
Transforming Growth Factor-Beta in Skeletal Muscle Wasting


{kind=link}
Abstract
:1. Introduction and Early Work
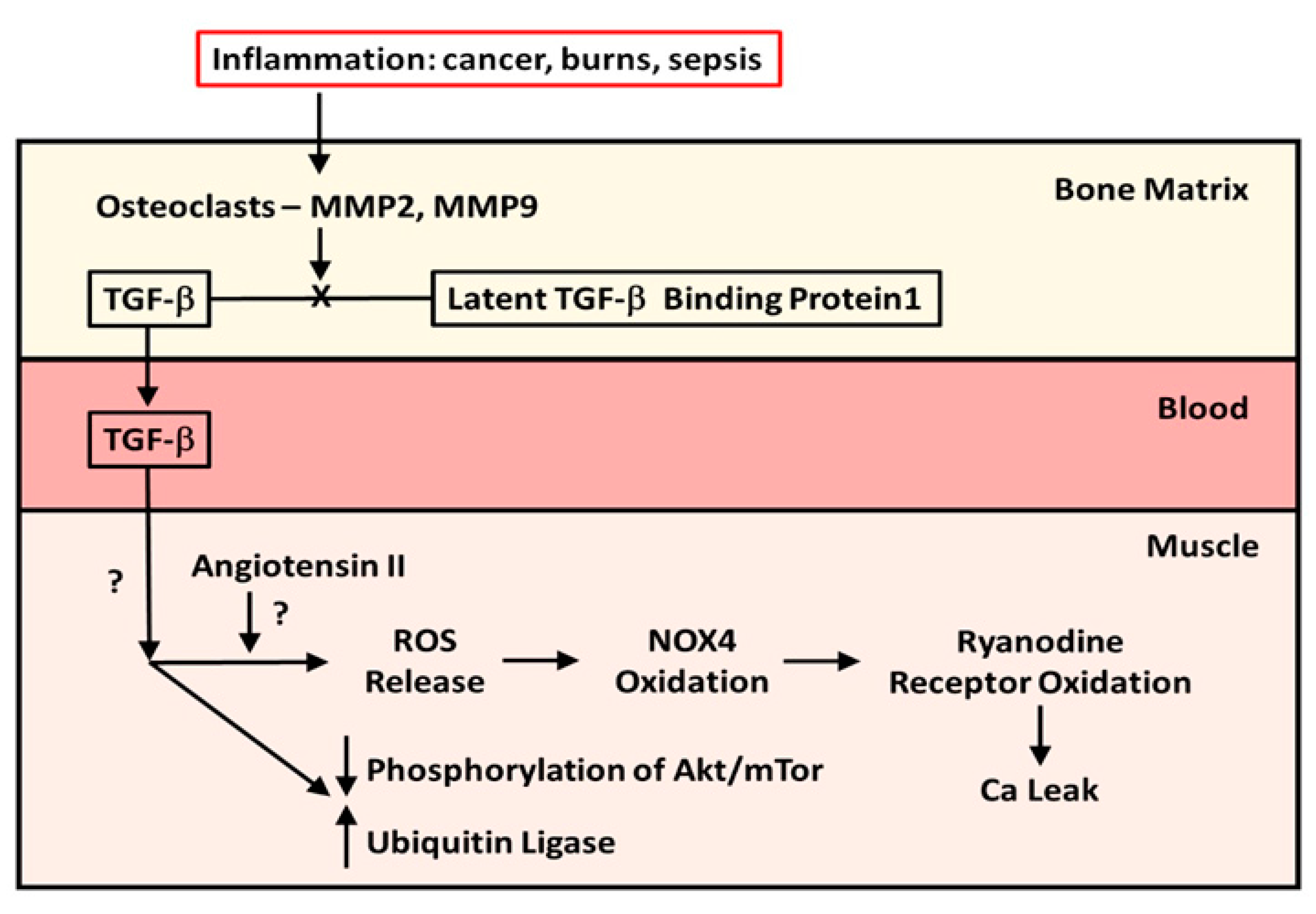
2. The Role of Bone
Expansion of the Role of Bone-Liberated TGF-β to Other Musculoskeletal Conditions
3. Evidence Suggesting That Immobilization Plays a Role in TGF-β-Mediated Muscle Wasting
4. Unanswered Questions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gosselin, L.E.; Williams, J.E.; Deering, M.; Brazeau, D.; Koury, S.; Martinez, D.A. Localization and early time course of TGF-beta 1 mRNA expression in dystrophic muscle. Muscle Nerve 2004, 30, 645–653. [Google Scholar] [CrossRef]
- Barros Maranhao, J.; de Oliveira Moreira, D.; Fogagnolo Mauricio, A.; Camacari de Carvalho, S.; Ferretti, R.; Alves Pereira, J.; Santo Neto, H.; Marques, M.J. Changes in calsequestrin, TNF-α, TGF-β, and MyoD levels during the progression of skeletal muscle dystrophy in mdx mice: A comparative analysis of the quadriceps, diaphragm, and intrinsic laryngeal muscles. Int. J. Exp. Pathol. 2015, 96, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Mazala, D.A.; Novak, J.S.; Hogarth, M.W.; Nearing, M.; Adusumalli, P.; Tully, C.B.; Habib, N.F.; Gordish-Dressman, H.; Chen, Y.; Jaiswal, J.K.; et al. TGF-beta-driven muscle degeneration and failed regeneration underlie disease onset in a DMD mouse model. JCI Insight 2020, 5, e135703. [Google Scholar] [CrossRef]
- Xu, D.; Li, S.; Wang, L.; Jiang, J.; Zhao, L.; Huang, X.; Sun, Z.; Li, C.; Sun, L.; Li, X.; et al. TAK1 inhibition improves myoblast differentiation and alleviates fibrosis in a mouse model of Duchenne muscular dystrophy. J. Cachexia Sarcopenia Muscle 2021, 12, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Contreras, O.; Cruz Soca, M.; Theret, M.; Soliman, H.; Tung, L.W.; Groppa, E.; Rossi, F.M.; Brandan, E. Cross-talk between TGF-β and PDGFR-α signaling pathways regulates the fate of stromal fibro-adipogenic precursors. J. Cell Sci. 2019, 132, jcs232157. [Google Scholar] [CrossRef]
- Gonzalez, D.; Contreras, O.; Rebolledo, D.L.; Espinoza, J.P.; van Zundert, B.; Brandan, E. ALS skeletal muscle shows enhanced TGF-β signaling, fibrosis, and fibroadipogenic progenitor markers. PLoS ONE. 2017, 12, e0177649. [Google Scholar] [CrossRef] [Green Version]
- Abrigo, J.; Campos, F.; Simon, F.; Riedel, C.; Cabrera, D.; Vilos, C.; Cabello-Verrugio, C. TGF-beta requires the activation of canonical and non-canonical signaling pathways to induce skeletal muscle atrophy. Biol. Chem. 2018, 399, 253–264. [Google Scholar] [CrossRef]
- Abrigo, J.; Simon, F.; Cabrera, D.; Cabello-Verrugio, C. Angiotensin-(1-7) prevents skeletal muscle atrophy induced by Transforming Growth Factor-type beta (TGF-beta) via Mas receptor activation. Cell Physiol. Biochem. 2016, 40, 27–38. [Google Scholar] [CrossRef]
