
A Deep Dive into VDAC1 Conformational Diversity Using All-Atom Simulations Provides New Insights into the Structural Origin of the Closed States



Abstract
:1. Introduction
2. Results
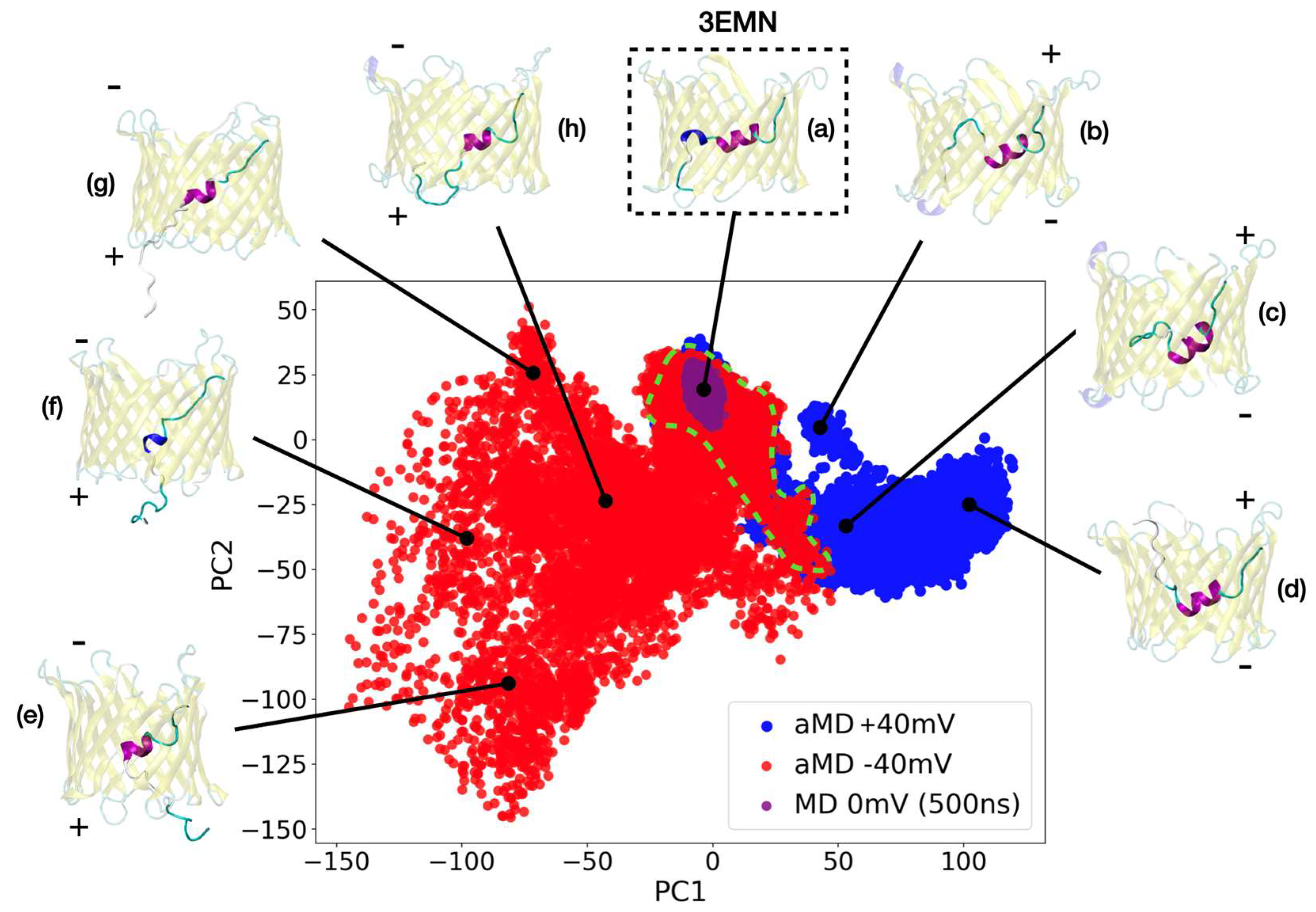
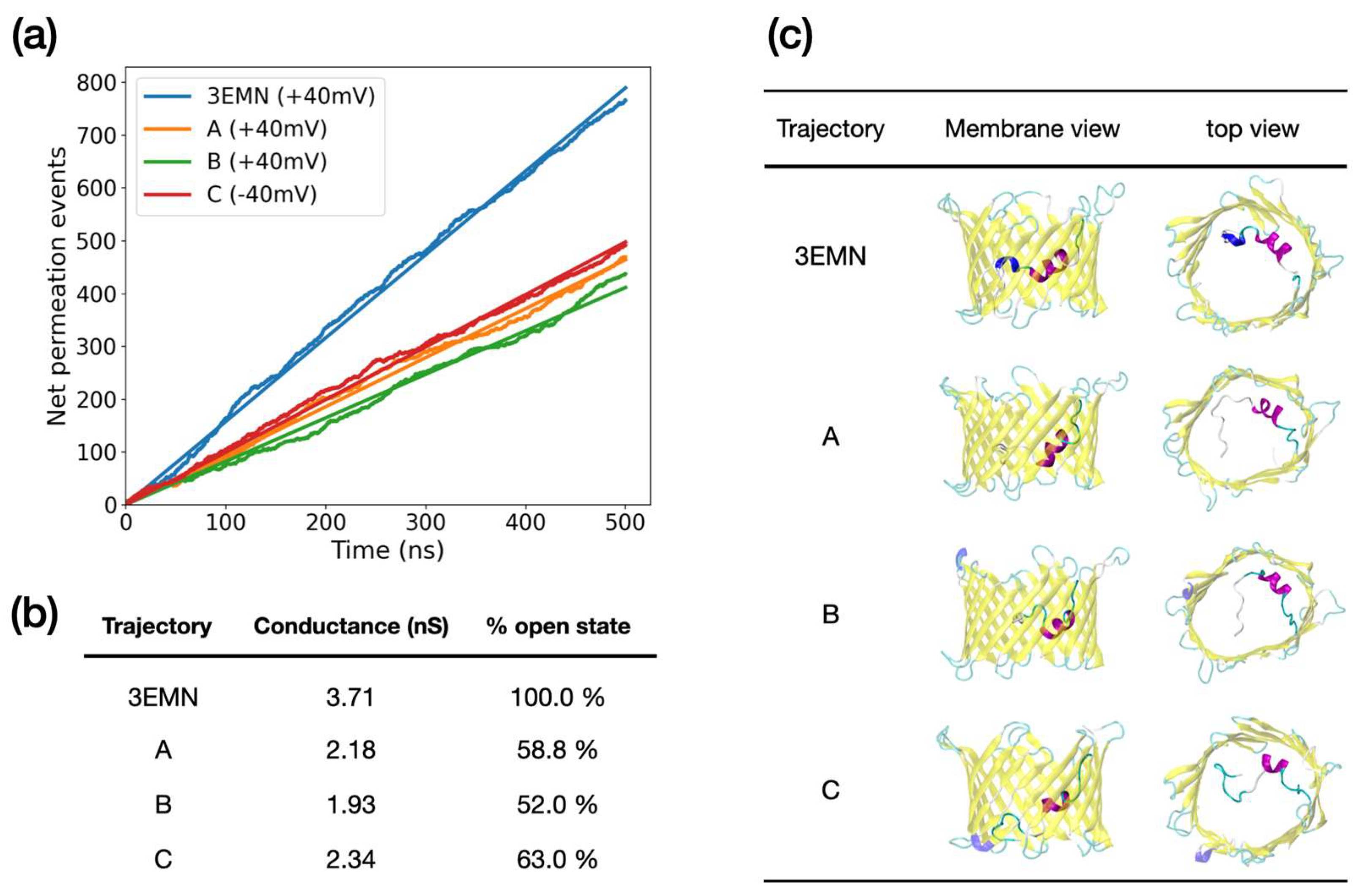
2.1. aMD Simulations
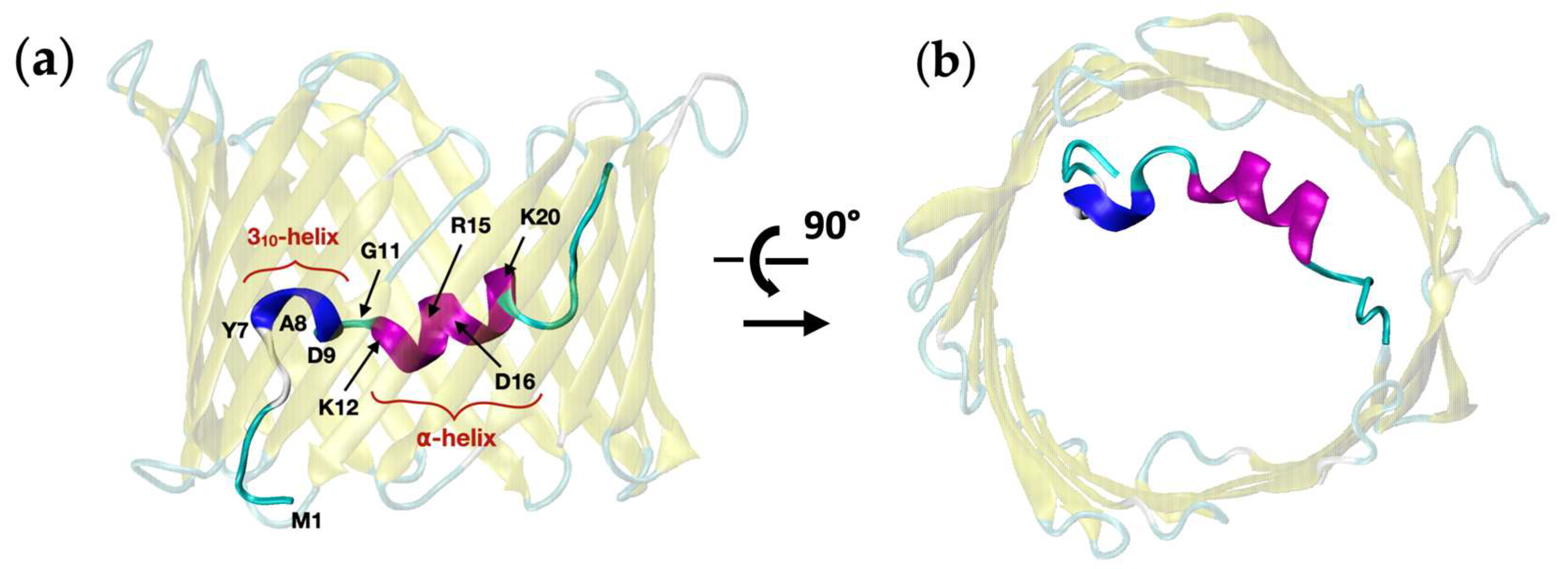
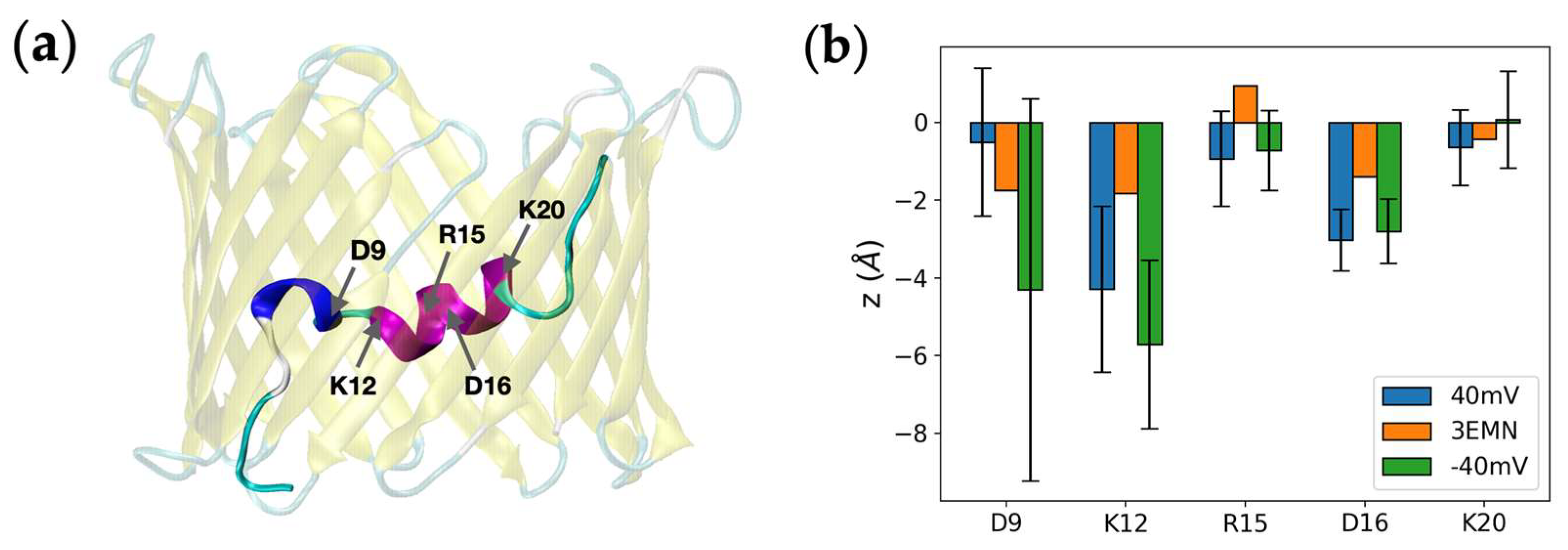
2.2. Investigating Subconducting States of mVDAC1
2.3. Refolding of the N-Terminus
3. Discussion
4. Materials and Methods
4.1. System Preparation
4.2. MD Software and Force Field
4.3. aMD Simulations
4.4. PB/PNP and GCMC/BD
4.5. Unbiased MD
4.6. Clustering Analysis
4.7. Molecular Visualization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More Than Just a Powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Youle, R.J. The Role of Mitochondria in Apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombini, M. VDAC: The Channel at the Interface between Mitochondria and the Cytosol. Mol. Cell. Biochem. 2004, 256, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Colombini, M. VDAC Closure Increases Calcium Ion Flux. Biochim. Biophys. Acta BBA Biomembr. 2007, 1768, 2510–2515. [Google Scholar] [CrossRef] [Green Version]
- Rosencrans, W.M.; Rajendran, M.; Bezrukov, S.M.; Rostovtseva, T.K. VDAC Regulation of Mitochondrial Calcium Flux: From Channel Biophysics to Disease. Cell Calcium 2021, 94, 102356. [Google Scholar] [CrossRef] [PubMed]
- Rostovtseva, T.; Colombini, M. VDAC Channels Mediate and Gate the Flow of ATP: Implications for the Regulation of Mitochondrial Function. Biophys. J. 1997, 72, 1954–1962. [Google Scholar] [CrossRef] [Green Version]
- Hodge, T.; Colombini, M. Regulation of Metabolite Flux through Voltage-Gating of VDAC Channels. J. Membr. Biol. 1997, 157, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Maldonado, E.N.; Krelin, Y. VDAC1 at the Crossroads of Cell Metabolism, Apoptosis and Cell Stress. CST 2017, 1, 11–36. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Shteinfer-Kuzmine, A.; Verma, A. VDAC1 at the Intersection of Cell Metabolism, Apoptosis, and Diseases. Biomolecules 2020, 10, 1485. [Google Scholar] [CrossRef]
