
Unravelling Mechanisms of Doxorubicin-Induced Toxicity in 3D Human Intestinal Organoids

 ,
,  and
and
Abstract
:1. Introduction
2. Results
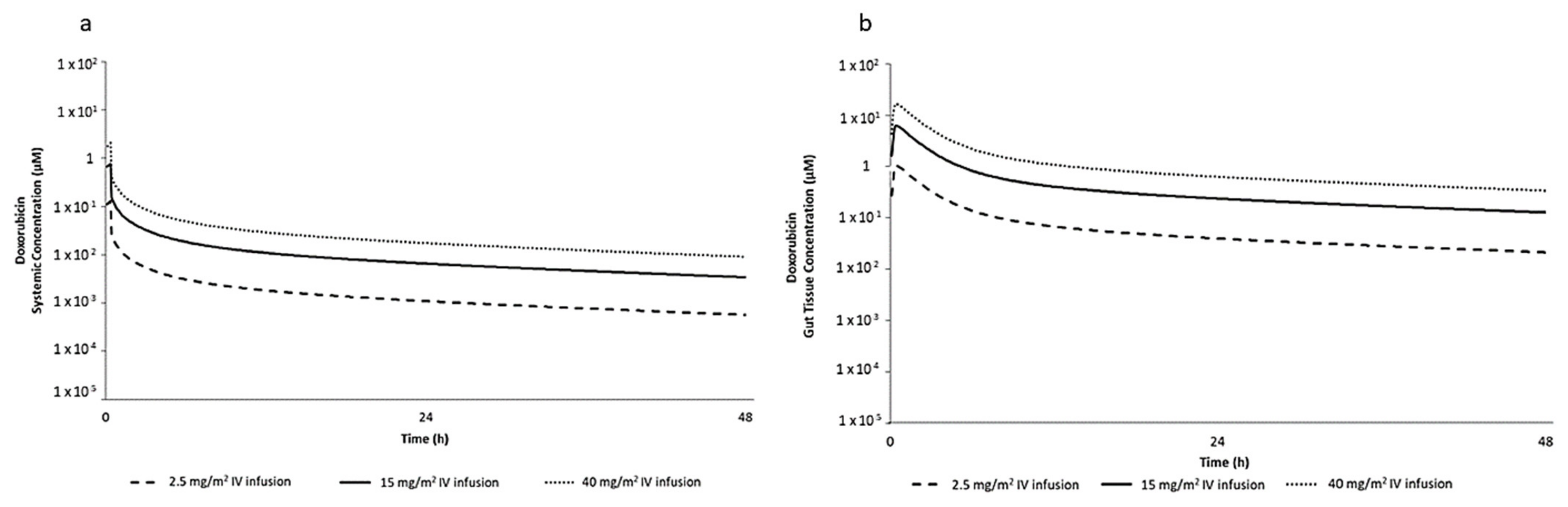
2.1. PBPK Simulation for Selection of DOX In Vitro Concentrations