- Klein, G.L.; Mills, I.H.; Wilson, R.J. Changes in renal function associated with the development of resistance of the renal vasculature to the arterial infusion of angiotensin. J. Physiol. 1971, 215, 43P–44P. [Google Scholar] [PubMed]
- Waning, D.L.; Mohammad, K.S.; Reiken, S.; Xie, W.; Andersson, D.C.; John, S.; Chiechi, A.; Wright, L.E.; Umanskaya, A.; Niewolna, M. Excess TGF-β mediates muscle weakness associated with bone metastases in mice. Nat. Med. 2015, 21, 1262–1271. [Google Scholar] [CrossRef]
- Dallas, S.L.; Rosser, J.L.; Mundy, G.R.; Bonewald, L.F. Proteolysis of latent transforming growth factor-beta (TGF-beta) binding protein 1 by osteoclasts. A cellular mechanism for release of TGF-beta from bone matrix. J. Biol. Chem. 2002, 277, 21352–21360. [Google Scholar] [CrossRef] [Green Version]
- Pin, F.; Bonewald, L.F.; Bonetto, A. Role of myokines and osteokines in cancer cachexia. Exp. Biol. Med. 2021, 246, 2118–2127. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Peng, Y.; Zhao, W.; Pan, J.; Ksiezak-Reding, H.; Cardozo, C.; Wu, Y.; Pajevic, P.D.; Bonewald, L.F.; Bauman, W.A.; et al. Myostatin inhibits osteoblastic differentiation by suppressing osteocyte-derived exosomal microsomal RNA 218: A novel mechanism in muscle-bone communication. J. Biol. Chem. 2017, 292, 11021–11033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.L.; Walton, K.L.; Hagg, A.; Colgan, T.D.; Johnson, K.; Qian, H.; Gregorevic, P.; Harrison, C.A. Specific targeting of TGF-β family ligands demonstrates distinct roles in the regulation of muscle mass in health and disease. Proc. Natl. Acad. Sci. USA 2017, 114, E5266–E5275. [Google Scholar] [CrossRef] [Green Version]
- Hain, B.A.; Jude, B.; Xu, H.; Smuin, D.M.; Fox, E.J.; Elfar, J.C.; Waning, D.L. Zoledronic acid improves muscle function in healthy mice treated with chemotherapy. J. Bone Miner. Res. 2020, 35, 368–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essex, A.L.; Pin, F.; Huot, J.R.; Bonewald, L.F.; Plotkin, L.I.; Bonetto, A. Bisphosphonate treatment ameliorates bone and muscle abnormalities in young mice. Front. Endocrinol. 2019, 10, 809. [Google Scholar] [CrossRef]
- Klein, G.L.; Wimalawansa, S.J.; Kulkarni, G.; Sherrard, D.J.; Sanford, A.P.; Herndon, D.N. The efficacy of the acute administration of pamidronate on the conservation of bone mass following severe burn injury in children: A double blind, randomized, controlled study. Osteoporos. Int. 2005, 16, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Borsheim, E.; Herndon, D.N.; Hawkins, H.K.; Suman, O.E.; Cotter, M.; Klein, G.L. Pamidronate attenuates muscle loss after pediatric burn injury. J. Bone Miner. Res. 2014, 29, 1369–1372. [Google Scholar] [CrossRef] [Green Version]
- Pin, F.; Bonetto, A.; Bonewald, L.F.; Klein, G.L. Molecular mechanisms responsible for the rescue effects of pamidronate on muscle atrophy in pediatric burn patients. Front. Endocrinol. 2019, 10, 543. [Google Scholar] [CrossRef] [Green Version]
