suzuki-Karasaki, Y. Hydrogen peroxide induces cell death in human TRAIL-resistant melanoma through intracellular superoxide generation. Int. J. Oncol. 2013, 42, 863–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohshima, Y.; Takata, N.; Suzuki-Karasaki, M.; Yoshida, Y.; Tokuhashi, Y.; Suzuki-Karasaki, Y. Disrupting mitochondrial Ca2+ homeostasis causes tumor-selective TRAIL sensitization through mitochondrial network abnormalities. Int. J. Oncol. 2017, 51, 1146–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaschler, M.M.; Hu, F.; Feng, H.; Linkermann, A.; Min, W.; Stockwell, B.R. Determination of the Subcellular Localization and Mechanism of Action of Ferrostatins in Suppressing Ferroptosis. ACS Chem. Biol. 2018, 13, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Quintana, A.; Schwarz, E.C.; Schwindling, C.; Lipp, P.; Kaestner, L.; Hoth, M. Sustained Activity of Calcium Release-activated Calcium Channels Requires Translocation of Mitochondria to the Plasma Membrane. J. Biol. Chem. 2006, 281, 40302–40309. [Google Scholar] [CrossRef] [Green Version]
- Hajnóczky, G.; Saotome, M.; Csordás, G.; Weaver, D.; Yi, M. Calcium Signalling and Mitochondrial Motility. Novartis Found. Symp. 2008, 287, 105–121. [Google Scholar] [CrossRef]
- Niescier, R.F.; Chang, K.T.; Min, K.-T. Miro, MCU, and calcium: Bridging our understanding of mitochondrial movement in axons. Front. Cell. Neurosci. 2013, 7, 148. [Google Scholar] [CrossRef] [Green Version]
- Madreiter-Sokolowski, C.T.; Ramadani-Muja, J.; Ziomek, G.; Burgstaller, S.; Bischof, H.; Koshenov, Z.; Gottschalk, B.; Malli, R.; Graier, W. Tracking intra- and inter-organelle signaling of mitochondria. FEBS J. 2019, 286, 4378–4401. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Sagua, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Muñoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER–mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mironov, S.L.; Ivannikov, M.V.; Johansson, M. [Ca2+] Signaling between Mitochondria and Endoplasmic Reticulum in Neurons Is Regulated by Microtubules. From mitochondrial permeability transition pore to Ca2+-induced Ca2+ release. J. Biol. Chem. 2005, 280, 715–721. [Google Scholar] [CrossRef] [Green Version]
- Debattisti, V.; Gerencser, A.A.; Saotome, M.; Das, S.; Hajnóczky, G. ROS Control Mitochondrial Motility through p38 and the Motor Adaptor Miro/Trak. Cell Rep. 2017, 21, 1667–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alshaabi, H.; Shannon, N.; Gravelle, R.; Milczarek, S.; Messier, T.; Cunniff, B. Miro1-mediated mitochondrial positioning supports subcellular redox status. Redox Biol. 2020, 38, 101818. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suzuki-Karasaki, M.; Ando, T.; Ochiai, Y.; Kawahara, K.; Suzuki-Karasaki, M.; Nakayama, H.; Suzuki-Karasaki, Y. Air Plasma-Activated Medium Evokes a Death-Associated Perinuclear Mitochondrial Clustering. Int. J. Mol. Sci. 2022, 23, 1124. https://doi.org/10.3390/ijms23031124
Suzuki-Karasaki M, Ando T, Ochiai Y, Kawahara K, Suzuki-Karasaki M, Nakayama H, Suzuki-Karasaki Y. Air Plasma-Activated Medium Evokes a Death-Associated Perinuclear Mitochondrial Clustering. International Journal of Molecular Sciences. 2022; 23(3):1124. https://doi.org/10.3390/ijms23031124
Chicago/Turabian StyleSuzuki-Karasaki, Manami, Takashi Ando, Yushi Ochiai, Kenta Kawahara, Miki Suzuki-Karasaki, Hideki Nakayama, and Yoshihiro Suzuki-Karasaki. 2022. "Air Plasma-Activated Medium Evokes a Death-Associated Perinuclear Mitochondrial Clustering" International Journal of Molecular Sciences 23, no. 3: 1124. https://doi.org/10.3390/ijms23031124
APA StyleSuzuki-Karasaki, M., Ando, T., Ochiai, Y., Kawahara, K., Suzuki-Karasaki, M., Nakayama, H., & Suzuki-Karasaki, Y. (2022). Air Plasma-Activated Medium Evokes a Death-Associated Perinuclear Mitochondrial Clustering. International Journal of Molecular Sciences, 23(3), 1124. https://doi.org/10.3390/ijms23031124