
Epigenomic and Transcriptomic Prioritization of Candidate Obesity-Risk Regulatory GWAS SNPs


Abstract
:1. Introduction
2. Results
2.1. Candidate Regulatory SNPs (Tier-1 SNPs) for Obesity Risk Were Prioritized from 50 Obesity-Related GWAS
2.2. Limited Power in Finding Best Candidate Regulatory SNPs with Several Commonly Used Statistical Methods
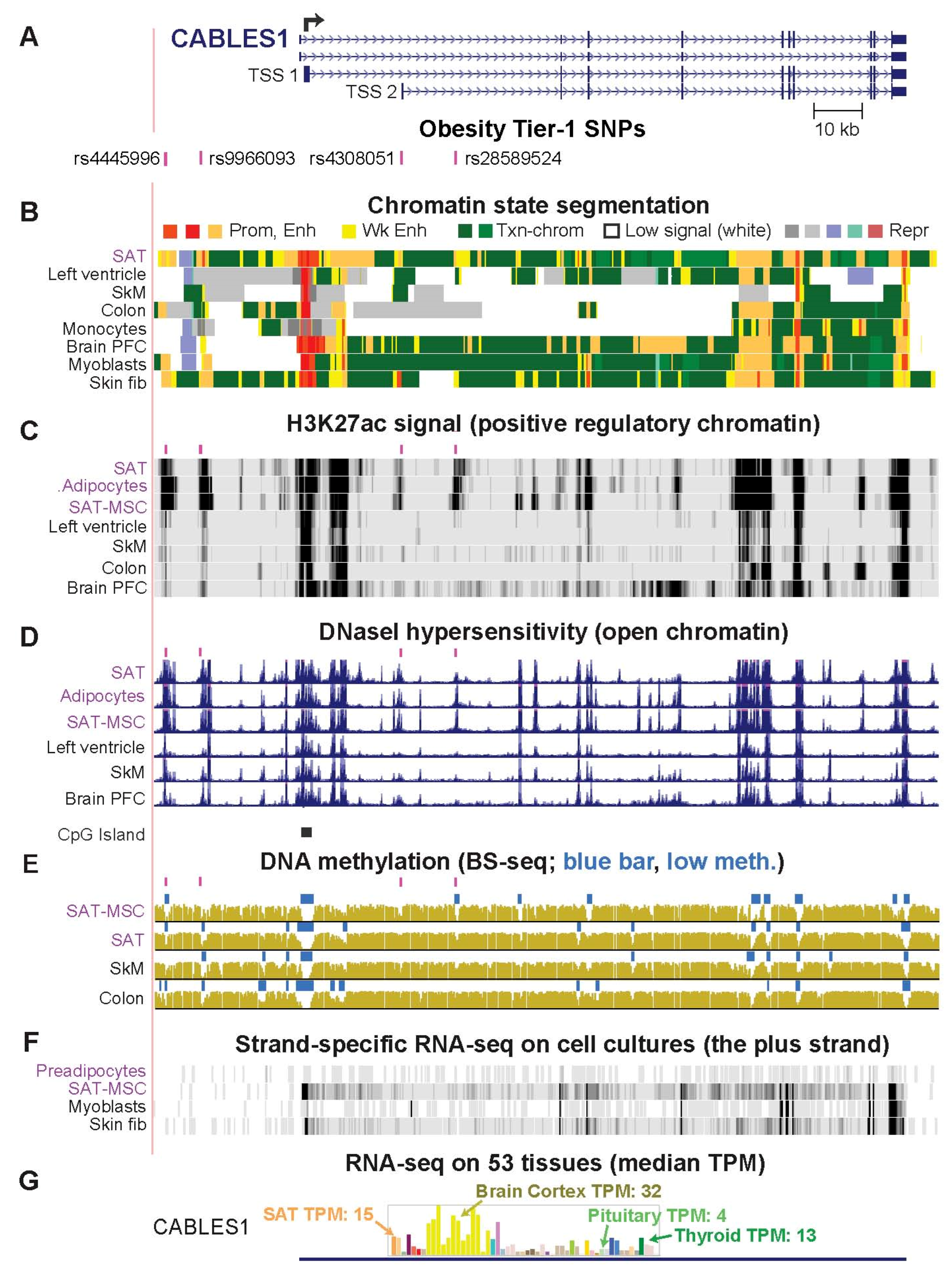
2.3. CABLES1, PEMT, and PC Genes Are Associated with Obesity Tier-1 SNPs That May Regulate Alternate Usage of Transcription Start Sites
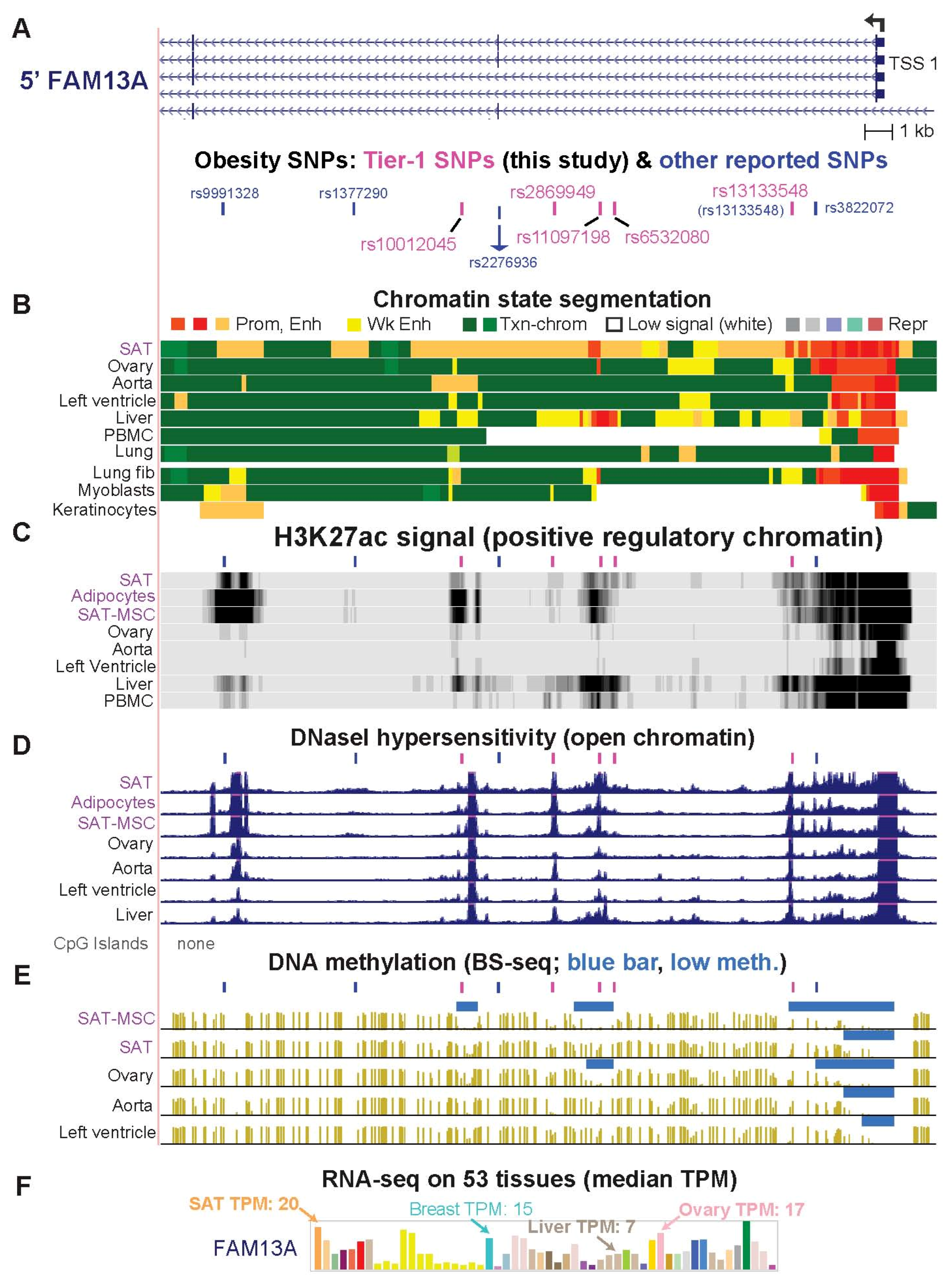
2.4. Only One of the Previously Prioritized FAM13A Obesity-Related Regulatory SNP Candidates Is among the Six Tier-1 FAM13A SNPs
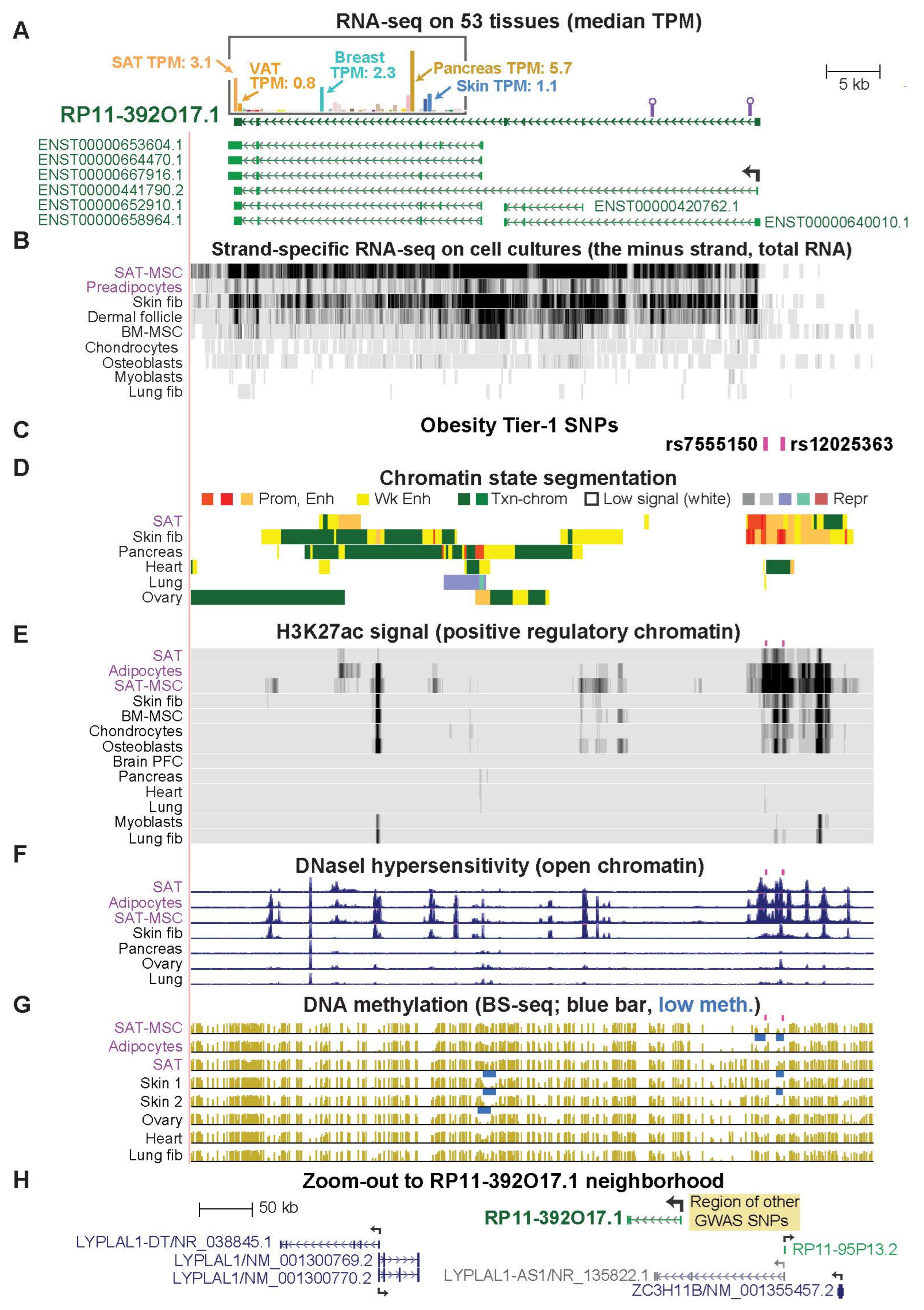
2.5. RP11-392O17.1, an Often-Overlooked SAT-Specific lncRNA Gene, Has Novel Tier-1 SNPs
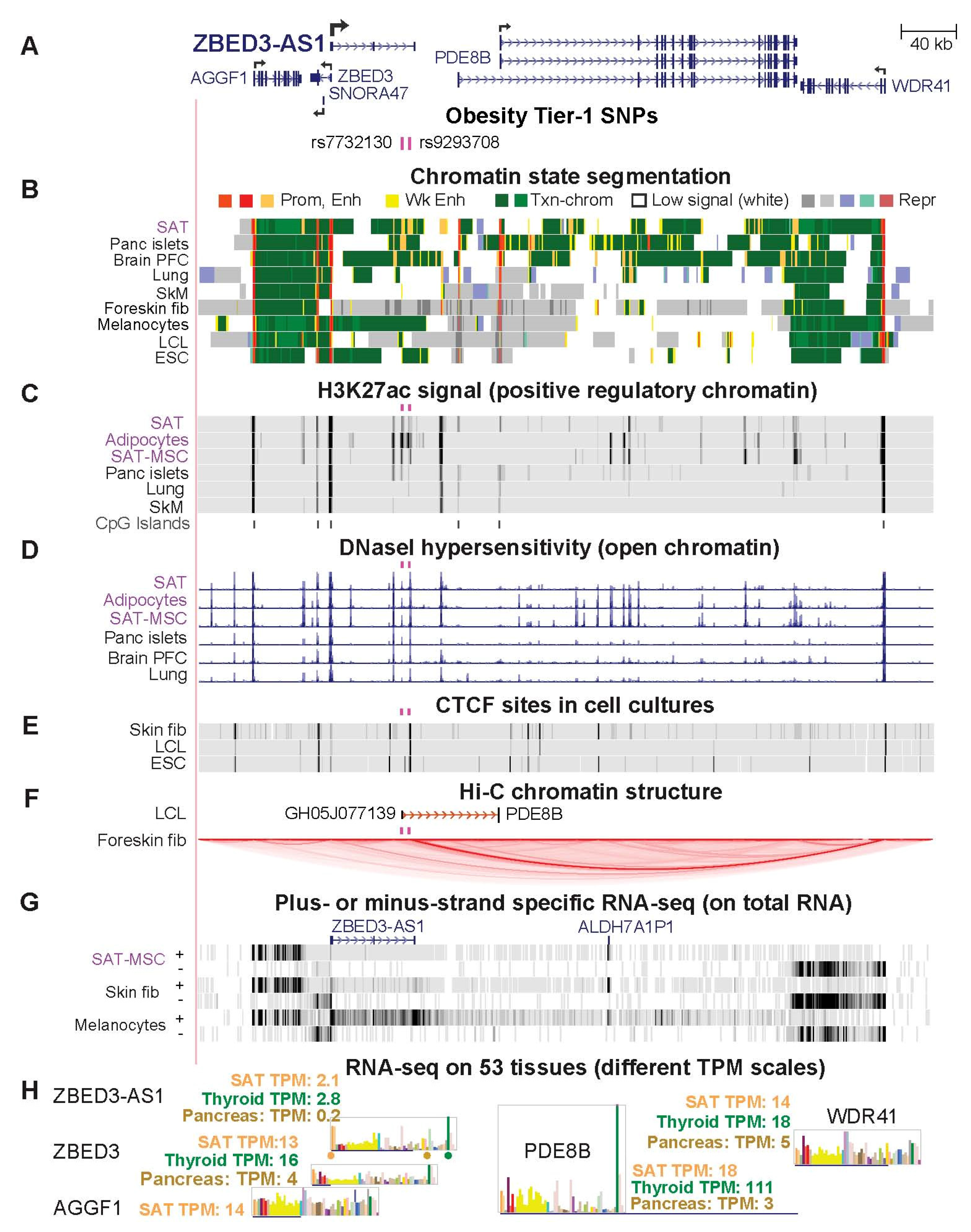
2.6. Two Obesity Tier-1 SNPs Located in ZBED3-AS1 Might Help Control Expression in SAT of Several Genes
3. Discussion
4. Methods
4.1. Obesity GWAS Data
4.2. Epigenomics
4.3. Transcriptomics
4.4. Excluding EnhPro SNPs Linked to Missense SNPs and Predicting Allele-Specific TFBS
4.5. Statistical Analyses to Look for Potential Causal Regulatory SNPs
4.6. Transcription from RP11-392O17.1 in SAT-MSC
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations:
| BS | bisulfite |
| CAGE | 5′ Cap analysis gene expression |
| CGI | CpG island |
| ChIP-seq | chromatin immunoprecipitation-sequencing |
| Enh | enhancer |
| FPKM | fragments per kilobase of exon per million reads mapped |
| H3K27ac | histone H3 lysine 27 acetylation |
| H3K4me1 | histone H3 lysine 4 monomethylation |
| H3K4me3 | histone H3 lysine 4 trimethylation |
| LCL | lymphoblastoid cell line |
| LMR | low methylated region |
| lncRNA | long non-coding RNA |
| Micro-C | chromatin capture analysis using micrococcal nuclease |
| NHLF | normal human lung fibroblasts |
| ORF | open reading frame |
| PBMC | peripheral blood mononuclear cells |
| SkM | skeletal muscle |
| TAD | topologically associating domain |
| TPM | transcripts per million |
| TSS | transcription start site |
| Txn | transcription |
References
- Ling, C.; Ronn, T. Epigenetics in human obesity and Type 2 diabetes. Cell Metab. 2019, 29, 1028–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohde, K.; Keller, M.; la Cour Poulsen, L.; Bluher, M.; Kovacs, P.; Bottcher, Y. Genetics and epigenetics in obesity. Metabolism 2019, 92, 37–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porro, S.; Genchi, V.A.; Cignarelli, A.; Natalicchio, A.; Laviola, L.; Giorgino, F.; Perrini, S. Dysmetabolic adipose tissue in obesity: Morphological and functional characteristics of adipose stem cells and mature adipocytes in healthy and unhealthy obese subjects. J. Endocrinol. Investig. 2021, 44, 921–941. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.F.; Clegg, D.J. The sexual dimorphism of obesity. Mol. Cell Endocrinol. 2015, 402, 113–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zacharia, A.; Saidemberg, D.; Mannully, C.T.; Kogan, N.M.; Shehadeh, A.; Sinai, R.; Zucker, A.; Bruck-Haimson, R.; Goldstein, N.; Haim, Y.; et al. Distinct infrastructure of lipid networks in visceral and subcutaneous adipose tissues in overweight humans. Am. J. Clin. Nutr. 2020, 112, 979–990. [Google Scholar] [CrossRef]