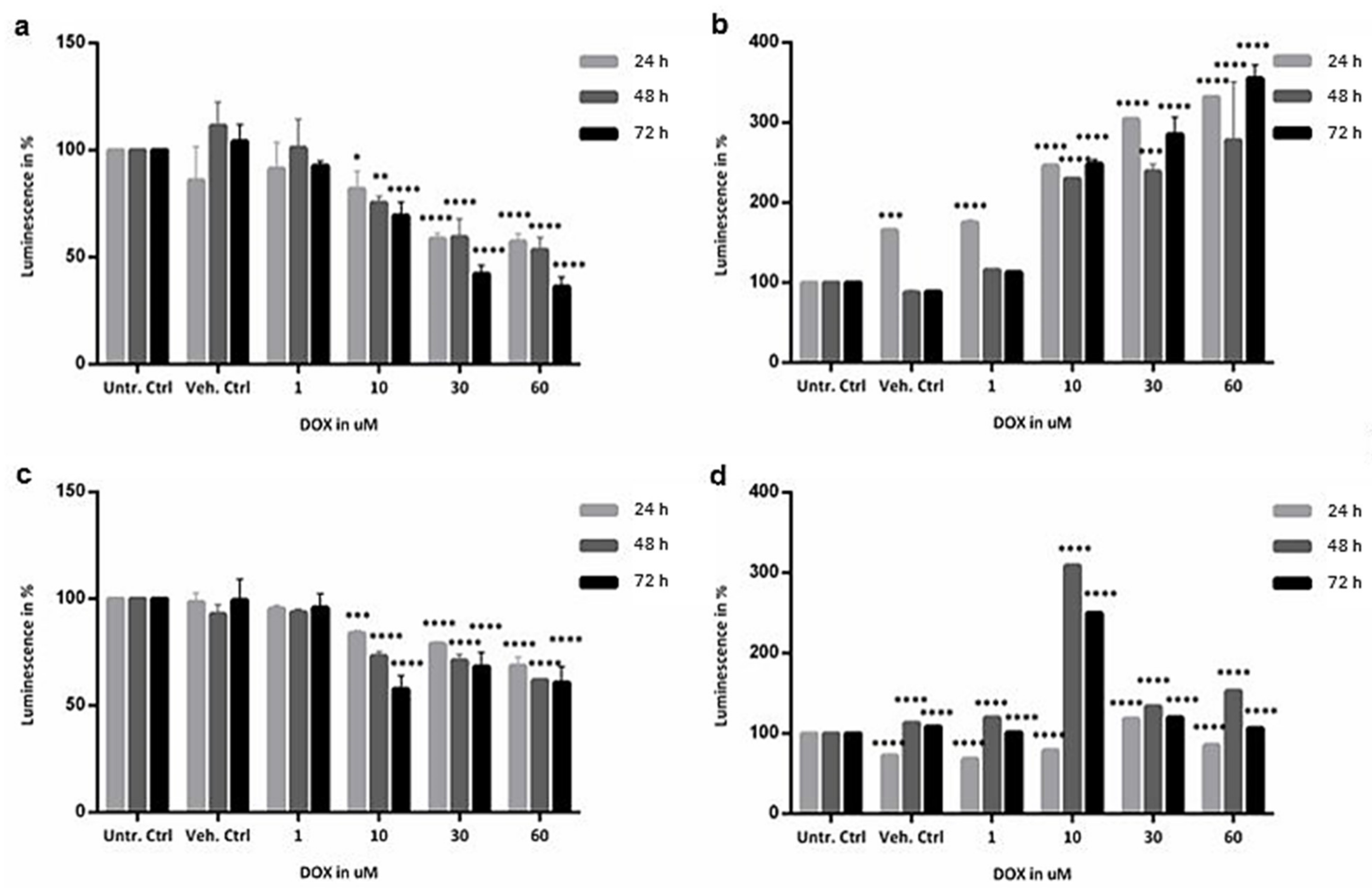
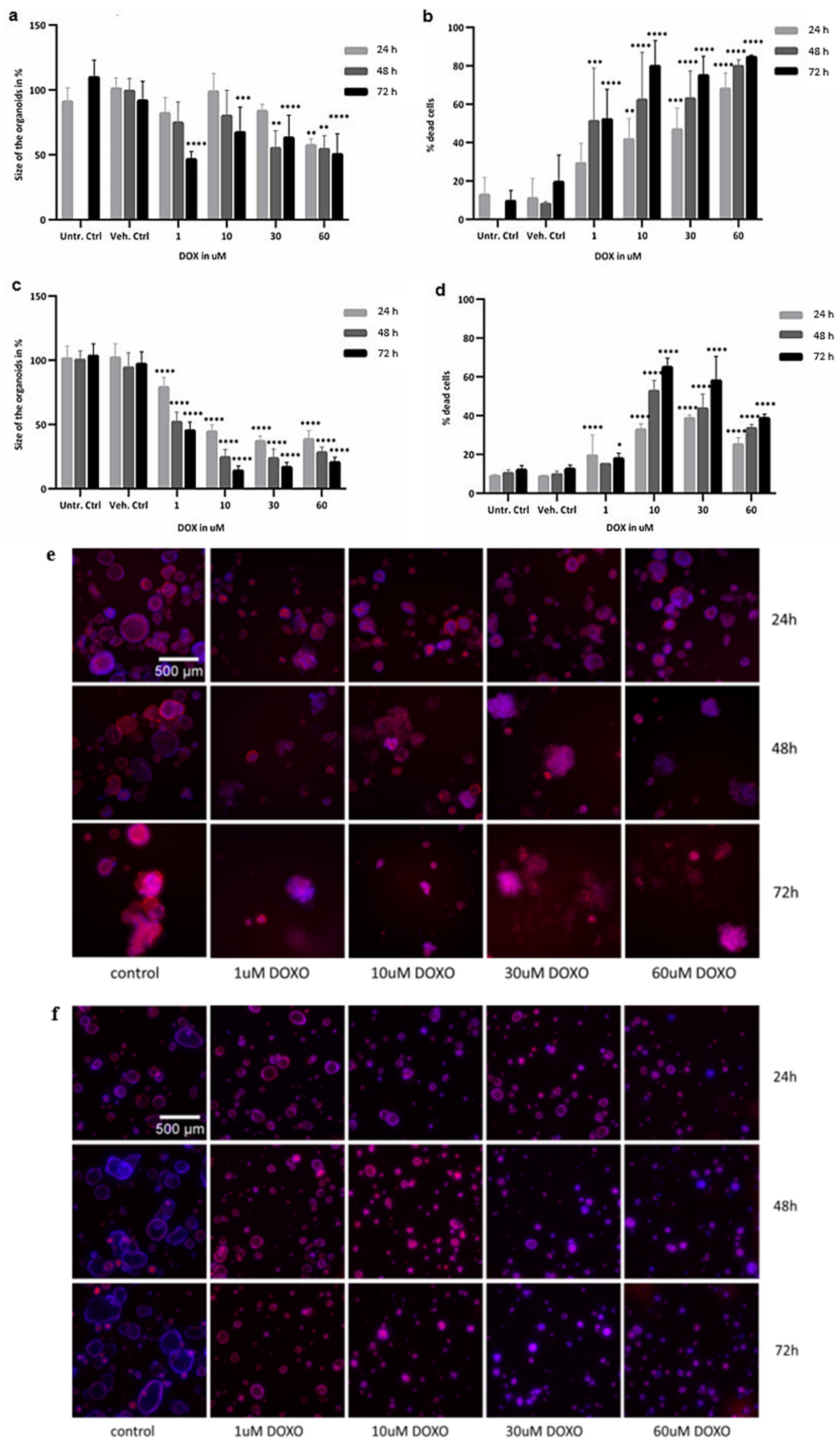
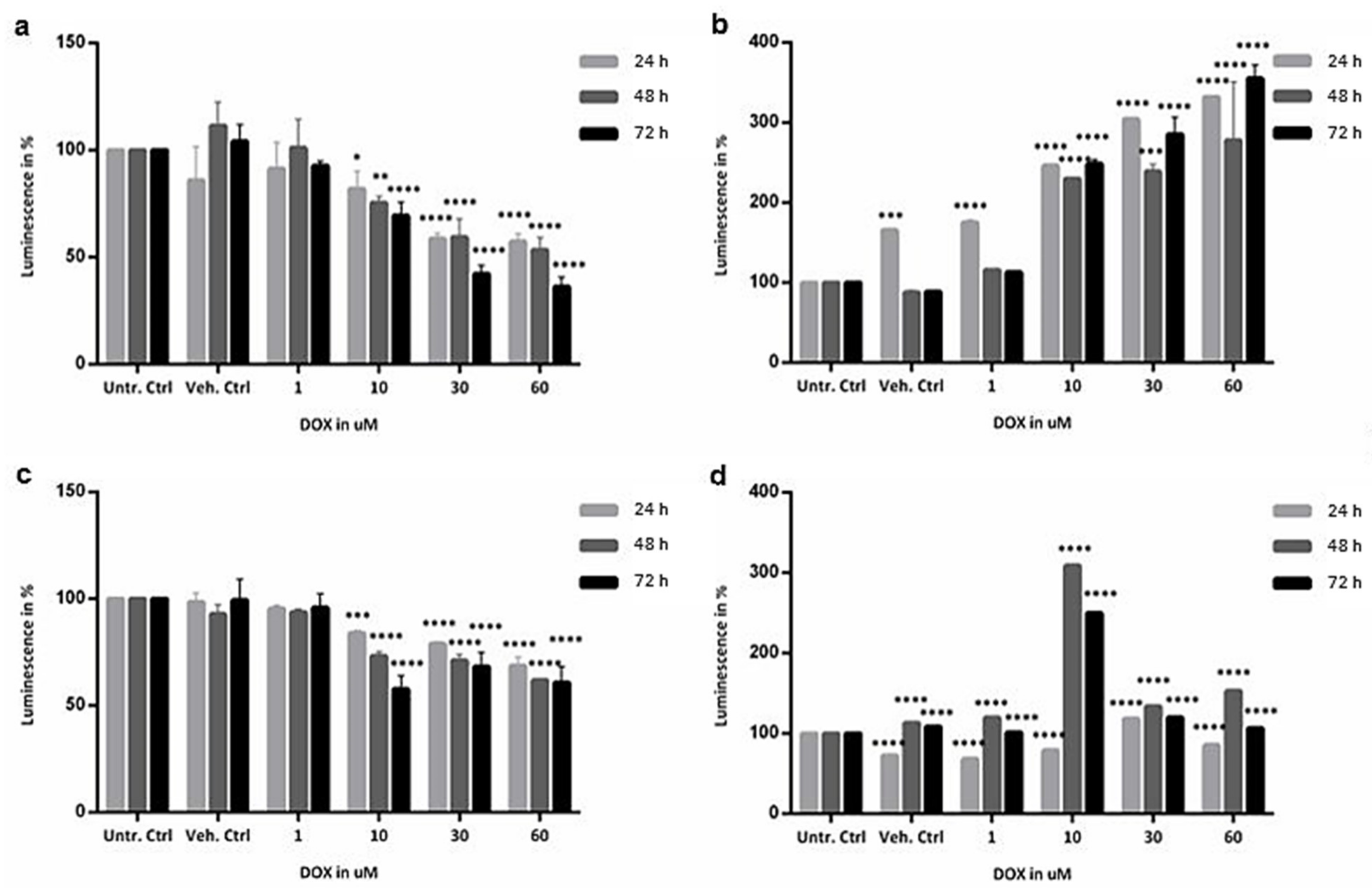
