
Meta-Analysis of APP Expression Modulated by SARS-CoV-2 Infection via the ACE2 Receptor



Abstract
:1. Introduction
2. Results
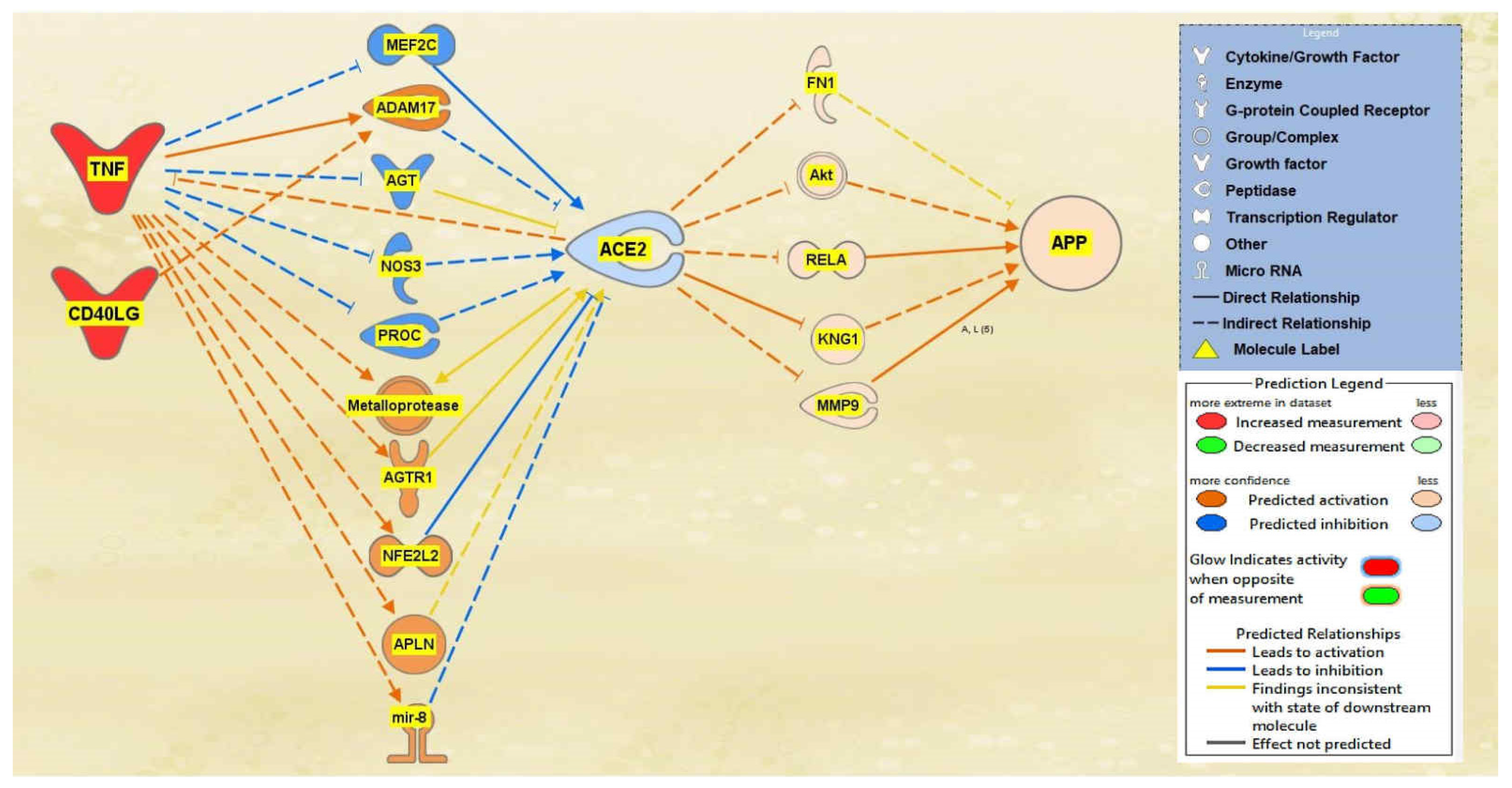
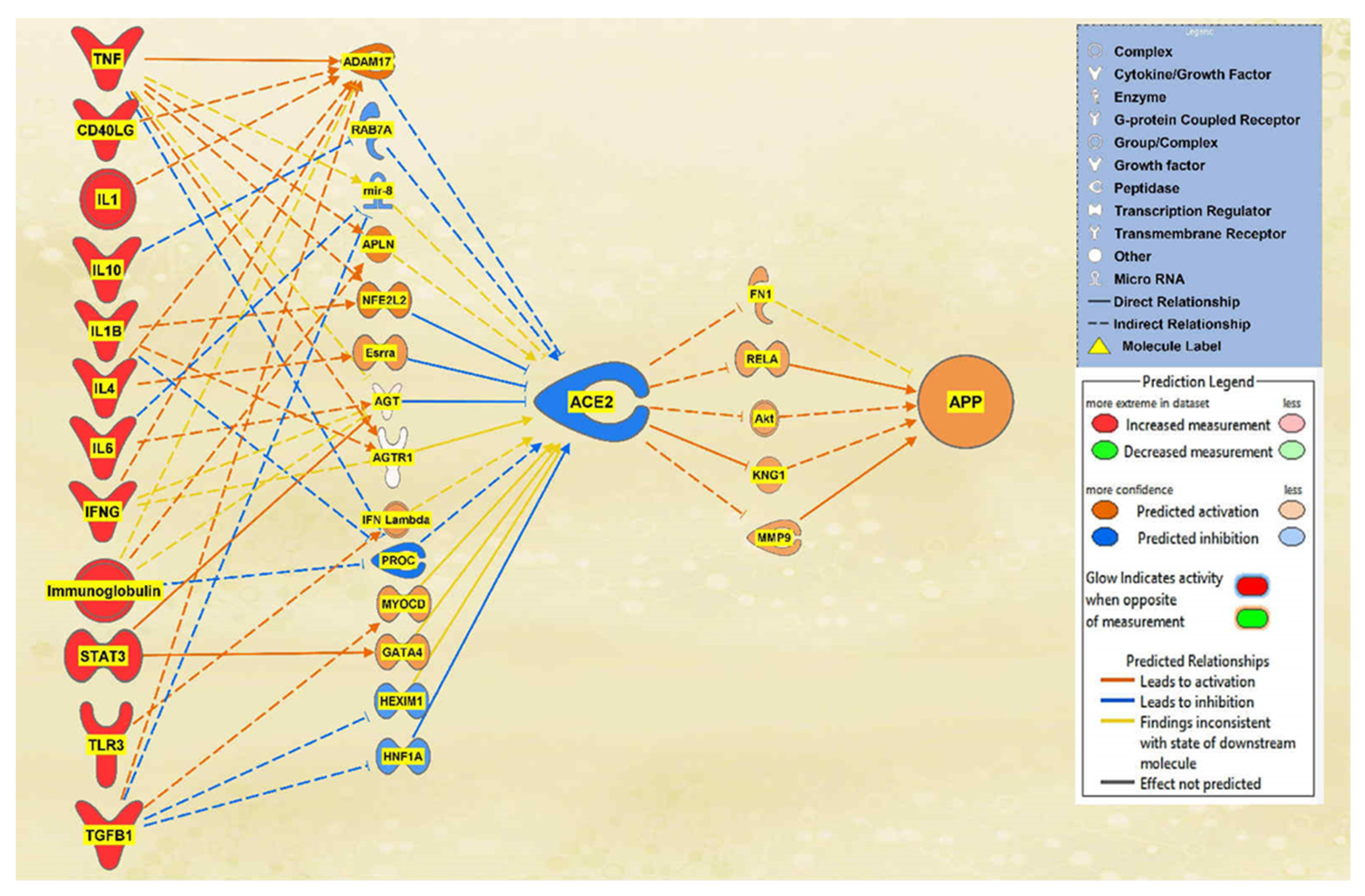
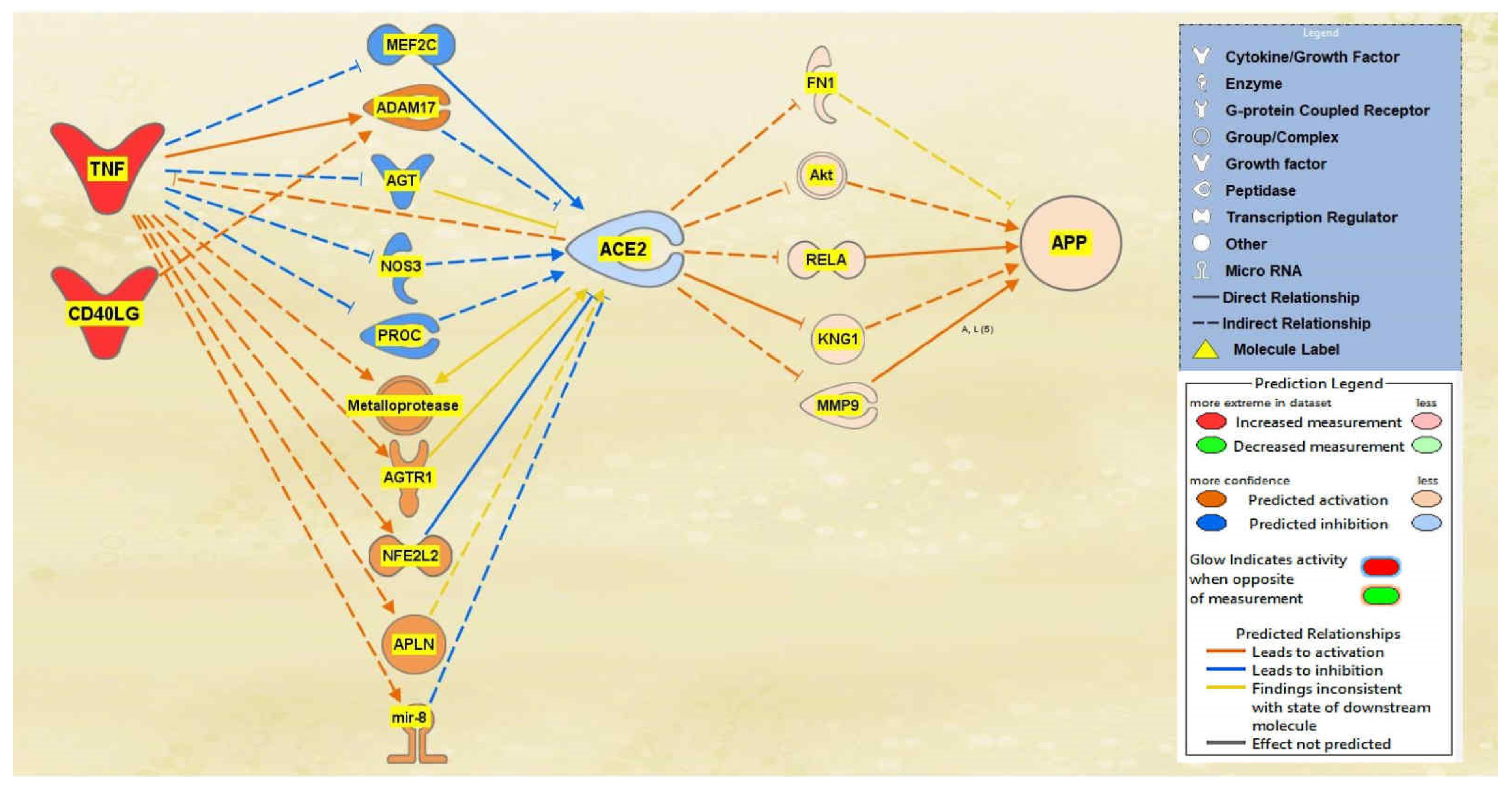
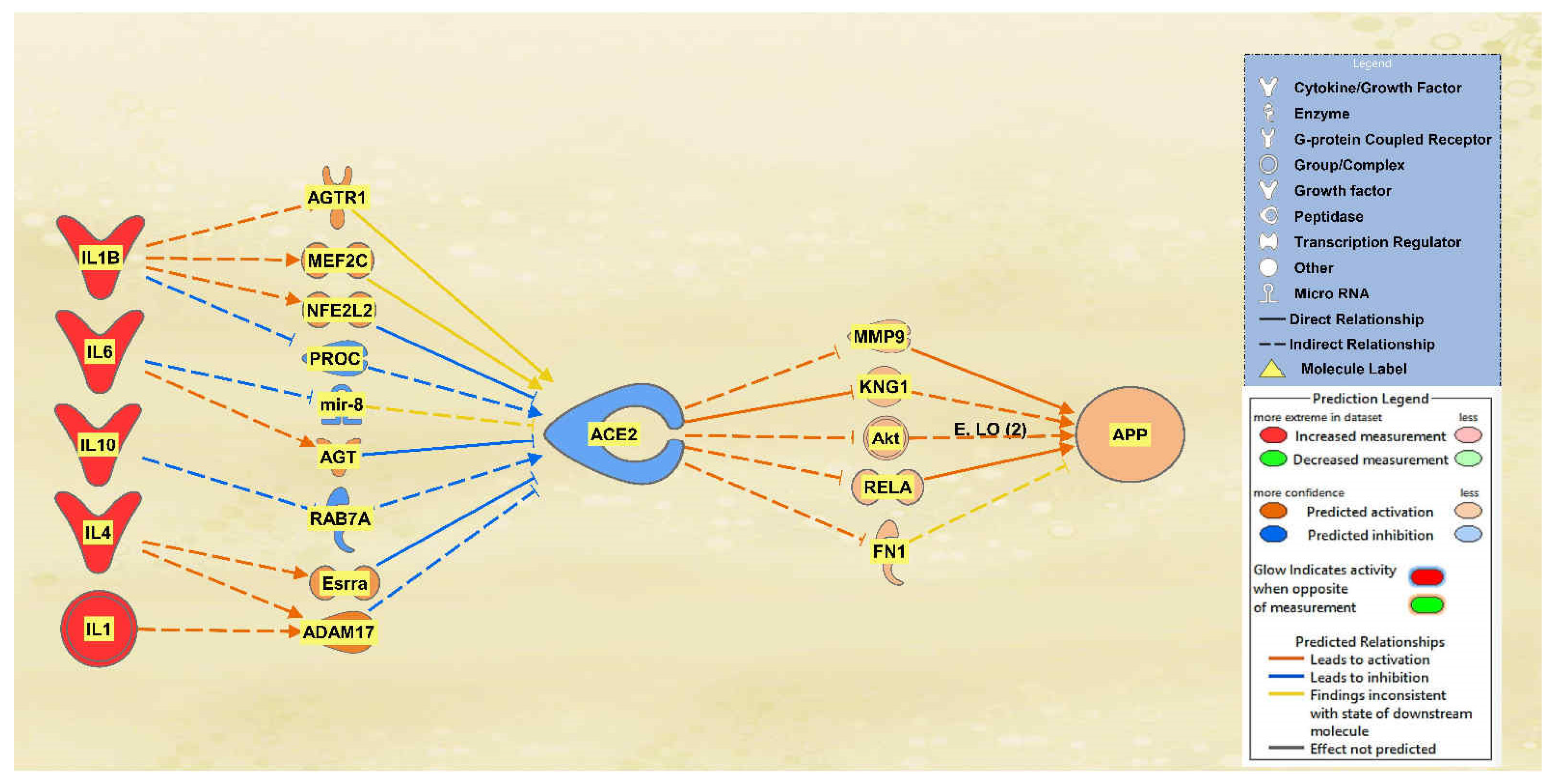
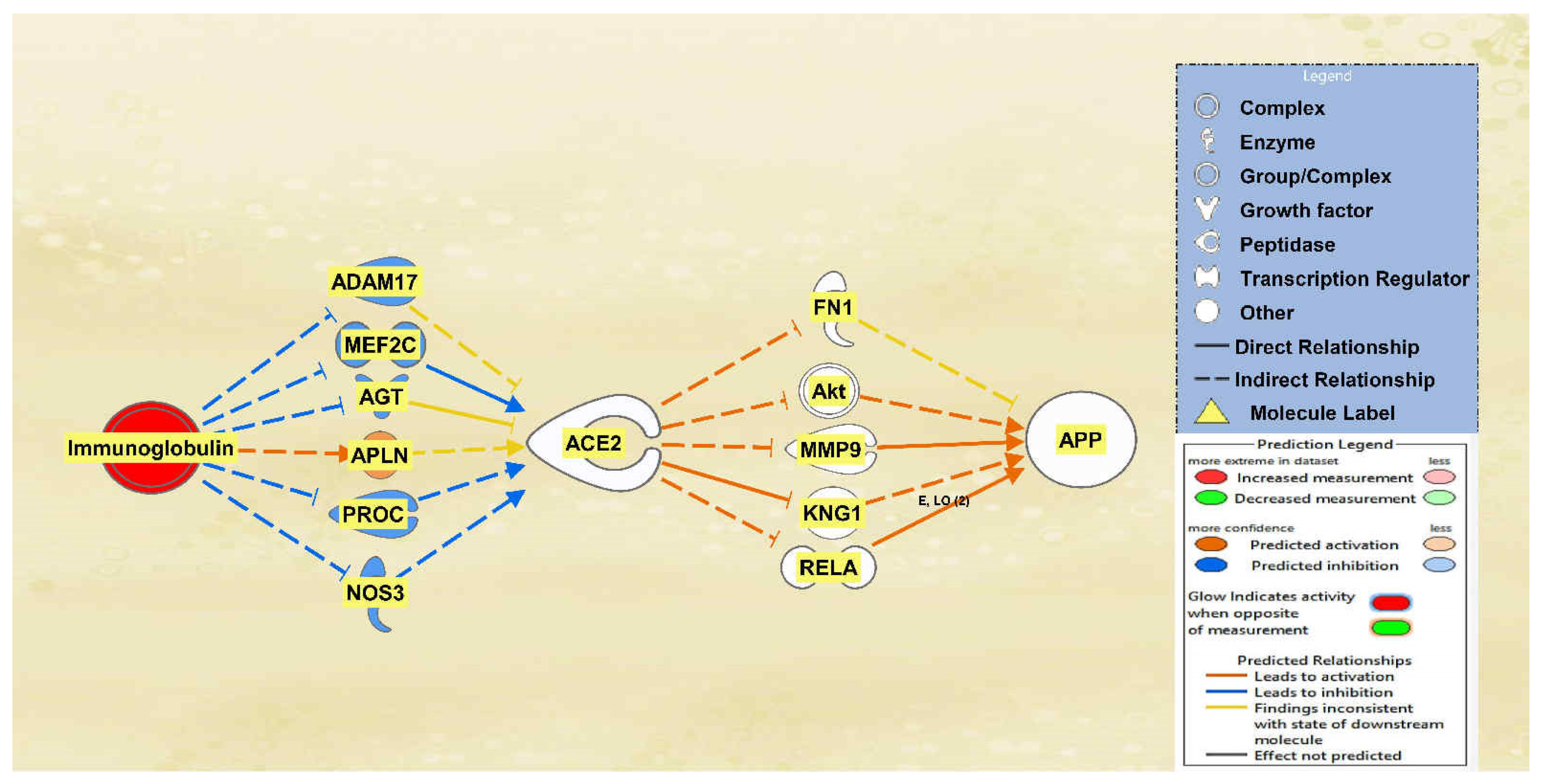
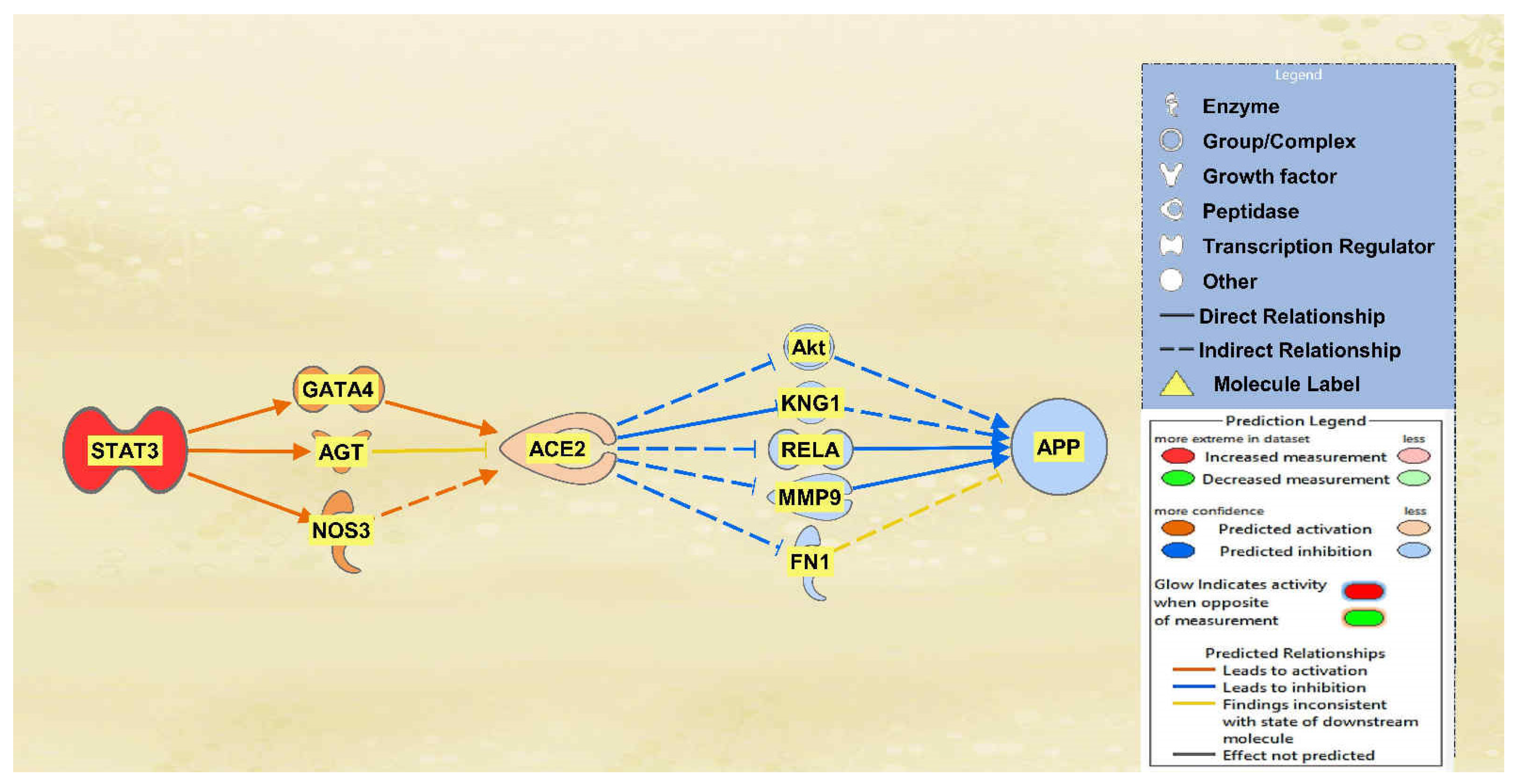
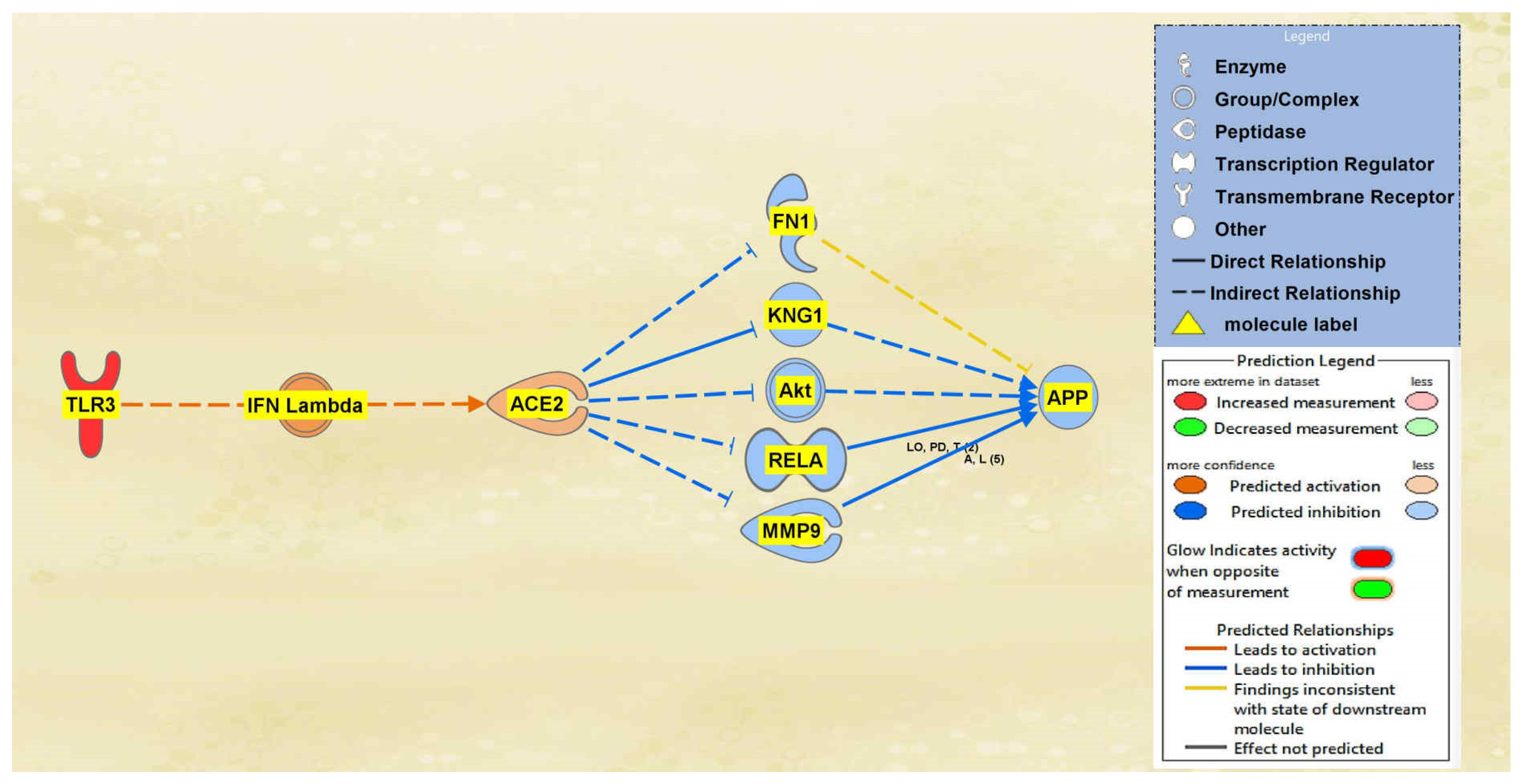
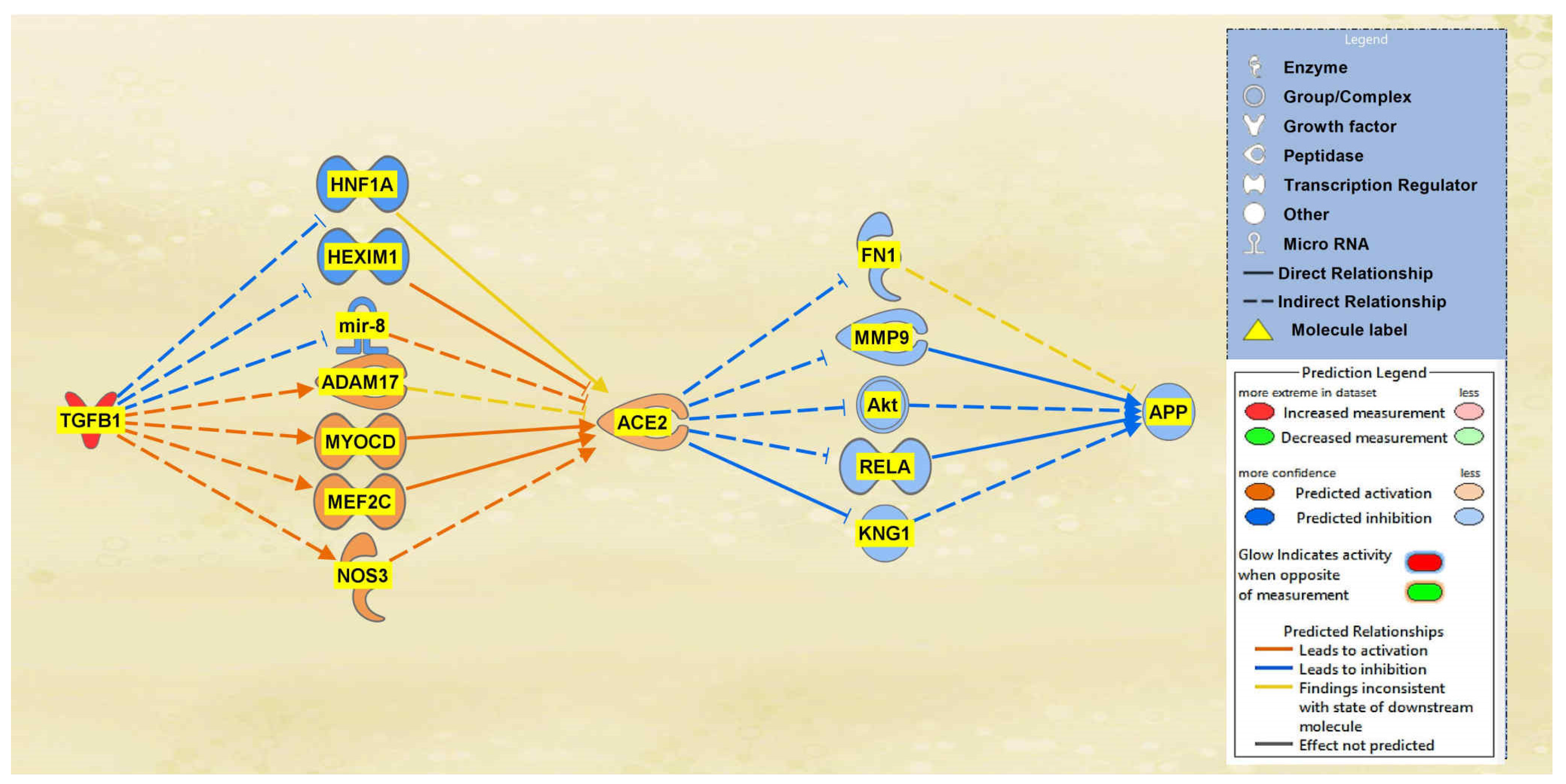
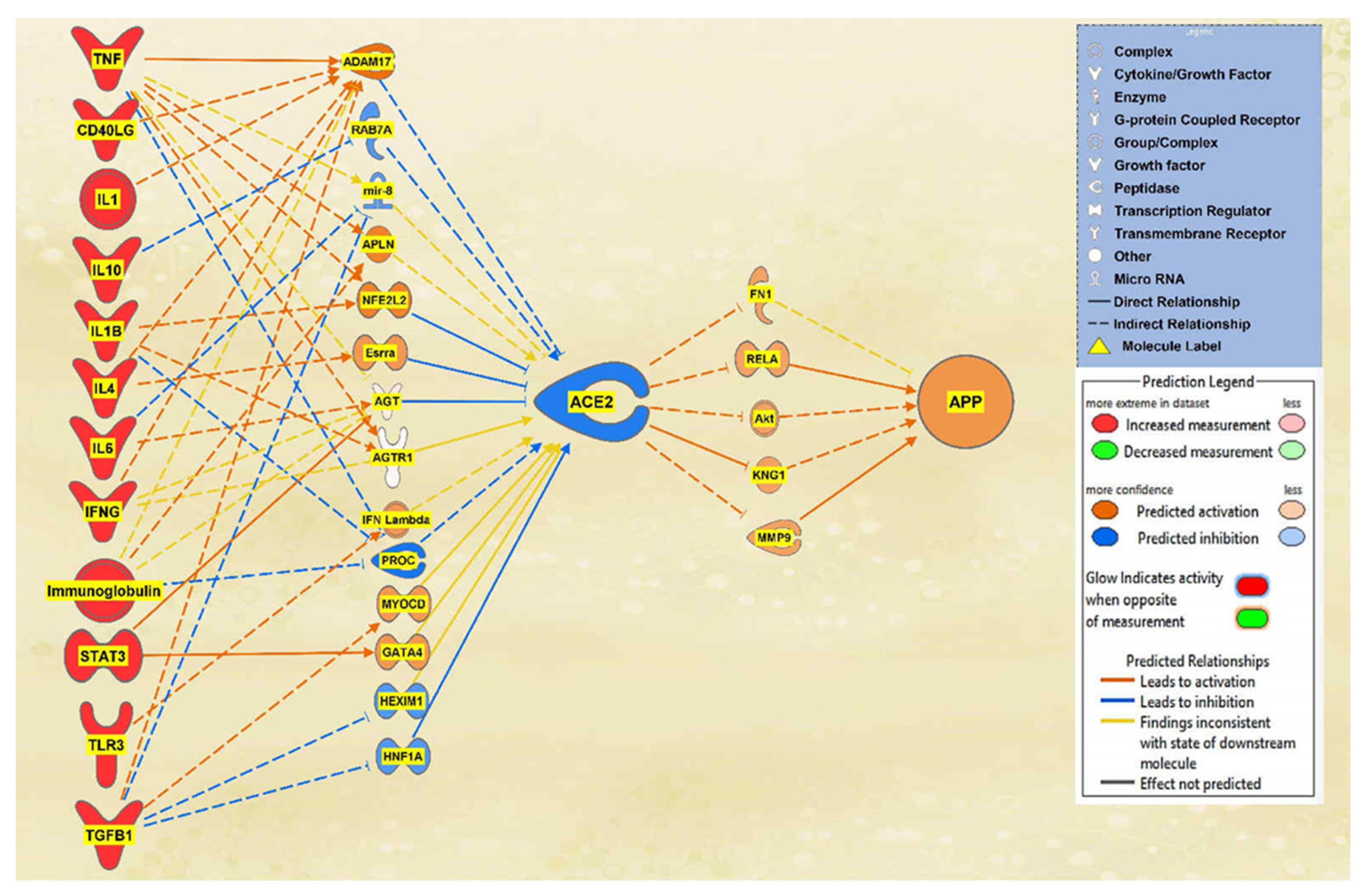
2.1. SARS-CoV Upstream Regulators
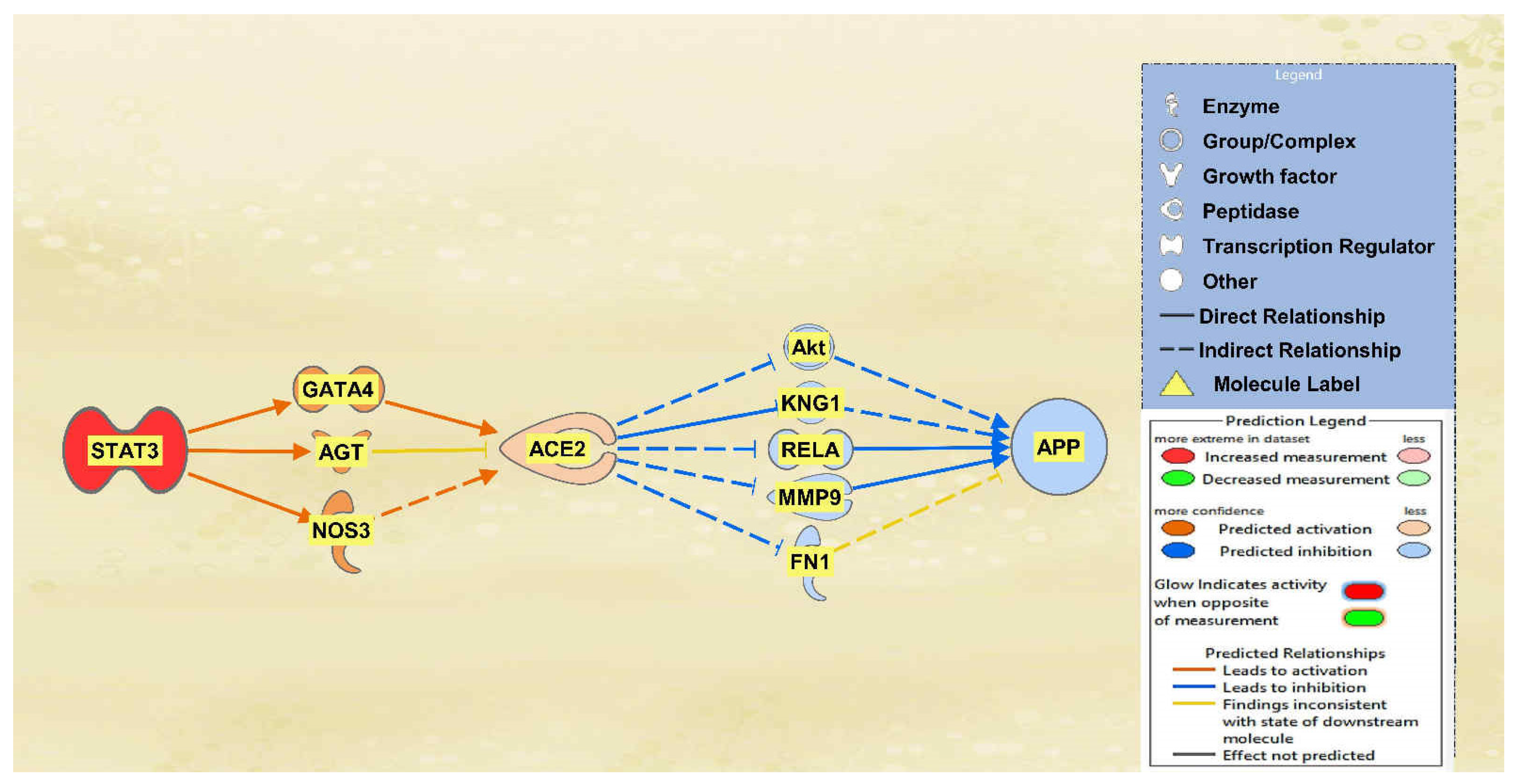
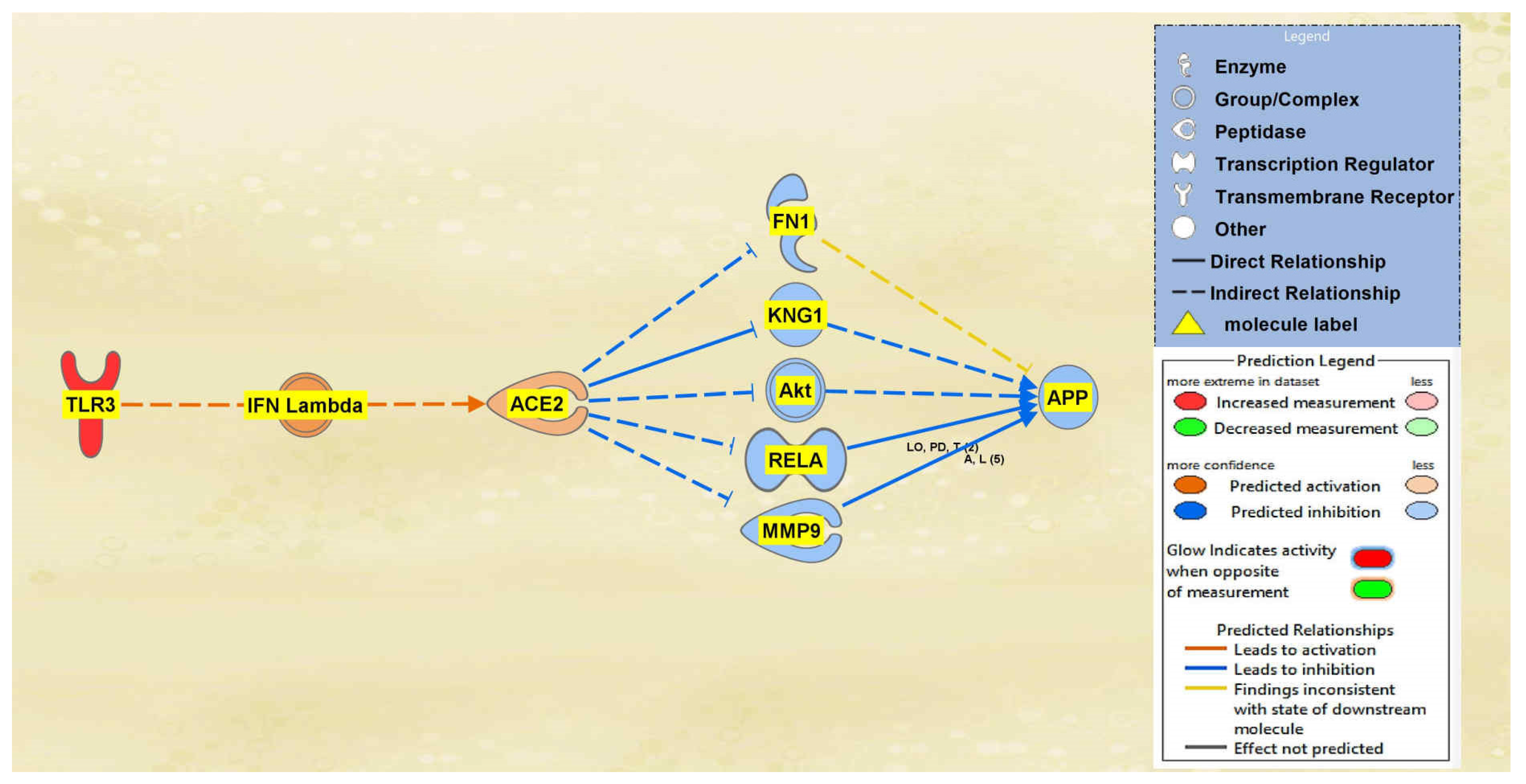
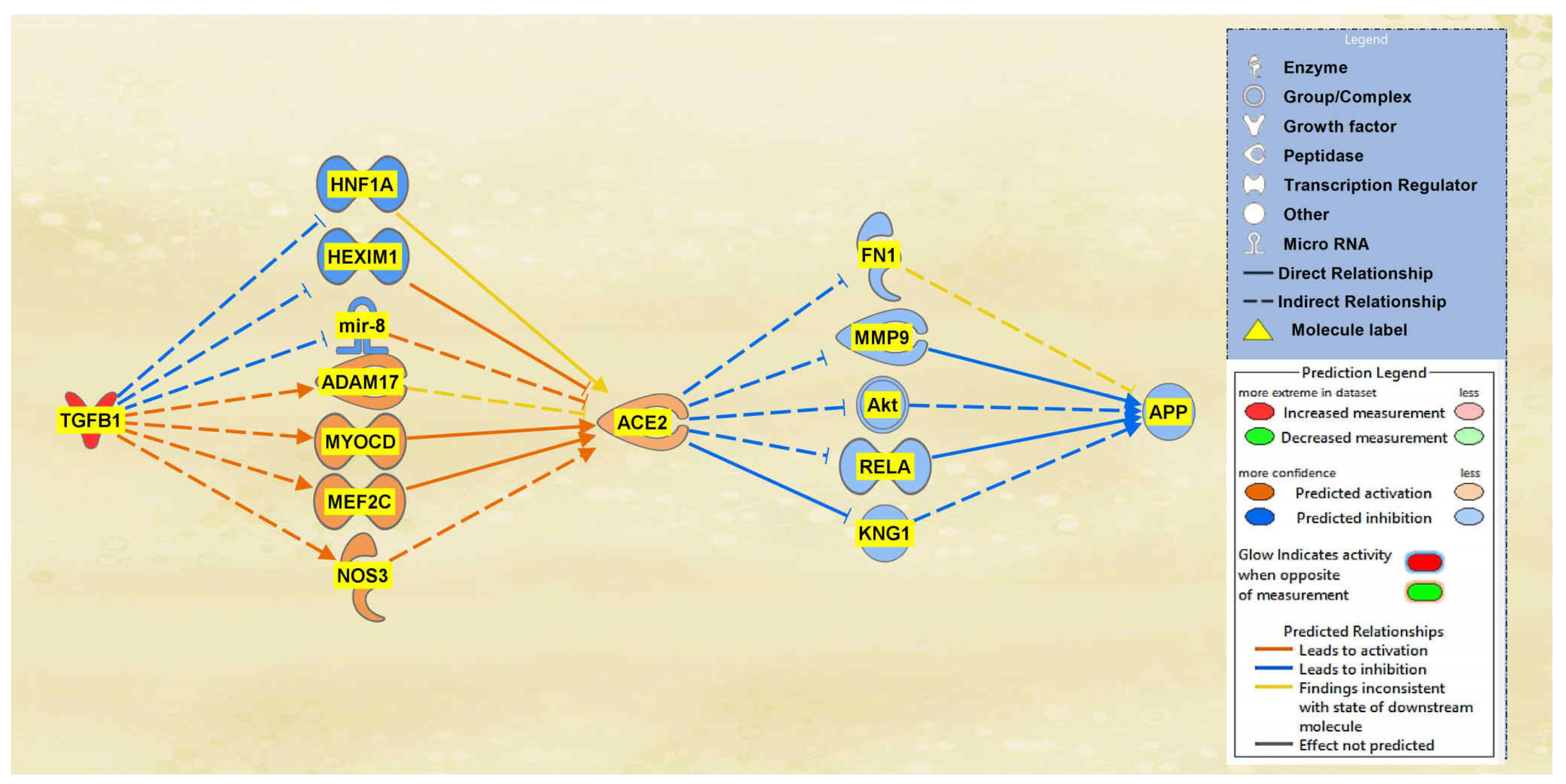
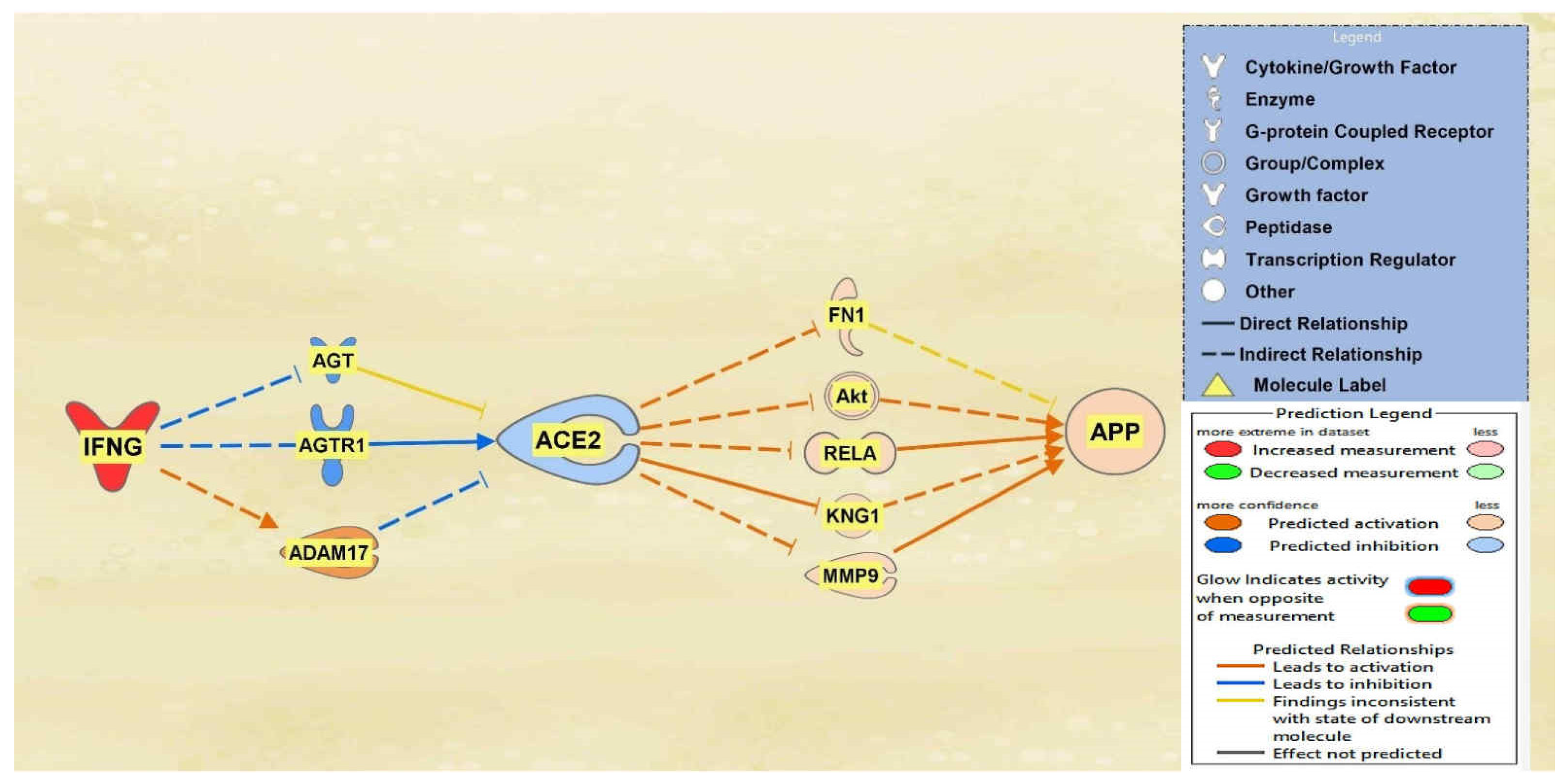
2.2. Positive Impact of Upstream Regulators on APP
2.3. Negative Impact of SARS-CoV Upstream Regulators on APP
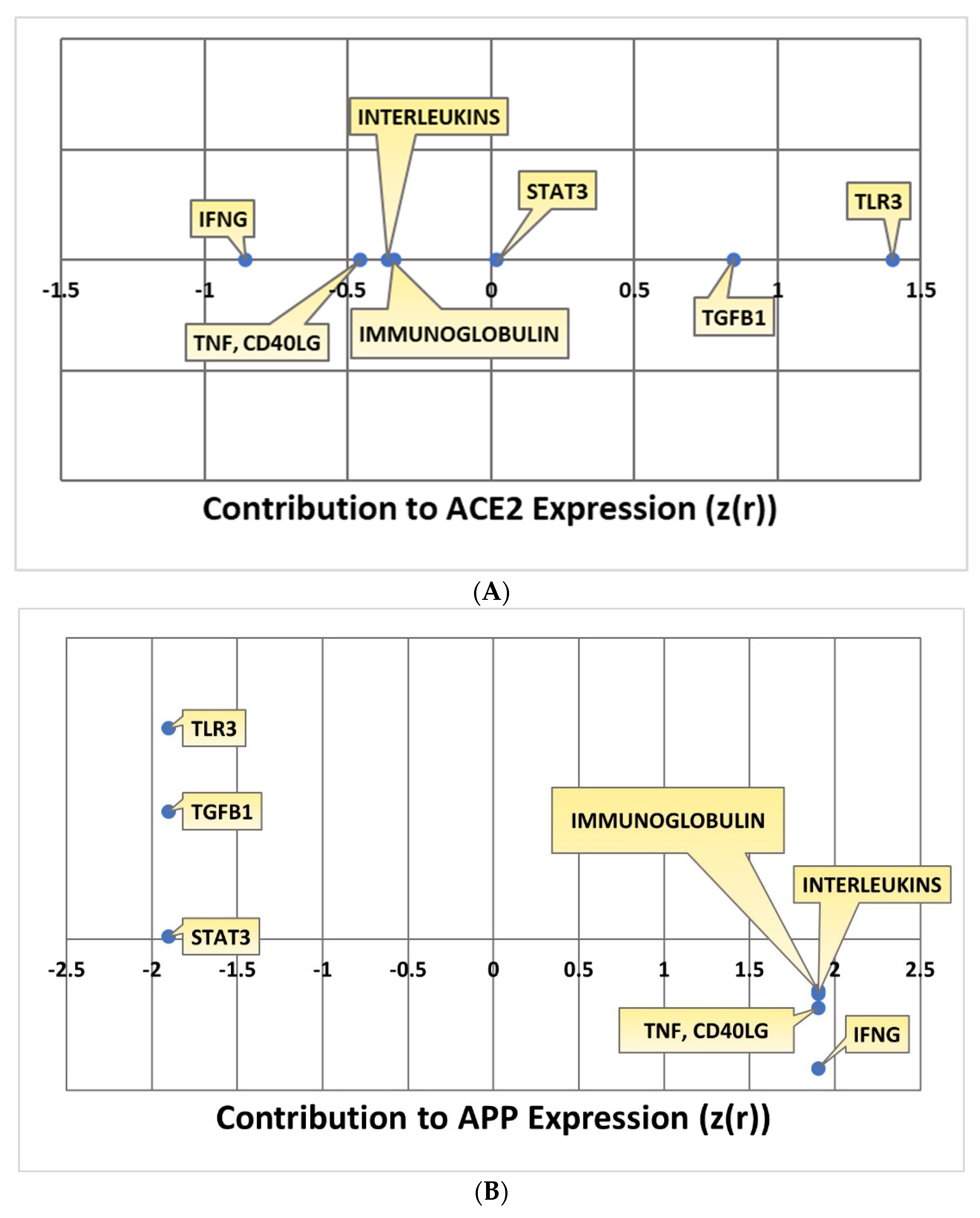
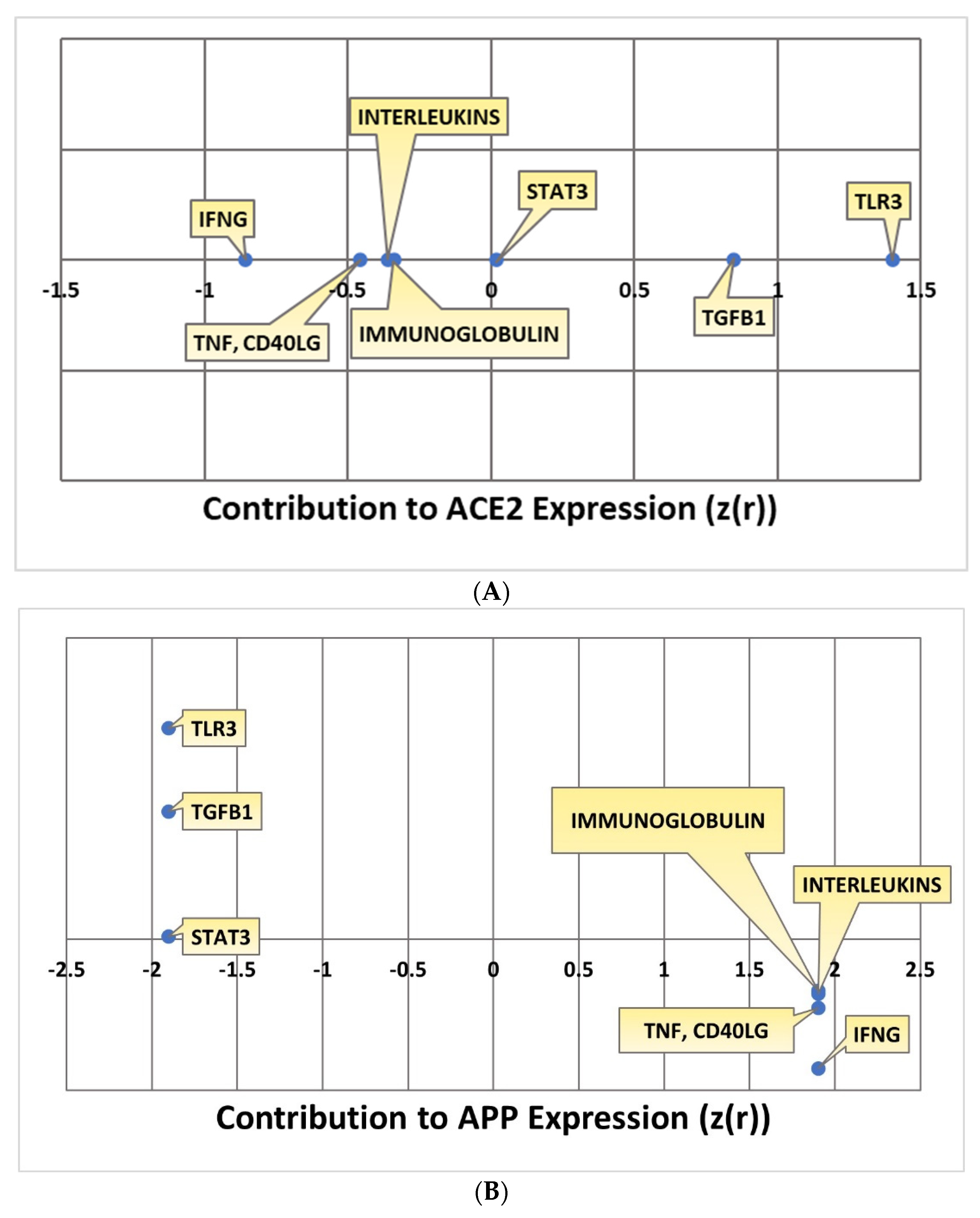
2.4. Quantitative Impact of SARS-CoV Upstream Regulators on ACE2 and APP
2.5. Overall Impact of Upstream Regulators on ACE2 and APP
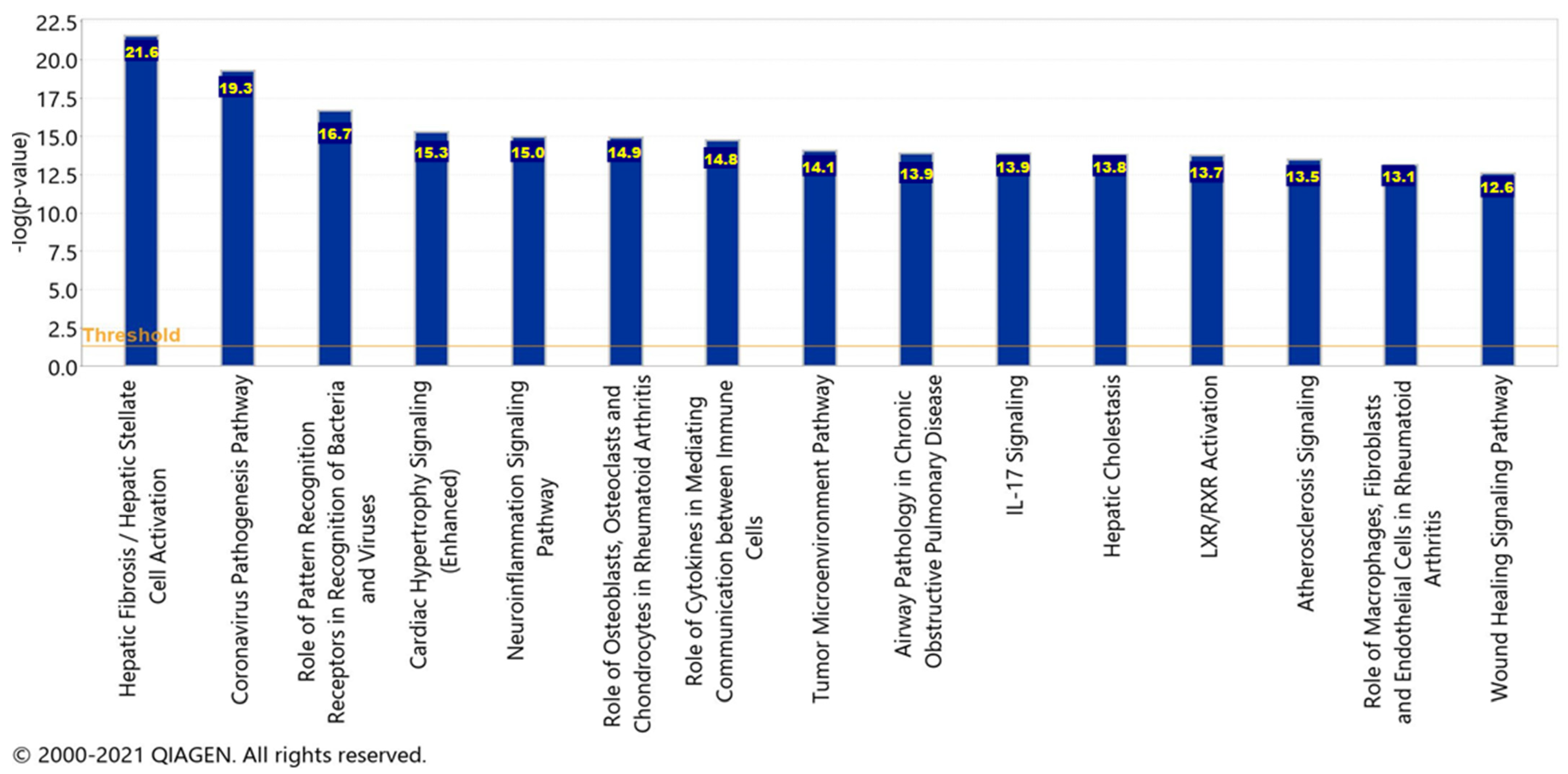
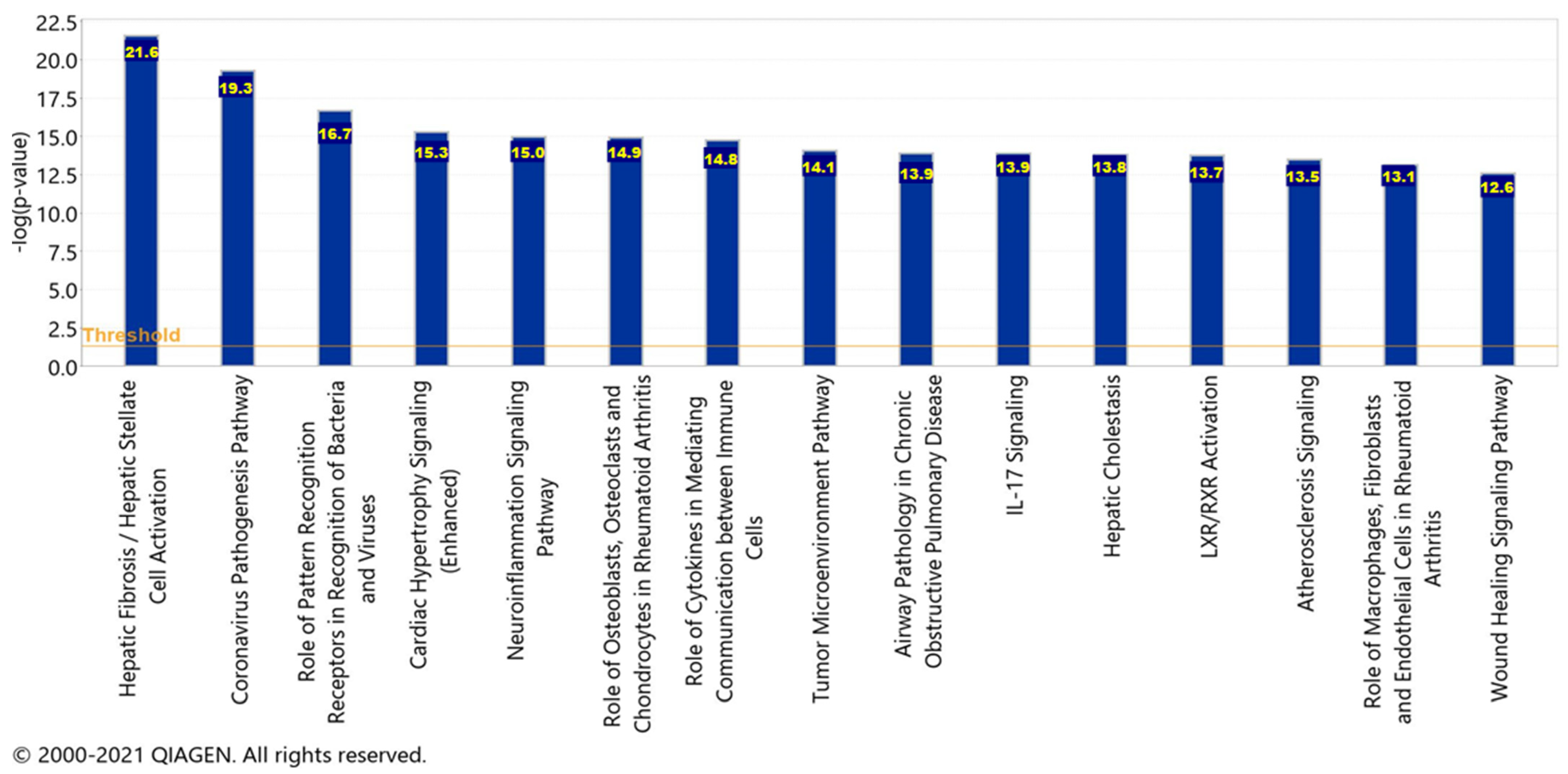
2.6. Canonical Pathways
3. Discussion
4. Materials and Methods
4.1. Ingenuity Pathway Analysis Software
4.2. Identification of SARS-CoV Upstream Regulators and Canonical Pathways
4.3. Connectivity and Molecule Activity Predictor (MAP)
4.4. Quantitative Analysis of the Influence of SARS-CoV Upstream Regulators on ACE2 and APP Expression
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abate, G.; Memo, M.; Uberti, D. Impact of COVID-19 on Alzheimer’s Disease Risk: Viewpoint for Research Action. Healthcare 2020, 8, 286. [Google Scholar] [CrossRef]
- Samavati, L.; Uhal, B.D. ACE2, Much More Than Just a Receptor for SARS-CoV-2. Front. Cell. Infect. Microbiol. 2020, 10, 317. [Google Scholar] [CrossRef] [PubMed]