- Loos, R.J. The genetics of adiposity. Curr. Opin. Genet. Dev. 2018, 50, 86–95. [Google Scholar] [CrossRef]
- Dong, S.S.; Zhang, Y.J.; Chen, Y.X.; Yao, S.; Hao, R.H.; Rong, Y.; Niu, H.M.; Chen, J.B.; Guo, Y.; Yang, T.L. Comprehensive review and annotation of susceptibility SNPs associated with obesity-related traits. Obes. Rev. 2018, 19, 917–930. [Google Scholar] [CrossRef]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Fathzadeh, M.; Li, J.; Rao, A.; Cook, N.; Chennamsetty, I.; Seldin, M.; Zhou, X.; Sangwung, P.; Gloudemans, M.J.; Keller, M.; et al. FAM13A affects body fat distribution and adipocyte function. Nat. Commun. 2020, 11, 1465. [Google Scholar] [CrossRef] [PubMed]
- Kempe-Teufel, D.; Machicao, F.; Machann, J.; Bohm, A.; Schick, F.; Fritsche, A.; Stefan, N.; Hrabe de Angelis, M.; Haring, H.U.; Staiger, H. A Polygenic Risk Score of Lipolysis-Increasing Alleles Determines Visceral Fat Mass and Proinsulin Conversion. J. Clin. Endocrinol. Metab. 2019, 104, 1090–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lumish, H.S.; O’Reilly, M.; Reilly, M.P. Sex differences in genomic drivers of adipose distribution and related cardiometabolic disorders: Opportunities for precision medicine. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Lundback, V.; Kulyte, A.; Strawbridge, R.J.; Ryden, M.; Arner, P.; Marcus, C.; Dahlman, I. FAM13A and POM121C are candidate genes for fasting insulin: Functional follow-up analysis of a genome-wide association study. Diabetologia 2018, 61, 1112–1123. [Google Scholar] [CrossRef] [Green Version]
- Spracklen, C.N.; Karaderi, T.; Yaghootkar, H.; Schurmann, C.; Fine, R.S.; Kutalik, Z.; Preuss, M.H.; Lu, Y.; Wittemans, L.B.L.; Adair, L.S.; et al. Exome-Derived Adiponectin-Associated Variants Implicate Obesity and Lipid Biology. Am. J. Hum. Genet. 2019, 105, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Kichaev, G.; Roytman, M.; Johnson, R.; Eskin, E.; Lindström, S.; Kraft, P.; Pasaniuc, B. Improved methods for multi-trait fine mapping of pleiotropic risk loci. Bioinformatics 2017, 33, 248–255. [Google Scholar] [CrossRef]