- Jude, B.; Tissier, F.; Dubourg, A.; Droguet, M.; Castel, T.; Leon, K.; Giroux-Metges, M.; Pennec, J. TGF-β pathway inhibition protects the diaphragm from sepsis-induced wasting and weakness in rat. Shock 2020, 53, 772–778. [Google Scholar] [CrossRef]
- Carlson, M.E.; Hsu, M.; Conboy, I.M. Imbalance between pSmad 3 and Notch induces CDK inhibitors in old muscle stem cells. Nature 2008, 454, 528–532. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; He, J.; Wang, F.; Gong, J.; Wang, L.; Wu, Q.; Li, W.; Liu, H.; Wang, J.; Zhang, K.; et al. Hemojuvelin is a novel suppressor for Duchenne muscular dystrophy and age-related muscle wastin. J. Cachexia Sarcopenia Muscle 2019, 10, 557–573. [Google Scholar] [CrossRef] [Green Version]
- Komaba, H.; Kakuta, T.; Fukagawa, M. Management of secondary hyperparathyroidism: How and why? Clin. Exp. Nephrol. 2017, 21 (Suppl. 1), 37–45. [Google Scholar] [CrossRef] [PubMed]
- Kimonis, V.E.; Mehta, S.G.; Fulchiero, E.C.; Thomasova, D.; Pasquali, M.; Boycott, K.; Neilan, E.G.; Kartashov, A.; Forman, M.S.; Tucker, S.; et al. Clinical studies in familial VCP myopathy associated with Paget’s disease of bone and frontotemporal dementia. Am. J. Med. Genet. Part A 2008, 146, 745–757. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Hsu, Y.-H.; Mo, C.; Abreu, E.; Kiel, D.P.; Bonewald, L.F.; Brotto, M.; Karasik, D. METTL21C is a potential pleiotropic gene for osteoporosis and sarcopenia acting through the modulation of the NFκB signaling pathway. J. Bone Miner. Res. 2014, 29, 1531–1540. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, B.T.; Bhardwaj, G.; Penniman, C.M.; Krumpoch, M.T.; Suarez Beltran, P.A.; Klaus, K.; Poro, K.; Li, M.; Pan, H.; Dreyfuss, J.M.; et al. FoxO transcription factors are critical regulators of diabetes-related muscle atrophy. Diabetes 2019, 68, 556–570. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, B.T.; Lee, K.Y.; Klaus, K.; Softic, S.; Krumpoch, M.T.; Fentz, J.; Stanford, K.I.; Robinson, M.M.; Cai, W.; Kleinridders, A.; et al. Insulin and IGF-1 receptors regulate FoxO-mediated signaling in muscle proteostasis. J. Clin. Investig. 2016, 126, 3433–3446. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, G.; Penniman, C.M.; Jena, J.; Suarez Beltran, P.A.; Foster, C.; Poro, K.; Junck, T.L.; Hinton, A.O., Jr.; Souvenir, R.; Fuqua, J.D.; et al. Insulin and IGF-1 receptors regulate complex I-dependent bioenergetics and supercomplexes via FoxOs in muscle. J. Clin. Investig. 2021, 131, e146415. [Google Scholar] [CrossRef]
- Almeida, M.; Han, L.; Ambrogini, E.; Weinstein, R.S.; Manolagas, S.C. Glucocorticoids and tumor necrosis factor alpha increase oxidative stress and suppress Wnt protein signaling in osteoblasts. J. Biol. Chem. 2011, 286, 44326–44335. [Google Scholar] [CrossRef] [Green Version]