- Miners, S.; Kehoe, P.G.; Love, S. Cognitive impact of COVID-19: Looking beyond the short term. Alzheimer’s Res. Ther. 2020, 12, 170. [Google Scholar] [CrossRef]
- Beyerstedt, S.; Casaro, E.B.; Rangel, É.B. COVID-19: Angiotensin-converting enzyme 2 (ACE2) expression and tissue susceptibility to SARS-CoV-2 infection. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Baig, A.M.; Khaleeq, A.; Ali, U.; Syeda, H. Evidence of the COVID-19 Virus Targeting the CNS: Tissue Distribution, Host-Virus Interaction, and Proposed Neurotropic Mechanisms. ACS Chem. Neurosci. 2020, 11, 995–998. [Google Scholar] [CrossRef] [Green Version]
- Overmyer, K.A.; Shishkova, E.; Miller, I.J.; Balnis, J.; Bernstein, M.N.; Peters-Clarke, T.M.; Meyer, J.G.; Quan, Q.; Muehlbauer, L.K.; Trujillo, E.A.; et al. Large-Scale Multi-omic Analysis of COVID-19 Severity. Cell Syst. 2021, 12, 23–40. [Google Scholar] [CrossRef]
- Xia, X.; Wang, Y.; Zheng, J. COVID-19 and Alzheimer’s disease: How one crisis worsens the other. Transl. Neurodegener. 2021, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Calabrò, M.; Rinaldi, C.; Santoro, G.; Crisafulli, C. The biological pathways of Alzheimer disease: A review. AIMS Neurosci. 2021, 8, 86–132. [Google Scholar] [CrossRef]
- Park, G.; Nhan, H.S.; Tyan, S.-H.; Kawakatsu, Y.; Zhang, C.; Navarro, M.; Koo, E.H. Caspase Activation and Caspase-Mediated Cleavage of APP Is Associated with Amyloid β-Protein-Induced Synapse Loss in Alzheimer’s Disease. Cell Rep. 2020, 31, 107839. [Google Scholar] [CrossRef]
- Ding, Q.; Shults, N.V.; Harris, B.T.; Suzuki, Y.J. Angiotensin-converting enzyme 2 (ACE2) is upregulated in Alzheimer’s disease brain. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ding, Q.; Shults, N.V.; Gychka, S.G.; Harris, B.T.; Suzuki, Y.J. Protein Expression of Angiotensin-Converting Enzyme 2 (ACE2) is Upregulated in Brains with Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 1687. [Google Scholar] [CrossRef] [PubMed]