- De Marchi, U.; Fernandez-Martinez, S.; Fuente, S.; Wiederkehr, A.; Santo-Domingo, J. Mitochondrial Ion Channels in Pancreatic Β-cells: Novel Pharmacological Targets for the Treatment of Type 2 Diabetes. Br. J. Pharmacol. 2020, 178, 2077–2095. [Google Scholar] [CrossRef] [Green Version]
- Shoshan-Barmatz, V.; Krelin, Y.; Shteinfer-Kuzmine, A.; Arif, T. Voltage-Dependent Anion Channel 1 As an Emerging Drug Target for Novel Anti-Cancer Therapeutics. Front. Oncol. 2017, 7, 154. [Google Scholar] [CrossRef]
- Reina, S.; De Pinto, V. Anti-Cancer Compounds Targeted to VDAC: Potential and Perspectives. Curr. Med. Chem. 2018, 24, 4447–4469. [Google Scholar] [CrossRef]
- Magrì, A.; Reina, S.; De Pinto, V. VDAC1 as Pharmacological Target in Cancer and Neurodegeneration: Focus on Its Role in Apoptosis. Front. Chem. 2018, 6, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshan-Barmatz, V.; Nahon-Crystal, E.; Shteinfer-Kuzmine, A.; Gupta, R. VDAC1, Mitochondrial Dysfunction, and Alzheimer’s Disease. Pharmacol. Res. 2018, 131, 87–101. [Google Scholar] [CrossRef]
- Colombini, M. Voltage Gating in the Mitochondrial Channel, VDAC. J. Membr. Biol. 1989, 111, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Teijido, O.; Rappaport, S.M.; Chamberlin, A.; Noskov, S.Y.; Aguilella, V.M.; Rostovtseva, T.K.; Bezrukov, S.M. Acidification Asymmetrically Affects Voltage-Dependent Anion Channel Implicating the Involvement of Salt Bridges. J. Biol. Chem. 2014, 289, 23670–23682. [Google Scholar] [CrossRef] [Green Version]
- Najbauer, E.E.; Becker, S.; Giller, K.; Zweckstetter, M.; Lange, A.; Steinem, C.; de Groot, B.L.; Griesinger, C.; Andreas, L.B. Structure, Gating and Interactions of the Voltage-Dependent Anion Channel. Eur. Biophys. J. 2021, 50, 159–172. [Google Scholar] [CrossRef]