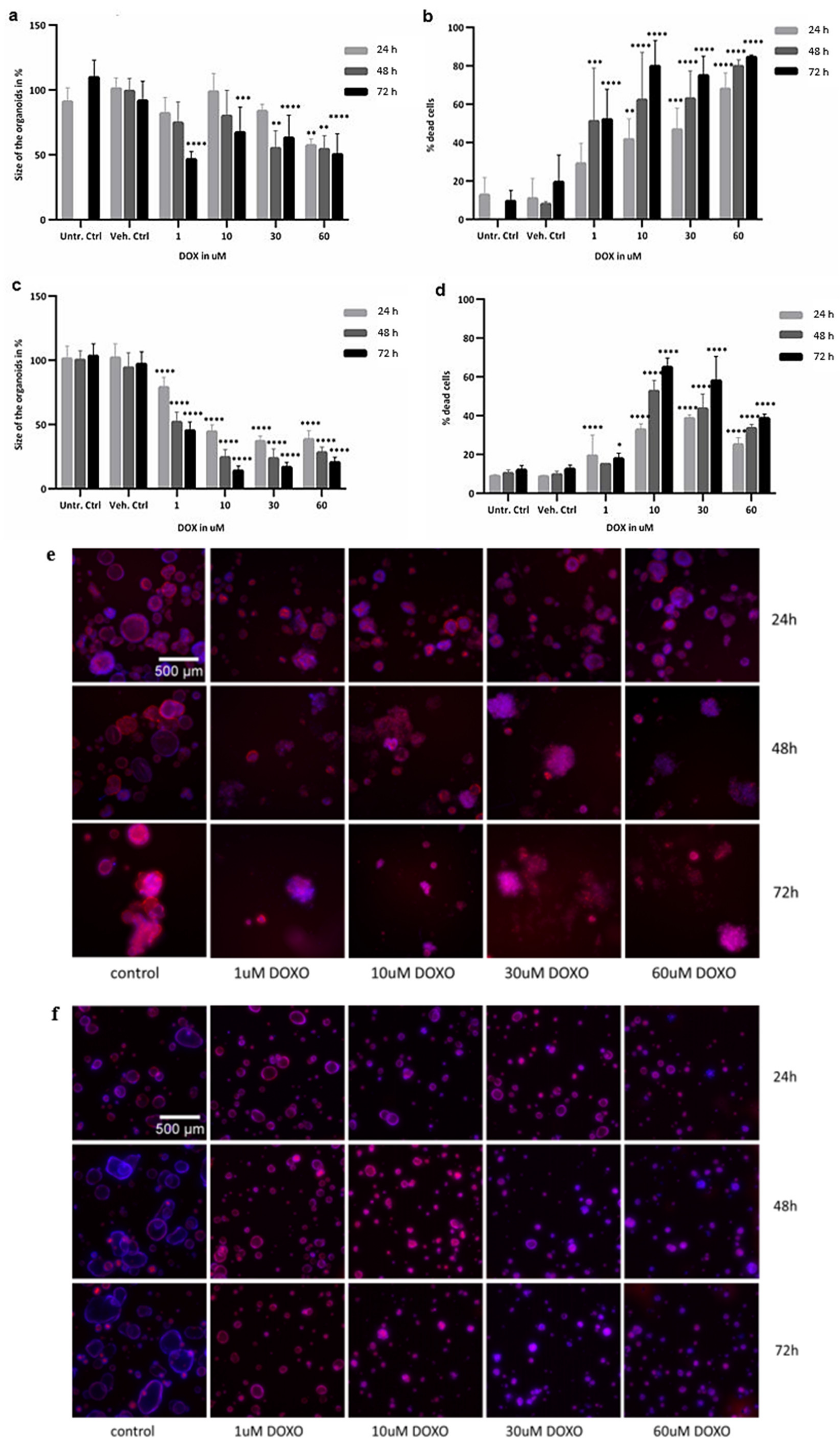
2.2. Cytotoxicity Evaluation of Colon and SI Organoids: Viability and