- Kehoe, P.G.; Wong, S.; Al Mulhim, N.; Palmer, L.E.; Miners, J.S. Angiotensin-converting enzyme 2 is reduced in Alzheimer’s disease in association with increasing amyloid-β and tau pathology. Alzheimer’s Res. Ther. 2016, 8, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahira, A.C.; Verjovski-Almeida, S.; Ferreira, S.T. Dementia is an age-independent risk factor for severity and death in COVID-19 inpatients. Alzheimers Dement. 2021, 17, 12352. [Google Scholar] [CrossRef]
- Yang, L.; Xie, X.; Tu, Z.; Fu, J.; Xu, D.; Zhou, Y. The signal pathways and treatment of cytokine storm in COVID-19. Signal Transduct. Target. Ther. 2021, 6, 255. [Google Scholar] [CrossRef] [PubMed]
- Haga, S.; Yamamoto, N.; Nakai-Murakami, C.; Osawa, Y.; Tokunaga, K.; Sata, T.; Yamamoto, N.; Sasazuki, T.; Ishizaka, Y. Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc. Natl. Acad. Sci. USA 2008, 105, 7809–7814. [Google Scholar] [CrossRef] [Green Version]
- Glowacka, I.; Bertram, S.; Herzog, P.; Pfefferle, S.; Steffen, I.; Muench, M.O.; Simmons, G.; Hofmann, H.; Kuri, T.; Weber, F.; et al. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J. Virol. 2010, 84, 1198–1205. [Google Scholar] [CrossRef] [Green Version]
- Shukla, N.; Roelle, S.M.; Suzart, V.G.; Bruchez, A.M.; Matreyek, K.A. Mutants of human ACE2 differentially promote SARS-CoV and SARS-CoV-2 spike mediated infection. PLoS Pathog. 2021, 17, e1009715. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, J.; Schiavon, C.R.; He, M.; Chen, L.; Shen, H.; Zhang, Y.; Yin, Q.; Cho, Y.; Andrade, L.; et al. SARS-CoV-2 Spike Protein Impairs Endothelial Function via Downregulation of ACE 2. Circ. Res. 2021, 128, 1323–1326. [Google Scholar] [CrossRef]