- Ujwal, R.; Cascio, D.; Colletier, J.-P.; Faham, S.; Zhang, J.; Toro, L.; Ping, P.; Abramson, J. The Crystal Structure of Mouse VDAC1 at 2.3 A Resolution Reveals Mechanistic Insights into Metabolite Gating. Proc. Natl. Acad. Sci. USA 2008, 105, 17742–17747. [Google Scholar] [CrossRef] [Green Version]
- Hosaka, T.; Okazaki, M.; Kimura-Someya, T.; Ishizuka-Katsura, Y.; Ito, K.; Yokoyama, S.; Dodo, K.; Sodeoka, M.; Shirouzu, M. Crystal Structural Characterization Reveals Novel Oligomeric Interactions of Human Voltage-Dependent Anion Channel 1: Novel Oligomeric Structure of Human VDAC1. Protein Sci. 2017, 26, 1749–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiller, S.; Garces, R.G.; Malia, T.J.; Orekhov, V.Y.; Colombini, M.; Wagner, G. Solution Structure of the Integral Human Membrane Protein VDAC-1 in Detergent Micelles. Science 2008, 321, 1206–1210. [Google Scholar] [CrossRef] [Green Version]
- Böhm, R.; Amodeo, G.F.; Murlidaran, S.; Chavali, S.; Wagner, G.; Winterhalter, M.; Brannigan, G.; Hiller, S. The Structural Basis for Low Conductance in the Membrane Protein VDAC upon β-NADH Binding and Voltage Gating. Structure 2019, 28, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Bayrhuber, M.; Meins, T.; Habeck, M.; Becker, S.; Giller, K.; Villinger, S.; Vonrhein, C.; Griesinger, C.; Zweckstetter, M.; Zeth, K. Structure of the Human Voltage-Dependent Anion Channel. Proc. Natl. Acad. Sci. USA 2008, 105, 15370–15375. [Google Scholar] [CrossRef] [Green Version]
- Martynowycz, M.W.; Khan, F.; Hattne, J.; Abramson, J.; Gonen, T. MicroED Structure of Lipid-Embedded Mammalian Mitochondrial Voltage-Dependent Anion Channel. Proc. Natl. Acad. Sci. USA 2020, 117, 32380–32385. [Google Scholar] [CrossRef]
