
Modulating mTOR Signaling as a Promising Therapeutic Strategy for Atherosclerosis

 ,
,  ,
, {kind=link}
Abstract
:1. Atherosclerosis
2. mTOR Signaling in Health
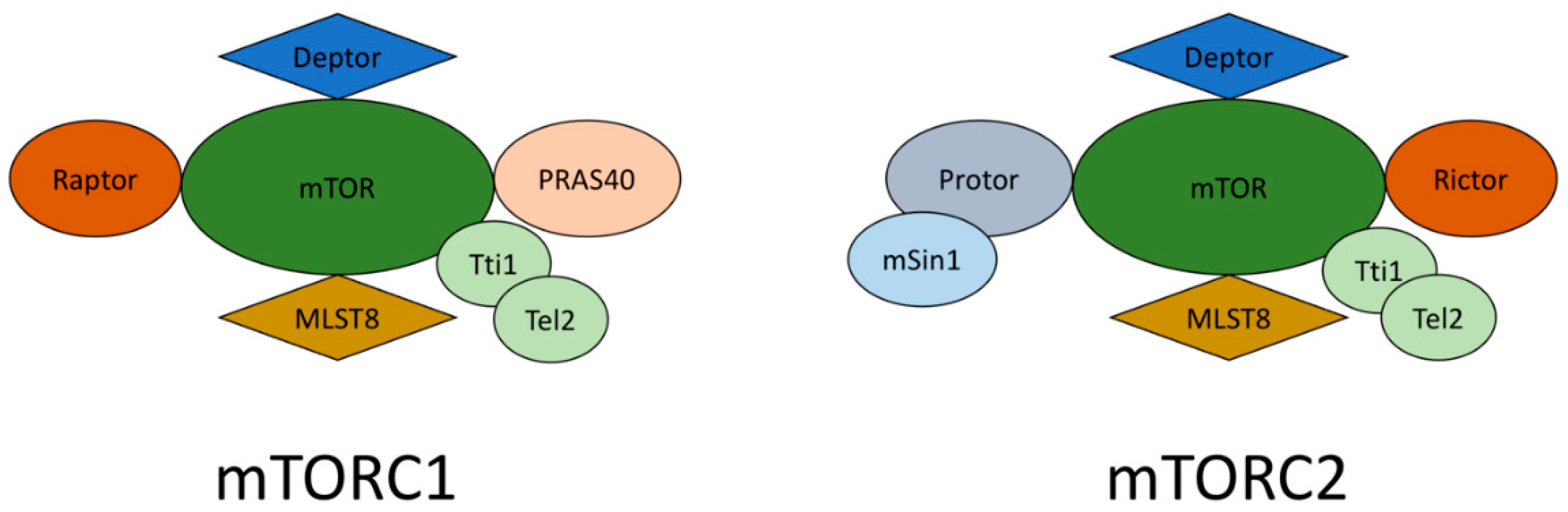
mTORC1 and mTORC2
3. mTOR Signaling in Atherosclerosis
3.1. mTOR Modulation in Atherosclerosis
3.2. mTORC1 Inhibition in Atherosclerosis
4. Clinical Suitability
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Poston, R.N. Atherosclerosis: Integration of its pathogenesis as a self-perpetuating propagating inflammation: A review. Cardiovasc. Endocrinol Metab. 2019, 2, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Lappalainen, J.; Yeung, N.; Nguyen, S.D.; Jauhiainen, M.; Kovanen, P.T.; Lee-Rueckert, M. Cholesterol loading suppresses the atheroinflammatory gene polarization of human macrophages induced by colony stimulating factors. Sci. Rep. 2021, 11, 4923. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.P. Is atherosclerosis a pediatric disease? [Updated 2020]. In Endotext [Internet]; Feingold, K.R., Anawalt, B., Boyce, A., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK395576/ (accessed on 17 August 2021).