- Miners, S.; Ashby, E.; Baig, S.; Harrison, R.; Tayler, H.; Speedy, E.; Prince, J.A.; Love, S.; Kehoe, P.G. Angiotensin-converting enzyme levels and activity in Alzheimer’s disease: Differences in brain and CSF ACE and association with ACE1 genotypes. Am. J. Transl. Res. 2009, 1, 163–177. [Google Scholar]
- Miners, J.S.; Ashby, E.; Van Helmond, Z.; Chalmers, K.A.; Palmer, L.E.; Love, S.; Kehoe, P.G. Angiotensin-converting enzyme (ACE) levels and activity in Alzheimer’s disease, and relationship of perivascular ACE-1 to cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. 2008, 34, 181–193. [Google Scholar] [CrossRef]
- Trougakos, I.P.; Stamatelopoulos, K.; Terpos, E.; Tsitsilonis, O.E.; Aivalioti, E.; Paraskevis, D.; Kastritis, E.; Pavlakis, G.N.; Dimopoulos, M.A. Insights to SARS-CoV-2 life cycle, pathophysiology, and rationalized treatments that target COVID-19 clinical complications. J. Biomed. Sci. 2021, 28, 9. [Google Scholar] [CrossRef] [PubMed]
- Mahmudpour, M.; Roozbeh, J.; Keshavarz, M.; Farrokhi, S.; Nabipour, I. COVID-19 cytokine storm: The anger of inflammation. Cytokine 2020, 133, 155151. [Google Scholar] [CrossRef]
- Cheng, F.; Fransson, L.-Å.; Mani, K. Proinflammatory cytokines induce accumulation of glypican-1-derived heparan sulfate and the C-terminal fragment of β-cleaved APP in autophagosomes of dividing neuronal cells. Glycobiology 2020, 30, 539–549. [Google Scholar] [CrossRef]
- Sun, Y.; Yao, J.; Kim, T.-W.; Tall, A.R. Expression of Liver X Receptor Target Genes Decreases Cellular Amyloid β Peptide Secretion*. J. Biol. Chem. 2003, 278, 27688–27694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebrahimi, H.; Naderian, M.; Sohrabpour, A.A. New Concepts on Pathogenesis and Diagnosis of Liver Fibrosis; A Review Article. Middle East J. Dig. Dis. 2016, 8, 166–178. [Google Scholar] [CrossRef] [Green Version]