- The GTEx Consortium; Ardlie, K.G.; Deluca, D.S.; Segrè, A.V.; Sullivan, T.J.; Young, T.R.; Gelfand, E.T.; Trowbridge, C.A.; Maller, J.B.; Tukiainen, T.; et al. The genotype-tissue expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [Green Version]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Ehrlich, K.C.; Yu, F.; Hu, X.; Meng, X.H.; Deng, H.W.; Shen, H.; Ehrlich, M. Osteoporosis- and obesity-risk interrelationships: An epigenetic analysis of GWAS-derived SNPs at the developmental gene TBX15. Epigenetics 2020, 15, 728–749. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, A.E.; Zheng, T.; Yi, C.; Leaderer, D.; Weidhaas, J.; Slack, F.; Zhang, Y.; Paranjape, T.; Zhu, Y. microRNA miR-196a-2 and breast cancer: A genetic and epigenetic association study and functional analysis. Cancer Res. 2009, 69, 5970–5977. [Google Scholar] [CrossRef] [Green Version]
- Divoux, A.; Xie, H.; Li, J.L.; Karastergiou, K.; Perera, R.J.; Chang, R.J.; Fried, S.K.; Smith, S.R. MicroRNA-196 regulates HOX gene expression in human gluteal adipose tissue. Obesity 2017, 25, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Ghanbari, M.; Sedaghat, S.; de Looper, H.W.; Hofman, A.; Erkeland, S.J.; Franco, O.H.; Dehghan, A. The association of common polymorphisms in miR-196a2 with waist to hip ratio and miR-1908 with serum lipid and glucose. Obesity 2015, 23, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Ferreira, T.; Morris, A.P.; Medland, S.E.; Genetic Investigation of ANthropometric Traits (GIANT) Consortium; DIAbetes Genetics Replication and Meta-analysis (DIAGRAM) Consortium; Madden, P.A.F.; Heath, A.C.; Martin, N.G.; Montgomery, G.W.; et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 2012, 44, 369–375. [Google Scholar] [CrossRef]
- Pulit, S.L.; Stoneman, C.; Morris, A.P.; Wood, A.R.; Glastonbury, C.A.; Tyrrell, J.; Yengo, L.; Ferreira, T.; Marouli, E.; Ji, Y.; et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum. Mol. Genet. 2019, 28, 166–174. [Google Scholar] [CrossRef] [Green Version]
- Hormozdiari, F.; van de Bunt, M.; Segrè, A.V.; Li, X.; Joo, J.W.J.; Bilow, M.; Sul, J.H.; Sankararaman, S.; Pasaniuc, B.; Eskin, E. Colocalization of GWAS and eQTL Signals Detects Target Genes. Am. J. Hum. Genet. 2016, 99, 1245–1260. [Google Scholar] [CrossRef] [Green Version]
- Greenwald, W.W.; Chiou, J.; Yan, J.; Qiu, Y.; Dai, N.; Wang, A.; Nariai, N.; Aylward, A.; Han, J.Y.; Kadakia, N.; et al. Pancreatic islet chromatin accessibility and conformation reveals distal enhancer networks of type 2 diabetes risk. Nat. Commun. 2019, 10, 2078. [Google Scholar] [CrossRef]
- Miguel-Escalada, I.; Bonas-Guarch, S.; Cebola, I.; Ponsa-Cobas, J.; Mendieta-Esteban, J.; Atla, G.; Javierre, B.M.; Rolando, D.M.Y.; Farabella, I.; Morgan, C.C.; et al. Human pancreatic islet three-dimensional chromatin architecture provides insights into the genetics of type 2 diabetes. Nat. Genet. 2019, 51, 1137–1148. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Roussel-Gervais, A.; Couture, C.; Langlais, D.; Takayasu, S.; Balsalobre, A.; Rueda, B.R.; Zukerberg, L.R.; Figarella-Branger, D.; Brue, T.; Drouin, J. The Cables1 Gene in Glucocorticoid Regulation of Pituitary Corticotrope Growth and Cushing Disease. J. Clin. Endocrinol. Metab. 2016, 101, 513–522. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; Pelizzola, M.; Kida, Y.S.; Hawkins, R.D.; Nery, J.R.; Hon, G.; Antosiewicz-Bourget, J.; O’Malley, R.; Castanon, R.; Klugman, S.; et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 2011, 471, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Zukerberg, L.R.; Patrick, G.N.; Nikolic, M.; Humbert, S.; Wu, C.L.; Lanier, L.M.; Gertler, F.B.; Vidal, M.; Van Etten, R.A.; Tsai, L.H. Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine phosphorylation, kinase upregulation, and neurite outgrowth. Neuron 2000, 26, 633–646. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.L.; Cai, C.; Yang, Z.Y.; Wu, Z.Y.; Wu, X.S.; Wang, X.F.; Dong, P.; Gong, W. DGCR5 is activated by PAX5 and promotes pancreatic cancer via targeting miR-3163/TOP2A and activating Wnt/beta-catenin pathway. Int. J. Biol. Sci. 2021, 17, 498–513. [Google Scholar] [CrossRef] [PubMed]
- Kawai, M.; Rosen, C.J. PPARgamma: A circadian transcription factor in adipogenesis and osteogenesis. Nat. Rev. Endocrinol. 2010, 6, 629–636. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Chung, J.W.; Mei, W.; Strack, S.; He, C.; Lau, G.W.; Yang, J. Regulation of nuclear-cytoplasmic shuttling and function of Family with sequence similarity 13, member A (Fam13a), by B56-containing PP2As and Akt. Mol. Biol. Cell. 2015, 26, 1160–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wardhana, D.A.; Ikeda, K.; Barinda, A.J.; Nugroho, D.B.; Qurania, K.R.; Yagi, K.; Miyata, K.; Oike, Y.; Hirata, K.I.; Emoto, N. Family with sequence similarity 13, member A modulates adipocyte insulin signaling and preserves systemic metabolic homeostasis. Proc. Natl. Acad. Sci. USA 2018, 115, 1529–1534. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Liou, Y.H.; Li, Y.; Gong, L.; Li, Y.; Hao, Y.; Pham, B.; Xu, S.; Jiang, Z.; Li, L.; et al. FAM13A Represses AMPK Activity and Regulates Hepatic Glucose and Lipid Metabolism. iScience 2020, 23, 100928. [Google Scholar] [CrossRef] [Green Version]
- Corvol, H.; Hodges, C.A.; Drumm, M.L.; Guillot, L. Moving beyond genetics: Is FAM13A a major biological contributor in lung physiology and chronic lung diseases? J. Med. Genet. 2014, 51, 646–649. [Google Scholar] [CrossRef]
- McDowell, I.C.; Pai, A.A.; Guo, C.; Vockley, C.M.; Brown, C.D.; Reddy, T.E.; Engelhardt, B.E. Many long intergenic non-coding RNAs distally regulate mRNA gene expression levels. bioRxiv 2016. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Fan, J.; Xu, H.; Fan, L.; Deng, L.; Li, J.; Li, D.; Li, H.; Zhang, F.; Zhao, R.C. Long noncoding RNA LYPLAL1-AS1 regulates adipogenic differentiation of human mesenchymal stem cells by targeting desmoplakin and inhibiting the Wnt/β-catenin pathway. Cell Death Discov. 2021, 7, 105. [Google Scholar] [CrossRef]