Apoptosis after Exposure to DOX
2.3. Image Analysis
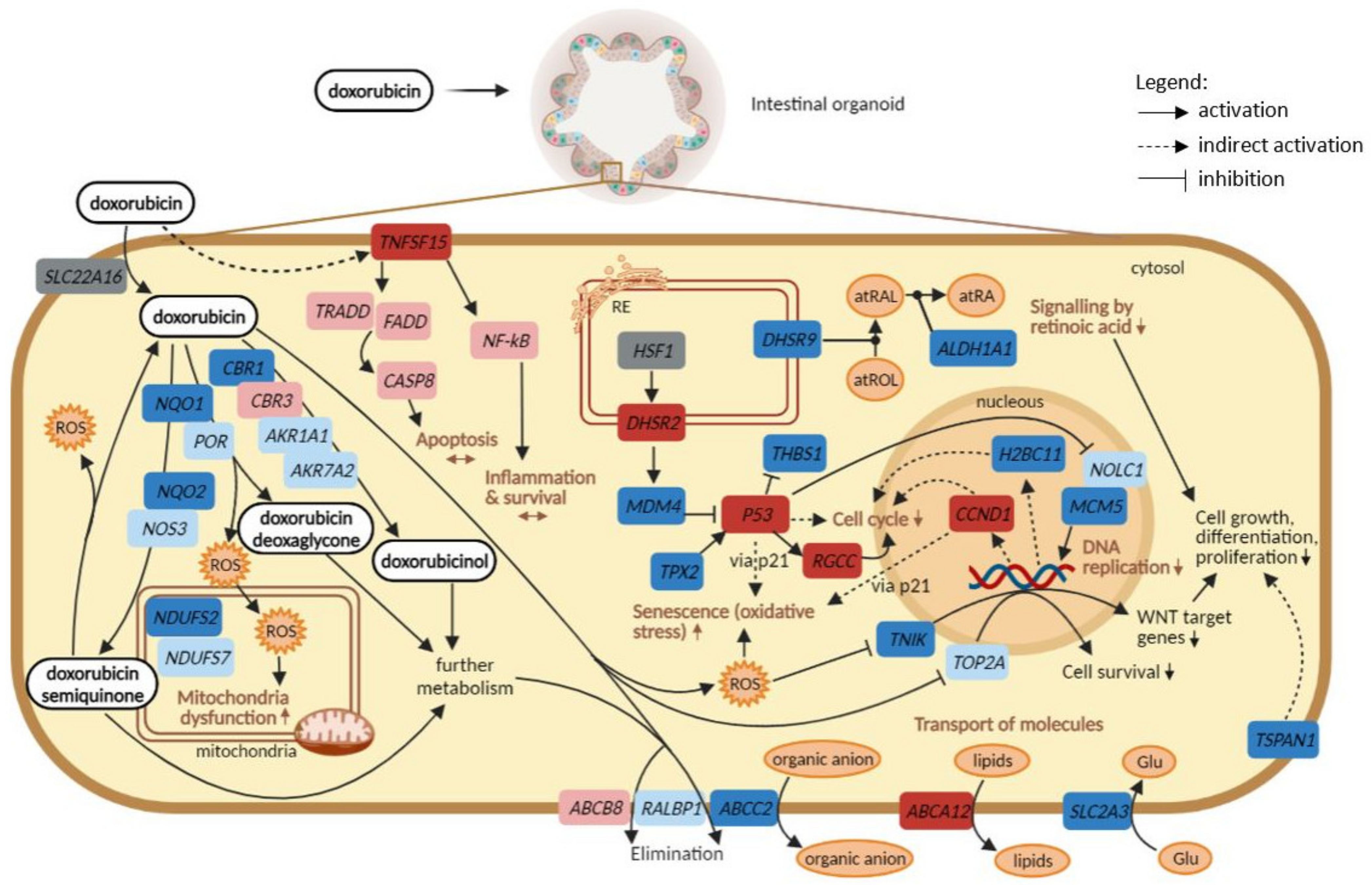
2.4. Identification of Biological