- Lee, K.I.; Rui, H.; Pastor, R.W.; Im, W. Brownian Dynamics Simulations of Ion Transport through the VDAC. Biophys. J. 2011, 100, 611–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhary, O.P.; Paz, A.; Adelman, J.L.; Colletier, J.-P.; Abramson, J.; Grabe, M. Structure-Guided Simulations Illuminate the Mechanism of ATP Transport through VDAC1. Nat. Struct. Mol. Biol. 2014, 21, 626–632. [Google Scholar] [CrossRef] [Green Version]
- Camara, A.K.S.; Zhou, Y.; Wen, P.-C.; Tajkhorshid, E.; Kwok, W.-M. Mitochondrial VDAC1: A Key Gatekeeper as Potential Therapeutic Target. Front. Physiol. 2017, 8, 460. [Google Scholar] [CrossRef] [Green Version]
- Hiller, S.; Wagner, G. The Role of Solution NMR in the Structure Determinations of VDAC-1 and Other Membrane Proteins. Curr. Opin. Struct. Biol. 2009, 19, 396–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zachariae, U.; Schneider, R.; Briones, R.; Gattin, Z.; Demers, J.-P.; Giller, K.; Maier, E.; Zweckstetter, M.; Griesinger, C.; Becker, S.; et al. β-Barrel Mobility Underlies Closure of the Voltage-Dependent Anion Channel. Structure 2012, 20, 1540–1549. [Google Scholar] [CrossRef] [Green Version]
- Villinger, S.; Briones, R.; Giller, K.; Zachariae, U.; Lange, A.; de Groot, B.L.; Griesinger, C.; Becker, S.; Zweckstetter, M. Functional Dynamics in the Voltage-Dependent Anion Channel. Proc. Natl. Acad. Sci. USA 2010, 107, 22546–22551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preto, J.; Krimm, I. The Intrinsically Disordered N-Terminus of the Voltage-Dependent Anion Channel. PLoS Comput. Biol. 2021, 17, e1008750. [Google Scholar] [CrossRef]
- Colombini, M. VDAC Structure, Selectivity, and Dynamics. Biochim. Biophys. Acta BBA Biomembr. 2012, 1818, 1457–1465. [Google Scholar] [CrossRef] [Green Version]
- Mertins, B.; Psakis, G.; Grosse, W.; Back, K.C.; Salisowski, A.; Reiss, P.; Koert, U.; Essen, L.-O. Flexibility of the N-Terminal MVDAC1 Segment Controls the Channel’s Gating Behavior. PLoS ONE 2012, 7, e47938. [Google Scholar] [CrossRef] [PubMed]