- Gheorghe, M.; Sandve, G.K.; Khan, A.; Cheneby, J.; Ballester, B.; Mathelier, A. A map of direct TF-DNA interactions in the human genome. Nucleic Acids Res. 2019, 47, e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Guo, Y.; Shi, H.; Liu, C.L.; Panganiban, R.A.; Chung, W.; O’Connor, L.J.; Himes, B.E.; Gazal, S.; Hasegawa, K.; et al. Shared genetic and experimental links between obesity-related traits and asthma subtypes in UK Biobank. J. Allergy Clin. Immunol. 2020, 145, 537–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funkhouser, S.A.; Vazquez, A.I.; Steibel, J.P.; Ernst, C.W.; Los Campos, G. Deciphering Sex-Specific Genetic Architectures Using Local Bayesian Regressions. Genetics 2020, 215, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Ballantyne, R.L.; Zhang, X.; Nunez, S.; Xue, C.; Zhao, W.; Reed, E.; Salaheen, D.; Foulkes, A.S.; Li, M.; Reilly, M.P. Genome-wide interrogation reveals hundreds of long intergenic noncoding RNAs that associate with cardiometabolic traits. Hum. Mol. Genet. 2016, 25, 3125–3141. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Yang, Y.; Fan, L.; Deng, L.; Fan, J.; Li, D.; Li, H.; Zhao, R.C. Lnc13728 facilitates human mesenchymal stem cell adipogenic differentiation via positive regulation of ZBED3 and downregulation of the WNT/beta-catenin pathway. Stem Cell Res. Ther. 2021, 12, 176. [Google Scholar] [CrossRef]
- Dov, A.; Abramovitch, E.; Warwar, N.; Nesher, R. Diminished phosphodiesterase-8B potentiates biphasic insulin response to glucose. Endocrinology 2008, 149, 741–748. [Google Scholar] [CrossRef]
- Westphal, N.J.; Seasholtz, A.F. CRH-BP: The regulation and function of a phylogenetically conserved binding protein. Front. Biosci. 2006, 11, 1878–1891. [Google Scholar] [CrossRef] [Green Version]
- Eraslan, G.; Drokhlyansky, E.; Anand, S.; Subramanian, A.; Fiskin, E.; Slyper, M.; Wang, J.; Wittenberghe, N.V.; Rouhana, J.M.; Waldman, J.; et al. Single-nucleus cross-tissue molecular reference maps to decipher disease gene function. bioRxiv 2021. [Google Scholar] [CrossRef]
- Mifsud, B.; Tavares-Cadete, F.; Young, A.N.; Sugar, R.; Schoenfelder, S.; Ferreira, L.; Wingett, S.W.; Andrews, S.; Grey, W.; Ewels, P.A.; et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet. 2015, 47, 598–606. [Google Scholar] [CrossRef]
- Krietenstein, N.; Abraham, S.; Venev, S.V.; Abdennur, N.; Gibcus, J.; Hsieh, T.S.; Parsi, K.M.; Yang, L.; Maehr, R.; Mirny, L.A.; et al. Ultrastructural details of mammalian chromosome architecture. Mol. Cell 2020, 78, 554–565.e7. [Google Scholar] [CrossRef]
- Banks, A.S.; McAllister, F.E.; Camporez, J.P.; Zushin, P.J.; Jurczak, M.J.; Laznik-Bogoslavski, D.; Shulman, G.I.; Gygi, S.P.; Spiegelman, B.M. An ERK/Cdk5 axis controls the diabetogenic actions of PPARgamma. Nature 2015, 517, 391–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Take, K.; Waki, H.; Sun, W.; Wada, T.; Yu, J.; Nakamura, M.; Aoyama, T.; Yamauchi, T.; Kadowaki, T. CDK5 Regulatory Subunit-Associated Protein 1-like 1 Negatively Regulates Adipocyte Differentiation through Activation of Wnt Signaling Pathway. Sci. Rep. 