Pathways and Gene Responses Affected by DOX
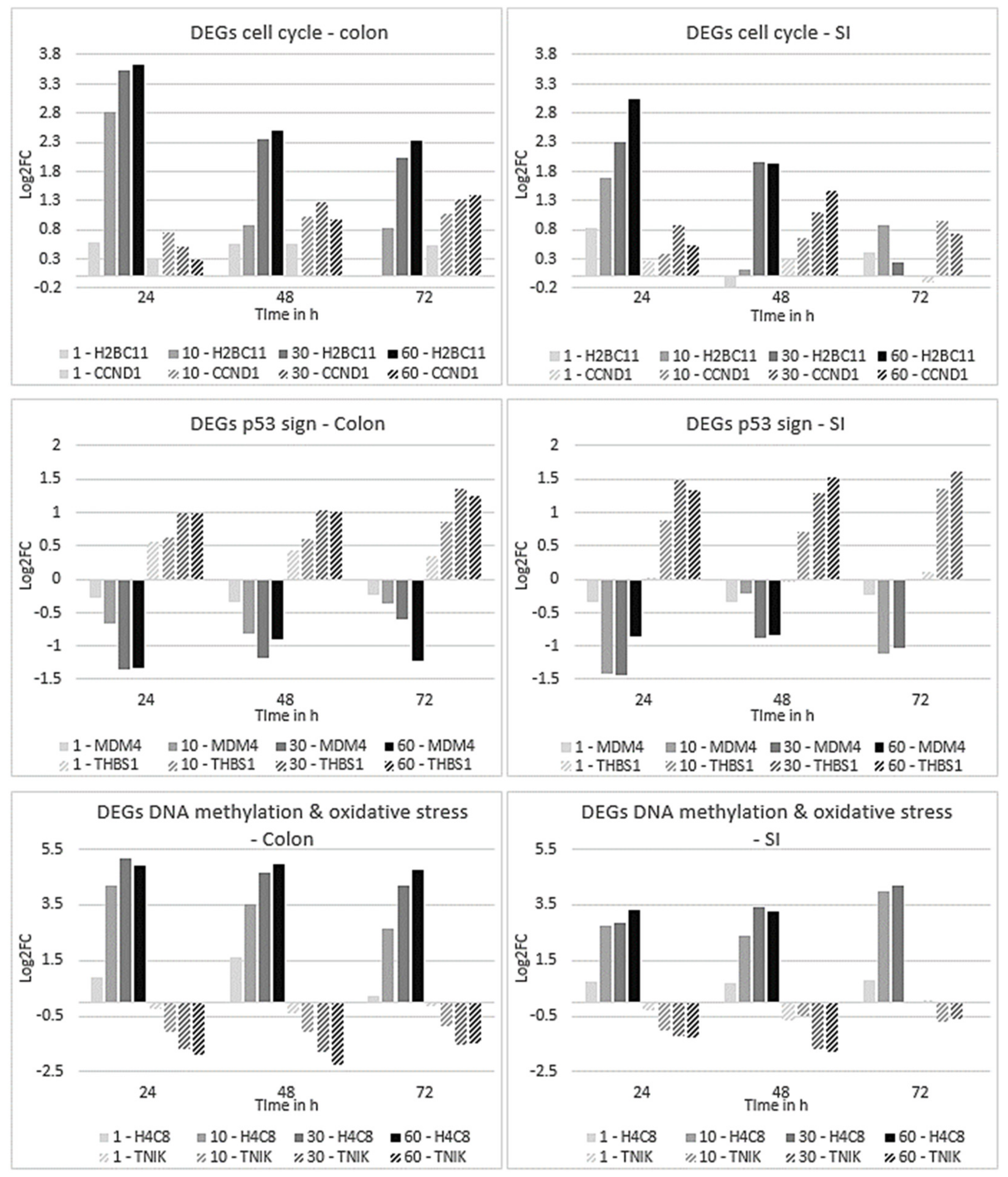
2.4.1. Pathway Analysis across Time