- Saidani, H.; Léonetti, M.; Kmita, H.; Homblé, F. The Open State Selectivity of the Bean Seed VDAC Depends on Stigmasterol and Ion Concentration. IJMS 2021, 22, 3034. [Google Scholar] [CrossRef]
- Song, D.; Luo, R.; Chen, H.-F. IDP-Specific Force Field Ff14IDPSFF Improves the Conformer Sampling of Intrinsically Disordered Proteins. J. Chem. Inf. Model. 2017, 57, 1166–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Im, W.; Seefeld, S.; Roux, B. A Grand Canonical Monte Carlo–Brownian Dynamics Algorithm for Simulating Ion Channels. Biophys. J. 2000, 79, 788–801. [Google Scholar] [CrossRef] [Green Version]
- Briones, R.; Weichbrodt, C.; Paltrinieri, L.; Mey, I.; Villinger, S.; Giller, K.; Lange, A.; Zweckstetter, M.; Griesinger, C.; Becker, S.; et al. Voltage Dependence of Conformational Dynamics and Subconducting States of VDAC-1. Biophys. J. 2016, 111, 1223–1234. [Google Scholar] [CrossRef] [Green Version]
- Colombini, M. Structure and Mode of Action of a Voltage Dependent Anion-Selective Channel (VDAC) Located in the Outer Mitochondrial Membrane. Ann. N. Y. Acad. Sci. 1980, 341, 552–563. [Google Scholar] [CrossRef]
- Souaille, M.; Roux, B. Extension to the Weighted Histogram Analysis Method: Combining Umbrella Sampling with Free Energy Calculations. Comput. Phys. Commun. 2001, 135, 40–57. [Google Scholar] [CrossRef]
- Pande, V.S.; Beauchamp, K.; Bowman, G.R. Everything You Wanted to Know about Markov State Models but Were Afraid to Ask. Methods 2010, 52, 99–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Queralt-Martín, M.; Hoogerheide, D.P.; Noskov, S.Y.; Berezhkovskii, A.M.; Rostovtseva, T.K.; Bezrukov, S.M. VDAC Gating Thermodynamics, but Not Gating Kinetics, Are Virtually Temperature Independent. Biophys. J. 2020, 119, 2584–2592. [Google Scholar] [CrossRef]
- Peng, S.; Blachly-Dyson, E.; Forte, M.; Colombini, M. Large Scale Rearrangement of Protein Domains Is Associated with Voltage Gating of the VDAC Channel. Biophys. J. 1992, 62, 123–135. [Google Scholar] [CrossRef] [Green Version]