- Asada, Y.; Yamashita, A.; Sato, Y.; Hatakeyama, K. Pathophysiology of atherothrombosis: Mechanisms of thrombus formation on disrupted atherosclerotic plaques. Pathol. Int. 2020, 70, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.V.; Grechko, A.V.; Orekhova, V.A.; Chegodaev, Y.S.; Wu, W.K.; Orekhov, A.N. Oxidative Stress and Antioxidants in Atherosclerosis Development and Treatment. Biology 2020, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Bobryshev, Y.V.; Ivanova, E.A.; Chistiakov, D.A.; Nikiforov, N.G.; Orekhov, A.N. Macrophages and Their Role in Atherosclerosis: Pathophysiology and Transcriptome Analysis. Biomed. Res. Int. 2016, 2016, 9582430. [Google Scholar] [CrossRef] [Green Version]
- Westman, J.; Grinstein, S.; Marques, P.E. Phagocytosis of Necrotic Debris at Sites of Injury and Inflammation. Front. Immunol. 2020, 10, 3030. [Google Scholar] [CrossRef]
- Hansson, G.K.; Libby, P.; Tabas, I. Inflammation and plaque vulnerability. J. Intern Med. 2015, 278, 483–493. [Google Scholar] [CrossRef]
- Toth, P.P.; Banach, M. Statins: Then and Now. Methodist Debakey Cardiovasc. J. 2019, 15, 23–31. [Google Scholar] [CrossRef]
- Mach, F.; Ray, K.K.; Wiklund, O.; Corsini, A.; Catapano, A.L.; Bruckert, E.; De Backer, G.; Hegele, R.A.; Hovingh, G.K.; Jacobson, T.A.; et al. European Atherosclerosis Society Consensus Panel. Adverse effects of statin therapy: Perception vs. the evidence-focus on glucose homeostasis, cognitive, renal and hepatic function, haemorrhagic stroke and cataract. Eur. Heart. J. 2018, 39, 2526–2539. [Google Scholar] [CrossRef]
- Cho, K.H.; Hong, Y.J. Proprotein convertase subtilisin/kexin type 9 inhibition in cardiovascular disease: Current status and future perspectives. Korean J. Intern. Med. 2020, 35, 1045–1058. [Google Scholar] [CrossRef]
- Kurdi, A.; De Meyer, G.R.; Martinet, W. Potential therapeutic effects of mTOR inhibition in atherosclerosis. Br. J. Clin. Pharmacol. 2016, 82, 1267–1279. [Google Scholar] [CrossRef] [Green Version]
- Angliker, N.; Burri, M.; Zaichuk, M.; Fritschy, J.M.; Rüegg, M.A. mTORC1 and mTORC2 have largely distinct functions in Purkinje cells. Eur. J. Neurosci. 2015, 42, 2595–2612. [Google Scholar] [CrossRef] [Green Version]
- Kezic, A.; Popovic, L.; Lalic, K. mTOR Inhibitor Therapy and Metabolic Consequences: Where Do We Stand? Oxid. Med. Cell. Longev. 2018, 2018, 2640342. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976, Erratum in Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [Green Version]
- Stallone, G.; Infante, B.; Prisciandaro, C.; Grandaliano, G. mTOR and Aging: An Old Fashioned Dress. Int. J. Mol. Sci. 2019, 20, 2774. [Google Scholar] [CrossRef] [Green Version]
- Yoon, M.S. The Role of Mammalian Target of Rapamycin (mTOR) in Insulin Signaling. Nutrients 2017, 9, 1176. [Google Scholar] [CrossRef]
- Chellappa, K.; Brinkman, J.A.; Mukherjee, S.; Morrison, M.; Alotaibi, M.I.; Carbajal, K.A.; Alhadeff, A.L.; Perron, I.J.; Yao, R.; Purdy, C.S.; et al. Hypothalamic mTORC2 is essential for metabolic health and longevity. Aging Cell. 2019, 18, e13014. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Felix, B.; Othmer, H.G. The Roles of Signaling in Cytoskeletal Changes, Random Movement, Direction-Sensing and Polarization of Eukaryotic Cells. Cells 2020, 9, 1437. [Google Scholar] [CrossRef]