and Concentration in Colon and SI Organoids
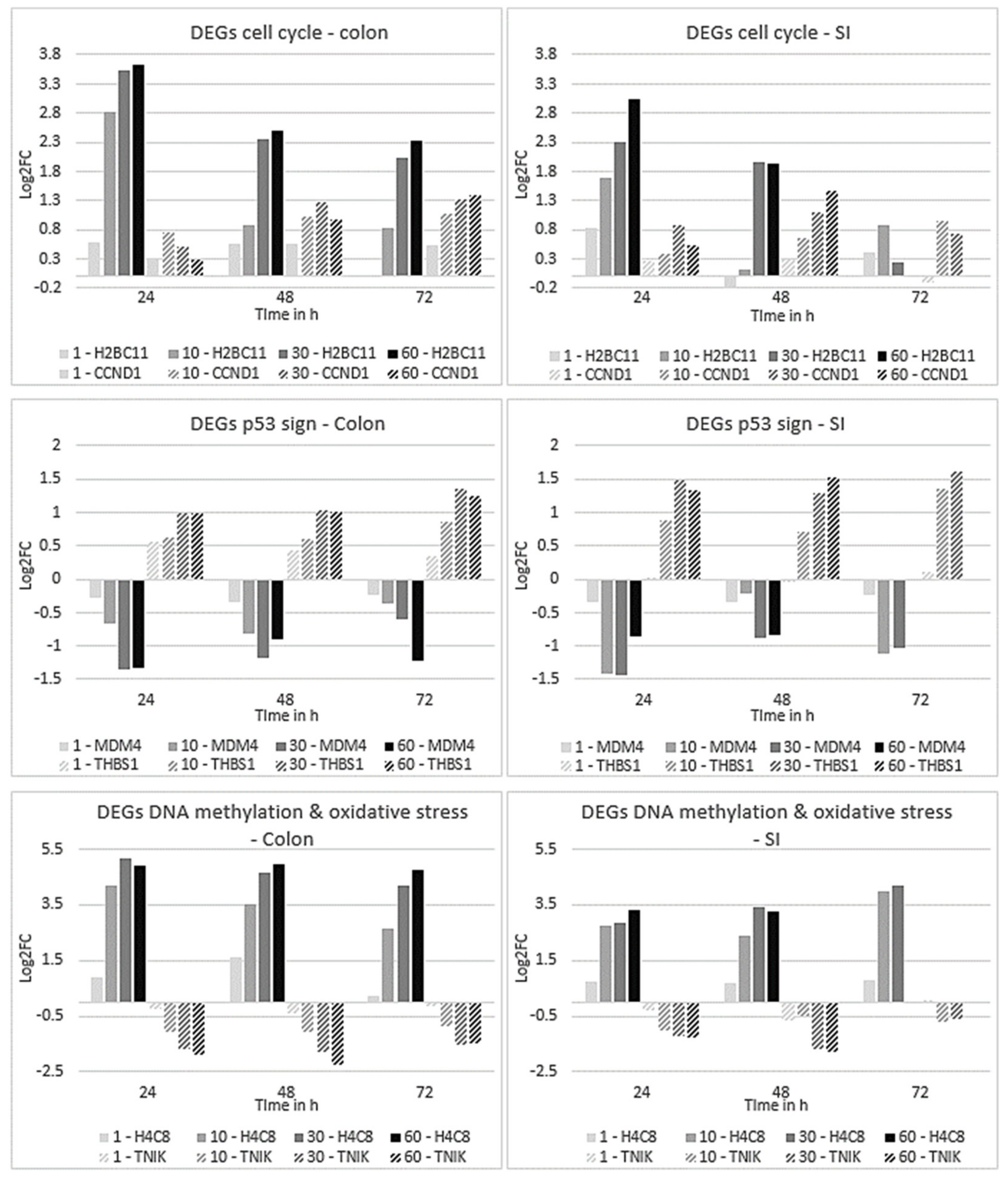
2.4.2. Expression Profiles of DEGs