- Blachly-Dyson, E.; Peng, S.; Colombini, M.; Forte, M. Selectivity Changes in Site-Directed Mutants of the VDAC Ion Channel: Structural Implications. Sci. New Ser. 1990, 247, 1233–1236. [Google Scholar] [CrossRef]
- Pavlov, E.; Grigoriev, S.M.; Dejean, L.M.; Zweihorn, C.L.; Mannella, C.A.; Kinnally, K.W. The Mitochondrial Channel VDAC Has a Cation-Selective Open State. Biochim. Biophys. Acta BBA Bioenerg. 2005, 1710, 96–102. [Google Scholar] [CrossRef] [Green Version]
- Cieplak, P.; Dupradeau, F.-Y.; Duan, Y.; Wang, J. Polarization Effects in Molecular Mechanical Force Fields. J. Phys. Condens. Matter 2009, 21, 333102. [Google Scholar] [CrossRef]
- Swails, J.M.; York, D.M.; Roitberg, A.E. Constant PH Replica Exchange Molecular Dynamics in Explicit Solvent Using Discrete Protonation States: Implementation, Testing, and Validation. J. Chem. Theory Comput. 2014, 10, 1341–1352. [Google Scholar] [CrossRef]
- Guardiani, C.; Scorciapino, M.A.; Amodeo, G.F.; Grdadolnik, J.; Pappalardo, G.; De Pinto, V.; Ceccarelli, M.; Casu, M. The N-Terminal Peptides of the Three Human Isoforms of the Mitochondrial Voltage-Dependent Anion Channel Have Different Helical Propensities. Biochemistry 2015, 54, 5646–5656. [Google Scholar] [CrossRef] [PubMed]
- Arbel, N.; Ben-Hail, D.; Shoshan-Barmatz, V. Mediation of the Antiapoptotic Activity of Bcl-XL Protein upon Interaction with VDAC1 Protein. J. Biol. Chem. 2012, 287, 23152–23161. [Google Scholar] [CrossRef] [Green Version]
- Azoulay-Zohar, H.; Israelson, A.; Abu-Hamad, S.; Shoshan-Barmatz, V. In Self-Defence: Hexokinase Promotes Voltage-Dependent Anion Channel Closure and Prevents Mitochondria-Mediated Apoptotic Cell Death. Biochem. J. 2004, 377, 347–355. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), 2019.01; Chemical Computing Group ULC: Montreal, QC, Canada, 2021.