- Saleiro, D.; Platanias, L.C. Intersection of mTOR and STAT signaling in immunity. Trends Immunol. 2015, 36, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Yao, C.A.; Ortiz-Vega, S.; Sun, Y.Y.; Chien, C.T.; Chuang, J.H.; Lin, Y. Association of mSin1 with mTORC2 Ras and Akt reveals a crucial domain on mSin1 involved in Akt phosphorylation. Oncotarget 2017, 8, 63392–63404. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Li, X.; Zhang, J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef] [Green Version]
- Oh, W.J.; Jacinto, E. mTOR complex 2 signaling and functions. Cell Cycle 2011, 10, 2305–2316. [Google Scholar] [CrossRef]
- Qin, X.; Jiang, B.; Zhang, Y. 4E-BP1, a multifactor regulated multifunctional protein. Cell Cycle 2016, 15, 781–786. [Google Scholar] [CrossRef] [Green Version]
- Kamran, H.; Kupferstein, E.; Sharma, N.; Karam, J.G.; Myers, A.K.; Youssef, I.; Sowers, J.R.; Gustafson, D.R.; Salifu, M.O.; McFarlane, S.I. Statins and New-Onset Diabetes in Cardiovascular and Kidney Disease Cohorts: A Meta-Analysis. Cardiorenal. Med. 2018, 8, 105–112. [Google Scholar] [CrossRef]
- Glynn, E.L.; Lujan, H.L.; Kramer, V.J.; Drummondm, M.J.; DiCarlo, S.E.; Rasmussen, B.B. A chronic increase in physical activity inhibits fed-state mTOR/S6K1 signaling and reduces IRS-1 serine phosphorylation in rat skeletal muscle. Appl. Physiol. Nutr. Metab. 2008, 33, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.; Bell, C.M.; Rothbart, S.B.; Moran, R.G. AMP-activated Protein Kinase (AMPK) Control of mTORC1 Is p53- and TSC2-independent in Pemetrexed-treated Carcinoma Cells. J. Biol. Chem. 2015, 290, 27473–27486. [Google Scholar] [CrossRef] [Green Version]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [Green Version]
- Zhu, P.J.; Chen, C.J.; Mays, J.; Stoica, L.; Costa-Mattioli, M. mTORC2, but not mTORC1, is required for hippocampal mGluR-LTD and associated behaviors. Nat. Neurosci. 2018, 21, 799–802. [Google Scholar] [CrossRef]
- Fu, W.; Hall, M.N. Regulation of mTORC2 Signaling. Genes 2020, 11, 1045. [Google Scholar] [CrossRef]
- Tsuchiya, A.; Kanno, T.; Nagaya, H.; Shimizu, T.; Tanaka, A.; Nishizaki, T. PTP1B inhibition causes Rac1 activation by enhancing receptor tyrosine kinase signaling. Cell Physiol. Biochem. 2014, 33, 1097–1105. [Google Scholar] [CrossRef]
- Dan, H.C.; Antonia, R.J.; Baldwin, A.S. PI3K/Akt promotes feedforward mTORC2 activation through IKKα. Oncotarget 2016, 7, 21064–21075. [Google Scholar] [CrossRef] [Green Version]
- Mok, K.W.; Mruk, D.D.; Lee, W.M.; Cheng, C.Y. Rictor/mTORC2 regulates blood-testis barrier dynamics via its effects on gap junction communications and actin filament network. FASEB J. 2013, 27, 1137–1152. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Hua, H.; Li, M.; Liu, S.; Kong, Q.; Shao, T.; Wang, J.; Luo, Y.; Wang, Q.; Luo, T.; et al. mTORC2 promotes type I insulin-like growth factor receptor and insulin receptor activation through the tyrosine kinase activity of mTOR. Cell Res. 2016, 26, 46–65. [Google Scholar] [CrossRef] [Green Version]
- Samidurai, A.; Kukreja, R.C.; Das, A. Emerging Role of mTOR Signaling-Related miRNAs in Cardiovascular Diseases. Oxid. Med. Cell Longev 2018, 2018, 6141902. [Google Scholar] [CrossRef]
- Xu, L.; Brink, M. mTOR, cardiomyocytes and inflammation in cardiac hypertrophy. Biochim. Biophys Acta 2016, 1863, 1894–1903. [Google Scholar] [CrossRef]
- Daneshgar, N.; Rabinovitch, P.S.; Dai, D.F. TOR Signaling Pathway in Cardiac Aging and Heart Failure. Biomolecules 2021, 11, 168. [Google Scholar] [CrossRef]