- Warner, F.J.; Rajapaksha, H.; Shackel, N.; Herath, C.B. ACE2: From protection of liver disease to propagation of COVID-19. Clin. Sci. 2020, 134, 3137–3158. [Google Scholar] [CrossRef] [PubMed]
- Wiese, O.J.; Allwood, B.W.; Zemlin, A.E. COVID-19 and the renin-angiotensin system (RAS): A spark that sets the forest alight? Med. Hypotheses 2020, 144, 110231. [Google Scholar] [CrossRef]
- Singh, K.D.; Karnik, S.S. Angiotensin Receptors: Structure, Function, Signaling and Clinical Applications. J. Cell Signal. 2016, 1, 111. [Google Scholar] [CrossRef]
- Grace, J.A.; Herath, C.B.; Mak, K.Y.; Burrell, L.M.; Angus, P.W. Update on new aspects of the renin–angiotensin system in liver disease: Clinical implications and new therapeutic options. Clin. Sci. 2012, 123, 225–239. [Google Scholar] [CrossRef] [Green Version]
- Rajapaksha, I.G.; Gunarathne, L.S.; Angus, P.W.; Herath, C.B. Update on New Aspects of the Renin-Angiotensin System in Hepatic Fibrosis and Portal Hypertension: Implications for Novel Therapeutic Options. J. Clin. Med. 2021, 10, 702. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metawea, M.I.; Yousif, W.I.; Moheb, I. COVID 19 and liver: An A–Z literature review. Dig. Liver Dis. 2021, 53, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, S.; Liu, H.; Li, W.; Lin, F.; Jiang, L.; Li, X.; Xu, P.; Zhang, L.; Zhao, L.; et al. SARS-CoV-2 infection of the liver directly contributes to hepatic impairment in patients with COVID-19. J. Hepatol. 2020, 73, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Nho, K.; Kueider-Paisley, A.; Ahmad, S.; MahmoudianDehkordi, S.; Arnold, M.; Risacher, S.L.; Louie, G.; Blach, C.; Baillie, R.; Han, X.; et al. Association of Altered Liver Enzymes with Alzheimer Disease Diagnosis, Cognition, Neuroimaging Measures, and Cerebrospinal Fluid Biomarkers. JAMA Netw. Open 2019, 2, e197978. [Google Scholar] [CrossRef]