2017, 7, 7326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jitrapakdee, S.; St Maurice, M.; Rayment, I.; Cleland, W.W.; Wallace, J.C.; Attwood, P.V. Structure, mechanism and regulation of pyruvate carboxylase. Biochem. J. 2008, 413, 369–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presa, N.; Dominguez-Herrera, A.; van der Veen, J.N.; Vance, D.E.; Gomez-Munoz, A. Implication of phosphatidylethanolamine N-methyltransferase in adipocyte differentiation. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165853. [Google Scholar] [CrossRef]
- Shank, R.P.; Bennett, G.S.; Freytag, S.O.; Campbell, G.L. Pyruvate carboxylase: An astrocyte-specific enzyme implicated in the replenishment of amino acid neurotransmitter pools. Brain Res. 1985, 329, 364–367. [Google Scholar] [CrossRef]
- Nuermaimaiti, N.; Liu, J.; Liang, X.; Jiao, Y.; Zhang, D.; Liu, L.; Meng, X.; Guan, Y. Effect of lncRNA HOXA11-AS1 on adipocyte differentiation in human adipose-derived stem cells. Biochem. Biophys. Res. Commun. 2018, 495, 1878–1884. [Google Scholar] [CrossRef]
- Singh, S.; Rajput, Y.S.; Barui, A.K.; Sharma, R.; Datta, T.K. Fat accumulation in differentiated brown adipocytes is linked with expression of Hox genes. Gene Expr. Patterns 2016, 20, 99–105. [Google Scholar] [CrossRef]
- Huang, J.R.; Tan, G.M.; Li, Y.; Shi, Z. The Emerging Role of Cables1 in Cancer and Other Diseases. Mol. Pharmacol. 2017, 92, 240–245. [Google Scholar] [CrossRef]
- Shi, Z.; Li, Z.; Li, Z.J.; Cheng, K.; Du, Y.; Fu, H.; Khuri, F.R. Cables1 controls p21/Cip1 protein stability by antagonizing proteasome subunit alpha type 3. Oncogene 2015, 34, 2538–2545. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.; Sun, J.G.; Wang, J.Q.; Li, B.; Liu, Q.; Ning, G.; Jin, W.; Yuan, Z. c-Abl inhibition mitigates diet-induced obesity through improving insulin sensitivity of subcutaneous fat in mice. Diabetologia 2017, 60, 900–910. [Google Scholar] [CrossRef] [Green Version]
- Ruffin, M.; Thompson, K.E.; Corvol, H.; Guillot, L. Two-hybrid screening of FAM13A protein partners in lung epithelial cells. BMC Res. Notes 2020, 12, 804. [Google Scholar] [CrossRef]
- Lin, X.; Li, Y.; Gong, L.; Yun, J.H.; Xu, S.; Tesfaigzi, Y.; Qiao, D.; Zhou, X. Tempo-spatial regulation of the Wnt pathway by FAM13A modulates the stemness of alveolar epithelial progenitors. EBioMedicine 2021, 69, 103463. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Wang, J. Wnt/beta-Catenin Signaling and Obesity. Front. Physiol. 2018, 9, 792. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Zhou, H.; Sahay, K.; Xu, W.; Yang, J.; Zhang, W.; Chen, W. Obesity-associated family with sequence similarity 13, member A (FAM13A) is dispensable for adipose development and insulin sensitivity. Int. J. Obes. 2019, 43, 1269–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, A.; Yu, Z.; Zhang, E.; Song, J. Long noncoding RNA ZBED3-AS1 restrains breast cancer progression by targeting the microRNA-513a-5p/KLF6 axis. Thorac. Cancer 2021, 12, 2719–2731. [Google Scholar] [CrossRef]