- Mera, P.; Laue, K.; Ferron, M.; Confavreux, C.; Wei, J.; Galan-Diez, M.; Lacampagne, A.; Mitchell, S.J.; Mattison, J.A.; Chen, Y.; et al. Osteocalcin signaling in myofibers is necessary and sufficient for optimum adaptation to exercise. Cell Metab. 2016, 23, 1078–1092. [Google Scholar] [CrossRef] [Green Version]
- Gugala, Z.; Cacciani, N.; Klein, G.L.; Larsson, L. Acute and severe trabecular bone loss in a rat model of critical illness myopathy. J. Orthop. Res. 2021. [Google Scholar] [CrossRef]
- Llano Diez, M.; Gustafson, A.M.; Olsson, C.; Goransson, H.; Larsson, L. Muscle wasting and the temporal gene expression pattern in a novel rat intensive care unit model. BMC Genom. 2011, 12, 602. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, G.; Dull, R.O.; Schwartz, D.E.; Hu, G. Autophagy in pulmonary macrophages mediates lung inflammatory injury via NLRP3 inflammasome activation during mechanical ventilation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L173–L185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burks, T.N.; Cohn, R.D. Role of TGF-β in inherited and acquired myopathies. Skelet. Muscle 2011, 1, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gumucio, J.P.; Mendias, C.L. Atrogin-1, MuRF-1 and sarcopenia. Endocrine 2013, 43, 12–21. [Google Scholar] [CrossRef]
- Klein, G.L.; Herndon, D.N.; Langman, C.B.; Rutan, T.C.; Young, W.E.; Pembleton, G.; Nusynowitz, M.; Barnett, J.L.; Broemeling, L.D.; Sailer, D.E.; et al. Long-term reduction in bone mass after servere burn inury in children. J. Pediatr. 1995, 126, 252–256. [Google Scholar] [CrossRef]
- Hundeshagen, G.; Herndon, D.N.; Clayton, R.P.; Wurzer, P.; McQuitty, A.; Jennings, K.; Branski, L.K.; Collins, V.N.; Marques, N.R.; Finnerty, C.C.; et al. Long-term effect of critical illness after severe paediatric burn injury on cardiac function in adolescent survivors: An observational study. Lancet Child Adolesc. Health 2017, 1, 293–301. [Google Scholar] [CrossRef]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.J.; et al. Fibroblast specific TGF-β-Smad 2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klein, G.L. Transforming Growth Factor-Beta in Skeletal Muscle Wasting. Int. J. Mol. Sci. 2022, 23, 1167. https://doi.org/10.3390/ijms23031167
Klein GL. Transforming Growth Factor-Beta in Skeletal Muscle Wasting. International Journal of Molecular Sciences. 2022; 23(3):1167. https://doi.org/10.3390/ijms23031167
Chicago/Turabian StyleKlein, Gordon L. 2022. "Transforming Growth Factor-Beta in Skeletal Muscle Wasting" International Journal of Molecular Sciences 23, no. 3: 1167. https://doi.org/10.3390/ijms23031167
APA StyleKlein, G. L. (2022). Transforming Growth Factor-Beta in Skeletal Muscle Wasting. International Journal of Molecular Sciences, 23(3), 1167. https://doi.org/10.3390/ijms23031167