- Vieira, C.; Nery, L.; Martins, L.; Jabour, L.; Dias, R.; Simões, E.S.A.C. Downregulation of Membrane-bound Angiotensin Converting Enzyme 2 (ACE2) Receptor has a Pivotal Role in COVID-19 Immunopathology. Curr. Drug Targets 2021, 22, 254–281. [Google Scholar] [CrossRef]
- Banu, N.; Panikar, S.S.; Leal, L.R.; Leal, A.R. Protective role of ACE2 and its downregulation in SARS-CoV-2 infection leading to Macrophage Activation Syndrome: Therapeutic implications. Life Sci. 2020, 256, 117905. [Google Scholar] [CrossRef]
- Silhol, F.; Sarlon, G.; Deharo, J.-C.; Vaïsse, B. Downregulation of ACE2 induces overstimulation of the renin–angiotensin system in COVID-19: Should we block the renin–angiotensin system? Hypertens. Res. 2020, 43, 854–856. [Google Scholar] [CrossRef]
- Estrada, L.D.; Ahumada, P.; Cabrera, D.; Arab, J.P. Liver Dysfunction as a Novel Player in Alzheimer’s Progression: Looking Outside the Brain. Front. Aging Neurosci. 2019, 11, 174. [Google Scholar] [CrossRef] [Green Version]
- Maarouf, C.L.; Walker, J.E.; Sue, L.I.; Dugger, B.N.; Beach, T.G.; Serrano, G.E. Impaired hepatic amyloid-beta degradation in Alzheimer’s disease. PLoS ONE 2018, 13, e0203659. [Google Scholar] [CrossRef]
- Kanekiyo, T.; Bu, G. The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 93. [Google Scholar] [CrossRef] [Green Version]
- Roher, A.E.; Esh, C.L.; Kokjohn, T.A.; Castaño, E.M.; Van Vickle, G.D.; Kalback, W.M.; Patton, R.L.; Luehrs, D.C.; Daugs, I.D.; Kuo, Y.-M.; et al. Amyloid beta peptides in human plasma and tissues and their significance for Alzheimer’s disease. Alzheimer’s Dement. 2009, 5, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Tontonoz, P. Liver X receptors in lipid signalling and membrane homeostasis. Nat. Rev. Endocrinol. 