- Li, F.; Wu, X.; Zhang, Y.; Wei, K. Plasma miR-513a-5p as a potential biomarker for diagnosis of nonalcoholic fatty liver disease in Type 2 diabetes mellitus patients. Clin. Lab. 2020, 66. [Google Scholar] [CrossRef]
- Doumatey, A.P.; Xu, H.; Huang, H.; Trivedi, N.S.; Lei, L.; Elkahloun, A.; Adeyemo, A.; Rotimi, C.N. Global gene expression profiling in omental adipose tissue of morbidly obese diabetic African Americans. J. Endocrinol. Metab. 2015, 5, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Boyle, A.P. Predicting functional variants in enhancer and promoter elements using RegulomeDB. Hum. Mutat. 2019, 40, 1292–1298. [Google Scholar] [CrossRef] [Green Version]
- Wan, S.; van der Veen, J.N.; Bakala N’Goma, J.C.; Nelson, R.C.; Vance, D.E.; Jacobs, R.L. Hepatic PEMT activity mediates liver health, weight gain, and insulin resistance. FASEB J. 2019, 33, 10986–10995. [Google Scholar] [CrossRef] [Green Version]
- Rahardini, E.P.; Ikeda, K.; Nugroho, D.B.; Hirata, K.I.; Emoto, N. Loss of Family with Sequence Similarity 13, Member a Exacerbates Pulmonary Fibrosis Potentially by Promoting Epithelial to Mesenchymal Transition. Kobe J. Med. Sci. 2020, 65, E100–E109. [Google Scholar]
- Pineault, K.M.; Song, J.Y.; Kozloff, K.M.; Lucas, D.; Wellik, D.M. Hox11 expressing regional skeletal stem cells are progenitors for osteoblasts, chondrocytes and adipocytes throughout life. Nat. Commun. 2019, 10, 3168. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Miao, Y.; Xie, J.; Lin, Y.; Yao, Q.; Cai, J.; Yang, X. Long noncoding RNA lysophospholipase-like 1–2 as ceRNA modulates glioma metastasis by regulating miR-217/YWHAG. Am. J. Transl. Res. 2020, 12, 4204–4215. [Google Scholar] [PubMed]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; Abecasis, G.R. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machiela, M.J.; Chanock, S.J. LDlink: A web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 2015, 31, 3555–3557. [Google Scholar] [CrossRef]
- Andersson, R.; Sandelin, A. Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet. 2020, 21, 71–87. [Google Scholar] [CrossRef]
- Zhang, X.; Deng, H.W.; Shen, H.; Ehrlich, M. Prioritization of osteoporosis-associated Genome-wide Association Study (GWAS) single-nucleotide polymorphisms (SNPs) using epigenomics and transcriptomics. JBMR Plus 2021, 5, e10481. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Tier-1 SNP for Obesity Association | Index/Proxy (I/P) or Imputed (Imp) SNP | Alleles (Ref or Alt) | Obesity Increasing Allele (Freq EUR) b | Chromatin State at the SNP in SAT c | Gene Location of SNP Relative to Gene | Predicted TF Binding d | Type of Additional Statistical Analysis That Predicts Assocn. e | |
|---|---|---|---|---|---|---|---|---|---|
| Ref Allele | Alt Allele | ||||||||