- Sciarretta, S.; Forte, M.; Frati, G.; Sadoshima, J. New Insights Into the Role of mTOR Signaling in the Cardiovascular System. Circ. Res. 2018, 122, 489–505. [Google Scholar] [CrossRef]
- Sciarretta, S.; Volpe, M.; Sadoshima, J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ. Res. 2014, 114, 549–564. [Google Scholar] [CrossRef] [Green Version]
- Sanches-Silva, A.; Testai, L.; Nabavi, S.F.; Battino, M.; Pandima Devi, K.; Tejada, S.; Sureda, A.; Xu, S.; Yousefi, B.; Majidinia, M.; et al. Therapeutic potential of polyphenols in cardiovascular diseases: Regulation of mTOR signaling pathway. Pharm. Res. 2020, 152, 104626. [Google Scholar] [CrossRef]
- Seto, B. Rapamycin and mTOR: A serendipitous discovery and implications for breast cancer. Clin. Transl. Med. 2012, 1, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.R.; Yoo, Y.J.; Ban, Y.H.; Yoon, Y.J. Biosynthesis of rapamycin and its regulation: Past achievements and recent progress. J. Antibiot. Tokyo 2010, 63, 434–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, Y.J.; Kim, H.; Park, S.R.; Yoon, Y.J. An overview of rapamycin: From discovery to future perspectives. J. Ind. Microbiol. Biotechnol. 2017, 44, 537–553. [Google Scholar] [CrossRef] [PubMed]
- Graziani, E.I.; Ritacco, F.V.; Summers, M.Y.; Zabriskie, T.M.; Yu, K.; Bernan, V.S.; Greenstein, M.; Carter, G.T. Novel sulfur-containing rapamycin analogs prepared by precursor-directed biosynthesis. Org. Lett. 2003, 5, 2385–2388. [Google Scholar] [CrossRef]
- Kolos, J.M.; Voll, A.M.; Bauder, M.; Hausch, F. FKBP Ligands-Where We Are and Where to Go? Front. Pharmacol. 2018, 9, 1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Jiang, X.; Li, B.; Yang, H.J.; Miller, M.; Yang, A.; Dhar, A.; Pavletich, N.P. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 2017, 552, 368–373. [Google Scholar] [CrossRef]
- Sato, K.; Yamashita, T.; Shirai, R.; Shibata, K.; Okano, T.; Yamaguchi, M.; Mori, Y.; Hirano, T.; Watanabe, T. Adropin Contributes to Anti-Atherosclerosis by Suppressing Monocyte-Endothelial Cell Adhesion and Smooth Muscle Cell Proliferation. Int. J. Mol. Sci. 2018, 19, 1293. [Google Scholar] [CrossRef] [Green Version]
- Mehla, K.; Singh, P.K. Metabolic Regulation of Macrophage Polarization in Cancer. Trends Cancer 2019, 5, 822–834. [Google Scholar] [CrossRef]
- Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuñiga, F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 122. [Google Scholar] [CrossRef]
- Manolis, A.S.; Manolis, A.A.; Manolis, T.A.; Apostolaki, N.E.; Apostolopoulos, E.J.; Melita, H.; Katsiki, N. Mitochondrial dysfunction in cardiovascular disease: Current status of translational research/clinical and therapeutic implications. Med. Res. Rev. 2021, 41, 275–313. [Google Scholar] [CrossRef] [PubMed]
- Gori, T. Vascular Wall Reactions to Coronary Stents-Clinical Implications for Stent Failure. Life Basel 2021, 11, 63. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, H.; Takemura, G.; Goto, K.; Maruyama, R.; Tsujimoto, A.; Ogino, A.; Takeyama, T.; Kawaguchi, T.; Watanabe, T.; Fujiwara, T.; et al. The role of autophagy emerging in postinfarction cardiac remodelling. Cardiovasc. Res. 2011, 9, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Houssaini, A.; Abid, S.; Mouraret, N.; Wan, F.; Rideau, D.; Saker, M.; Marcos, E.; Tissot, C.M.; Dubois-Randé, J.L.; Amsellem, V.; et al. Rapamycin reverses pulmonary artery smooth muscle cell proliferation in pulmonary hypertension. Am. J. Respir. Cell. Mol. Biol. 2013, 48, 568–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef] [PubMed]