2018, 14, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Rivest, S. Lipid Metabolism and Neuroinflammation in Alzheimer’s Disease: A Role for Liver X Receptors. Endocr. Rev. 2012, 33, 715–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouzat, K.; Chudinova, A.; Polge, A.; Kantar, J.; Camu, W.; Raoul, C.; Lumbroso, S. Regulation of Brain Cholesterol: What Role Do Liver X Receptors Play in Neurodegenerative Diseases? Int. J. Mol. Sci. 2019, 20, 3858. [Google Scholar] [CrossRef] [Green Version]
- Sodhi, R.K.; Singh, N. Liver X receptors: Emerging therapeutic targets for Alzheimer’s disease. Pharmacol. Res. 2013, 72, 45–51. [Google Scholar] [CrossRef]
- Ito, A.; Hong, C.; Rong, X.; Zhu, X.; Tarling, E.J.; Hedde, P.N.; Gratton, E.; Parks, J.; Tontonoz, P. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. Elife 2015, 4, e08009. [Google Scholar] [CrossRef]
- Zotova, E.; Nicoll, J.A.R.; Kalaria, R.; Holmes, C.; Boche, D. Inflammation in Alzheimer’s disease: Relevance to pathogenesis and therapy. Alzheimers Res. Ther. 2010, 2, 1. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; Khoury, J.E.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Jiang, L. Neuroinflammation in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2015, 11, 243–256. [Google Scholar] [CrossRef] [Green Version]
- Nakhlband, A.; Fakhari, A.; Azizi, H. Interferon-beta offers promising avenues to COVID-19 treatment: A systematic review and meta-analysis of clinical trial studies. Naunyn. Schmiedebergs Arch. Pharmacol. 2021, 394, 829–838. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Upstream Regulator | Molecule Type | p-Value of Overlap |
|---|---|---|
| IFNG | cytokine | 9.57 × 10−73 |
| TNF | cytokine | 4.57 × 10−67 |
| IL1B | cytokine | 8.24 × 10−65 |