| CABLES1 | rs4445996 | Imp | A/G | G (0.91) | Str enh | 5′ to TSS 1 | DNTTIP1 | MAX | None |
| “ | rs9966093 | Imp | C/T | T (0.91) | Str enh | 5′ to TSS 1 | ZXDA, ZXDB | RXRA | None |
| “ | rs4308051 *,† | I/P & Imp | T/G | G (0.79) | Str enh | Near TSS 2 | BHLHE40/41 | KLF4 | None |
| “ | rs28589524 *,† | I/P | G/A | NA (0.79) | Str enh | Near TSS 2 | NFKB1, RELA | SMAD2 | None |
| FAM13A | rs10012045 *,† | I/P | C/T | NA (0.56) | Str enh | Intron 2 f | EPAS1 | None | None |
| “ | rs2869949 *,† | I/P | A/G | NA (0.51) | Str enh | Intron 1 | ZNF333 | None | SMR&HEIDI |
| “ | rs11097198 *,† | I/P | C/T | NA (0.51) | Str enh | Intron 1 | STAT1/3/4/5B | None | SMR&HEIDI |
| “ | rs6532080 *,† | I/P | A/C | NA (0.56) | Str enh | Intron 1 | SOX4 | None | None |
| “ | rs13133548 *,† | I/P & Imp | G/A | A (0.48) | Str enh/prom | Intron 1 | None | POU3F3 | PAINTOR; SMR&HEIDI |
| “ | rs7660000 | I/P & Imp | C/T | C (0.70) | Str enh | 5′ to TSS 1 | ZBTB44 | None | None |
| HOXA11 | rs530375207 | I/P | T/A | NA (0.91) | Str enh | 3′-UTR | ZNF254 | PRRX2, DLX5 | None |
| PC | rs17147932 | I/P & Imp | G/T | T (0.05) | Str enh | Near TSS 2 | KLF2/6, SP2/3 | None | None |
| PEMT | rs8078513 * | I/P & Imp | C/A | A (0.07) | Str enh | End of intron 2 | SMAD3 | None | None |
| “ | rs8070432 * | Imp | T/C | C (0.07) | Str enh | Near TSS 3 | None | RUNX1 | None |
| RP11-392O17.1 | rs7555150 *,† | I/P & Imp | A/G | G (0.61) | Str enh/prom | Near prom | None | NFIB | None |
| “ | rs12025363 *,† | I/P & Imp | G/A | A (0.61) | Str enh | Prom region | GTF2IRD1 | None | None |
| ZBED3-AS1 | rs7732130 | I/P & Imp | G/A | G (0.33) | Str enh | Near 3′ end | RFX7 | None | None |
| “ | rs9293708† | I/P & Imp | C/T | C (0.43) | Str enh | Near 3′ end | SOX18 | None | PAINTOR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Li, T.-Y.; Xiao, H.-M.; Ehrlich, K.C.; Shen, H.; Deng, H.-W.; Ehrlich, M. Epigenomic and Transcriptomic Prioritization of Candidate Obesity-Risk Regulatory GWAS SNPs. Int. J. Mol. Sci. 2022, 23, 1271. https://doi.org/10.3390/ijms23031271
Zhang X, Li T-Y, Xiao H-M, Ehrlich KC, Shen H, Deng H-W, Ehrlich M. Epigenomic and Transcriptomic Prioritization of Candidate Obesity-Risk Regulatory GWAS SNPs. International Journal of Molecular Sciences. 2022; 23(3):1271. https://doi.org/10.3390/ijms23031271
Chicago/Turabian StyleZhang, Xiao, Tian-Ying Li, Hong-Mei Xiao, Kenneth C. Ehrlich, Hui Shen, Hong-Wen Deng, and Melanie Ehrlich. 2022. "Epigenomic and Transcriptomic Prioritization of Candidate Obesity-Risk Regulatory GWAS SNPs" International Journal of Molecular Sciences 23, no. 3: 1271. https://doi.org/10.3390/ijms23031271
APA StyleZhang, X., Li, T.-Y., Xiao, H.-M., Ehrlich, K. C., Shen, H., Deng, H.-W., & Ehrlich, M. (2022). Epigenomic and Transcriptomic Prioritization of Candidate Obesity-Risk Regulatory GWAS SNPs. International Journal of Molecular Sciences, 23(3), 1271. https://doi.org/10.3390/ijms23031271