- Jo, S.; Kim, T.; Im, W. Automated Builder and Database of Protein/Membrane Complexes for Molecular Dynamics Simulations. PLoS ONE 2007, 2, e880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM Database and PPM Web Server: Resources for Positioning of Proteins in Membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef]
- Horvath, S.E.; Daum, G. Lipids of Mitochondria. Prog. Lipid Res. 2013, 52, 590–614. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham, T.E.; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a Package of Computer Programs for Applying Molecular Mechanics, Normal Mode Analysis, Molecular Dynamics and Free Energy Calculations to Simulate the Structural and Energetic Properties of Molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, C.J.; Madej, B.D.; Skjevik, Å.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The Amber Lipid Force Field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef]
- Karplus, M.; McCammon, J.A. Molecular Dynamics Simulations of Biomolecules. Nat. Struct. Biol. 2002, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Im, W.; Roux, B. Ion Permeation and Selectivity of OmpF Porin: A Theoretical Study Based on Molecular Dynamics, Brownian Dynamics, and Continuum Electrodiffusion Theory. J. Mol. Biol. 2002, 322, 851–869. [Google Scholar] [CrossRef]
- Lee, K.I.; Jo, S.; Rui, H.; Egwolf, B.; Roux, B.; Pastor, R.W.; Im, W. Web Interface for Brownian Dynamics Simulation of Ion Transport and Its Applications to Beta-Barrel Pores. J. Comput. Chem. 2012, 33, 331–339. [Google Scholar] [CrossRef] [Green Version]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
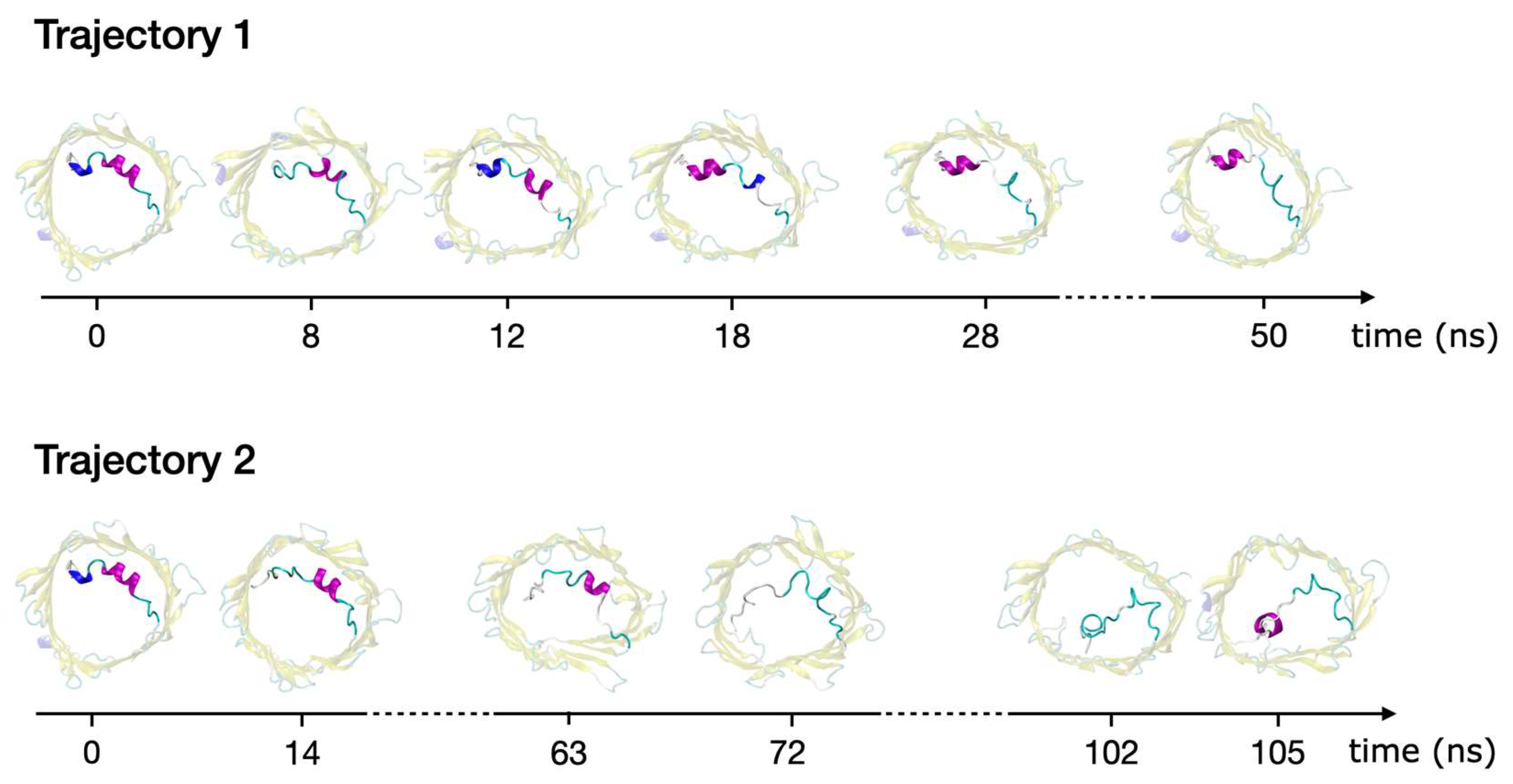

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
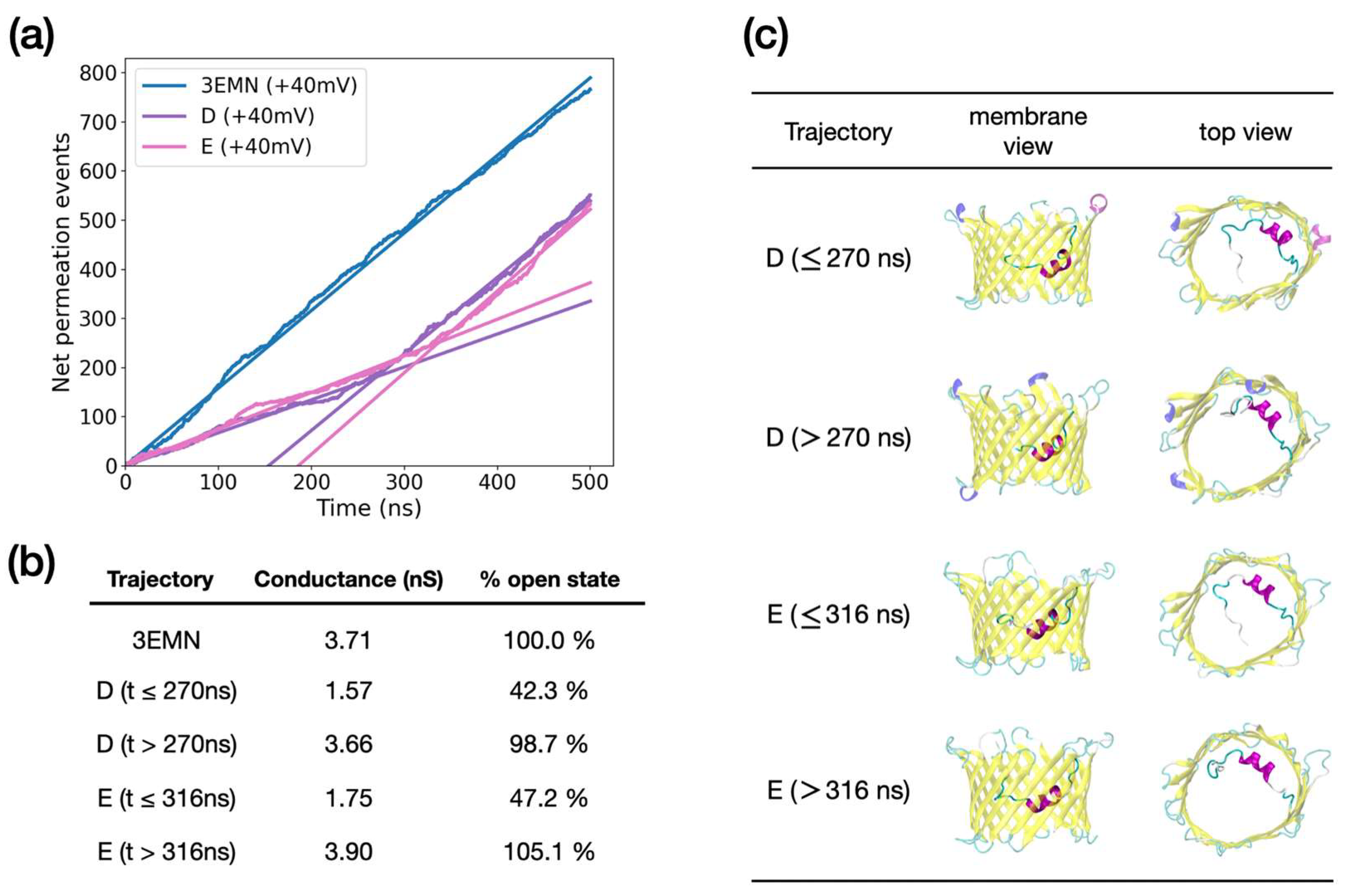
| Trajectory | Conductance (nS) | |
|---|---|---|
| 3EMN | 3.71 | 2.21 |
| A | 2.18 | 4.33 |
| B | 1.93 | 3.09 |
| C | 2.34 | 2.80 |
| D (t ≤ 270 ns) | 1.57 | 3.20 |
| D (t > 270 ns) | 3.66 | 2.25 |
| D (t ≤ 316 ns) | 1.75 | 3.27 |
| E (t > 316 ns) | 3.90 | 2.36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Preto, J.; Gorny, H.; Krimm, I. A Deep Dive into VDAC1 Conformational Diversity Using All-Atom Simulations Provides New Insights into the Structural Origin of the Closed States. Int. J. Mol. Sci. 2022, 23, 1175. https://doi.org/10.3390/ijms23031175
Preto J, Gorny H, Krimm I. A Deep Dive into VDAC1 Conformational Diversity Using All-Atom Simulations Provides New Insights into the Structural Origin of the Closed States. International Journal of Molecular Sciences. 2022; 23(3):1175. https://doi.org/10.3390/ijms23031175
Chicago/Turabian StylePreto, Jordane, Hubert Gorny, and Isabelle Krimm. 2022. "A Deep Dive into VDAC1 Conformational Diversity Using All-Atom Simulations Provides New Insights into the Structural Origin of the Closed States" International Journal of Molecular Sciences 23, no. 3: 1175. https://doi.org/10.3390/ijms23031175