| Immunoglobulin | complex | 2.99 × 10−57 |
| STAT1 | transcription regulator | 6.56 × 10−55 |
| Interferon alpha | Group | 1.3 × 10−54 |
| STAT3 | transcription regulator | 1.3 × 10−52 |
| APP | Other | 6.25 × 10−48 |
| IL6 | cytokine | 1.52 × 10−45 |
| IRF1 | transcription regulator | 1.36 × 10−44 |
| IL10 | cytokine | 1.56 × 10−44 |
| IL2 | cytokine | 1.31 × 10−43 |
| TLR3 | transmembrane receptor | 1.36 × 10−43 |
| NONO | transcription regulator | 2.13 × 10−42 |
| IL4 | cytokine | 6.99 × 10−42 |
| TGFB1 | growth factor | 6.35 × 10−41 |
| IL1 | Group | 4.68 × 10−40 |
| CD40LG | cytokine | 6.99 × 10−40 |
| IFNA2 | cytokine | 2.64 × 10−39 |
| IFN Beta | Group | 1.5 × 10−37 |
| IRF7 | transcription regulator | 1.7 × 10−37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caradonna, A.; Patel, T.; Toleska, M.; Alabed, S.; Chang, S.L. Meta-Analysis of APP Expression Modulated by SARS-CoV-2 Infection via the ACE2 Receptor. Int. J. Mol. Sci. 2022, 23, 1182. https://doi.org/10.3390/ijms23031182
Caradonna A, Patel T, Toleska M, Alabed S, Chang SL. Meta-Analysis of APP Expression Modulated by SARS-CoV-2 Infection via the ACE2 Receptor. International Journal of Molecular Sciences. 2022; 23(3):1182. https://doi.org/10.3390/ijms23031182
Chicago/Turabian StyleCaradonna, Alyssa, Tanvi Patel, Matea Toleska, Sedra Alabed, and Sulie L. Chang. 2022. "Meta-Analysis of APP Expression Modulated by SARS-CoV-2 Infection via the ACE2 Receptor" International Journal of Molecular Sciences 23, no. 3: 1182. https://doi.org/10.3390/ijms23031182
APA StyleCaradonna, A., Patel, T., Toleska, M., Alabed, S., & Chang, S. L. (2022). Meta-Analysis of APP Expression Modulated by SARS-CoV-2 Infection via the ACE2 Receptor. International Journal of Molecular Sciences, 23(3), 1182. https://doi.org/10.3390/ijms23031182