- Serruys, P.W.; Regar, E.; Carter, A.J. Rapamycin eluting stent: The onset of a new era in interventional cardiology. Heart 2002, 87, 305–307. [Google Scholar] [CrossRef] [Green Version]
- Stone, G.W.; Kedhi, E.; Kereiakes, D.J.; Parise, H.; Fahy, M.; Serruys, P.W.; Smits, P.C. Differential clinical responses to everolimus-eluting and Paclitaxel-eluting coronary stents in patients with and without diabetes mellitus. Circulation 2011, 124, 893–900. [Google Scholar] [CrossRef] [Green Version]
- Martinet, W.; De Loof, H.; De Meyer, G.R.Y. mTOR inhibition: A promising strategy for stabilization of atherosclerotic plaques. Atherosclerosis 2014, 233, 601–607. [Google Scholar] [CrossRef]
- Hoshii, T.; Tadokoro, Y.; Naka, K.; Ooshio, T.; Muraguchi, T.; Sugiyama, N.; Soga, T.; Araki, K.; Yamamura, K.; Hirao, A. mTORC1 is essential for leukemia propagation but not stem cell self-renewal. J. Clin. Investig. 2012, 122, 2114–2129. [Google Scholar] [CrossRef]
- Sergin, I.; Razani, B. Self-eating in the plaque: What macrophage autophagy reveals about atherosclerosis. Trends Endocrinol. Metab. 2014, 25, 225–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libby, P. Inflammation in Atherosclerosis-No Longer a Theory. Clin. Chem. 2021, 67, 131–142. [Google Scholar] [CrossRef]
- You, G.; Long, X.; Song, F.; Huang, J.; Tian, M.; Xiao, Y.; Deng, S.; Wu, Q. Metformin Activates the AMPK-mTOR Pathway by Modulating lncRNA TUG1 to Induce Autophagy and Inhibit Atherosclerosis. Drug. Des. Devel. Ther. 2020, 14, 457–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grünwald, V.; Weikert, S.; Pavel, M.E.; Hörsch, D.; Lüftner, D.; Janni, W.; Geberth, M.; Weber, M.M. Practical management of everolimus-related toxicities in patients with advanced solid tumors. Onkologie 2013, 36, 295–302. [Google Scholar] [CrossRef] [PubMed]
- MacKeigan, J.P.; Krueger, D.A. Differentiating the mTOR inhibitors everolimus and sirolimus in the treatment of tuberous sclerosis complex. Neuro. Oncol. 2015, 17, 1550–1559. [Google Scholar] [CrossRef]
- Puranik, A.S.; Dawson, E.R.; Peppas, N.A. Recent advances in drug eluting stents. Int. J. Pharm. 2013, 441, 665–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, M.; Saxena, A.; Kingswood, J.C. Management of everolimus-associated adverse events in patients with tuberous sclerosis complex: A practical guide. Orphanet. J. Rare Dis. 2017, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Mueller, M.A.; Beutner, F.; Teupser, D.; Ceglarek, U.; Thiery, J. Prevention of atherosclerosis by the mTOR inhibitor everolimus in LDLR-/- mice despite severe hypercholesterolemia. Atherosclerosis 2008, 198, 39–48. [Google Scholar] [CrossRef]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride Metabolism in the Liver. Compr. Physiol. 2017, 8, 1–8. [Google Scholar] [CrossRef]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef] [Green Version]
- Linton, M.R.F.; Yancey, P.G.; Davies, S.S.; Jerome, W.G.; Linton, E.F.; Song, W.L.; Doran, A.C.; Vickers, K.C.; Feingold, K.R.; Anawalt, B.; et al. The role of lipids and lipoproteins in atherosclerosis. [Updated 2019 Jan 3]. In Endotext [Internet]; Feingold, K.R., Anawalt, B., Boyce, A., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK343489/ (accessed on 17 August 2021).
- Lamming, D.W.; Sabatini, D.M. A Central role for mTOR in lipid homeostasis. Cell. Metab. 2013, 18, 465–469. [Google Scholar] [CrossRef] [Green Version]
- Blagosklonny, M.V. Fasting and rapamycin: Diabetes versus benevolent glucose intolerance. Cell. Death Dis. 2019, 10, 607. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.; Tomasiewicz, J.L.; Yang, S.E.; Miller, B.R.; Wakai, M.H.; Sherman, D.S.; Cummings, N.E.; Baar, E.L.; Brinkman, J.A.; Syed, F.A.; et al. Calorie-Restriction-Induced Insulin Sensitivity Is Mediated by Adipose mTORC2 and Not Required for Lifespan Extension. Cell. Rep. 2019, 29, 236–248.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Ye, S. Rapamycin improves insulin resistance and hepatic steatosis in type 2 diabetes rats through activation of autophagy. Cell. Biol. Int. 2018, 42, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Ding, T.; Cyrus, T.; Cheng, Y.; Tian, H.; Ma, M.; Falotico, R.; Praticò, D. Low-dose oral sirolimus reduces atherogenesis, vascular inflammation and modulates plaque composition in mice lacking the LDL receptor. Br. J. Pharmacol. 2009, 156, 774–785. [Google Scholar] [CrossRef] [Green Version]
- Ong, P.S.; Wang, L.Z.; Dai, X.; Tseng, S.H.; Loo, S.J.; Sethi, G. Judicious Toggling of mTOR Activity to Combat Insulin Resistance and Cancer: Current Evidence and Perspectives. Front. Pharmacol. 2016, 7, 395. [Google Scholar] [CrossRef] [PubMed]
- Faes, S.; Demartines, N.; Dormond, O. Resistance to mTORC1 Inhibitors in Cancer Therapy: From Kinase Mutations to Intratumoral Heterogeneity of Kinase Activity. Oxid. Med. Cell. Longev. 2017, 2017, 1726078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurdi, A.; De Doncker, M.; Leloup, A.; Neels, H.; Timmermans, J.P.; Lemmens, K.; Apers, S.; De Meyer, G.R.Y.; Martinet, W. Continuous administration of the mTORC1 inhibitor everolimus induces tolerance and decreases autophagy in mice. Br. J. Pharmacol. 2016, 173, 3359–3371. [Google Scholar] [CrossRef] [Green Version]
- Klop, B.; Elte, J.W.; Cabezas, M.C. Dyslipidemia in obesity: Mechanisms and potential targets. Nutrients 2013, 5, 1218–1240. [Google Scholar] [CrossRef] [Green Version]
- Luo, F.; Guo, Y.; Ruan, G.Y.; Long, J.K.; Zheng, X.L.; Xia, Q.; Zhao, S.P.; Peng, D.Q.; Fang, Z.F.; Li, X.P. Combined use of metformin and atorvastatin attenuates atherosclerosis in rabbits fed a high-cholesterol diet. Sci. Rep. 2017, 7, 2169. [Google Scholar] [CrossRef]
- Myasoedova, V.A.; Di Minno, A.; Songia, P.; Massaiu, I.; Alfieri, V.; Valerio, V.; Moschetta, D.; Andreini, D.; Alamanni, F.; Pepi, M.; et al. Sex-specific differences in age-related aortic valve calcium load: A systematic review and meta-analysis. Ageing Res. Rev. 2020, 61, 101077. [Google Scholar] [CrossRef]
- Howell, J.J.; Hellberg, K.; Turner, M.; Talbott, G.; Kolar, M.J.; Ross, D.S.; Hoxhaj, G.; Saghatelian, A.; Shaw, R.J.; Manning, B.D. Metformin Inhibits Hepatic mTORC1 Signaling via Dose-Dependent Mechanisms Involving AMPK and the TSC Complex. Cell. Metab. 2017, 25, 463–471. [Google Scholar] [CrossRef] [Green Version]
- Blagosklonny, M.V. Rapamycin for longevity: Opinion article. Aging Albany NY 2019, 11, 8048–8067. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poznyak, A.V.; Sukhorukov, V.N.; Zhuravlev, A.; Orekhov, N.A.; Kalmykov, V.; Orekhov, A.N. Modulating mTOR Signaling as a Promising Therapeutic Strategy for Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 1153. https://doi.org/10.3390/ijms23031153
Poznyak AV, Sukhorukov VN, Zhuravlev A, Orekhov NA, Kalmykov V, Orekhov AN. Modulating mTOR Signaling as a Promising Therapeutic Strategy for Atherosclerosis. International Journal of Molecular Sciences. 2022; 23(3):1153. https://doi.org/10.3390/ijms23031153
Chicago/Turabian StylePoznyak, Anastasia V., Vasily N. Sukhorukov, Alexander Zhuravlev, Nikolay A. Orekhov, Vladislav Kalmykov, and Alexander N. Orekhov. 2022. "Modulating mTOR Signaling as a Promising Therapeutic Strategy for Atherosclerosis" International Journal of Molecular Sciences 23, no. 3: 1153. https://doi.org/10.3390/ijms23031153