Affected in Colon and SI Organoids
2.5. Time-Dependent Gene Clustering Analysis
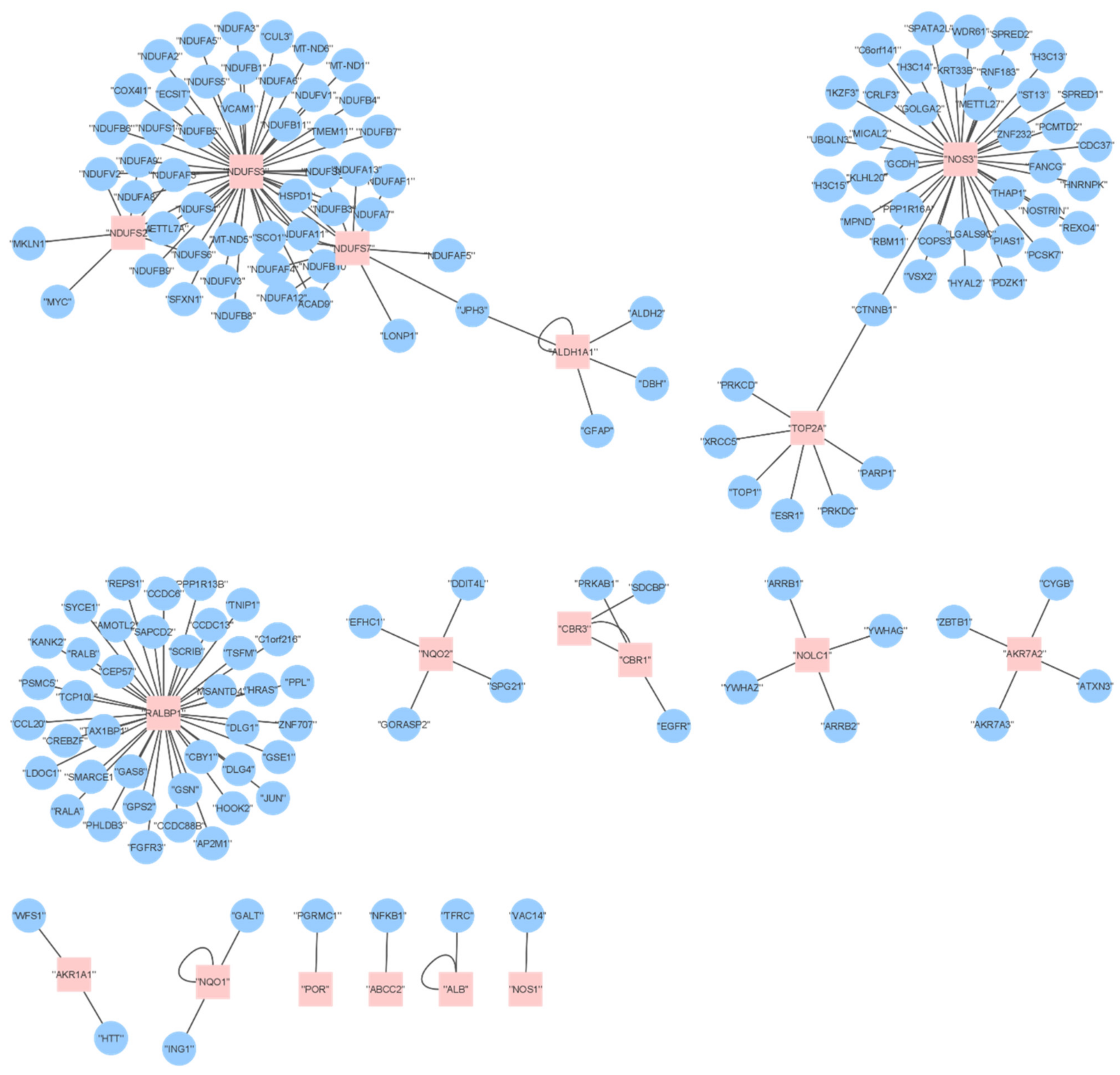
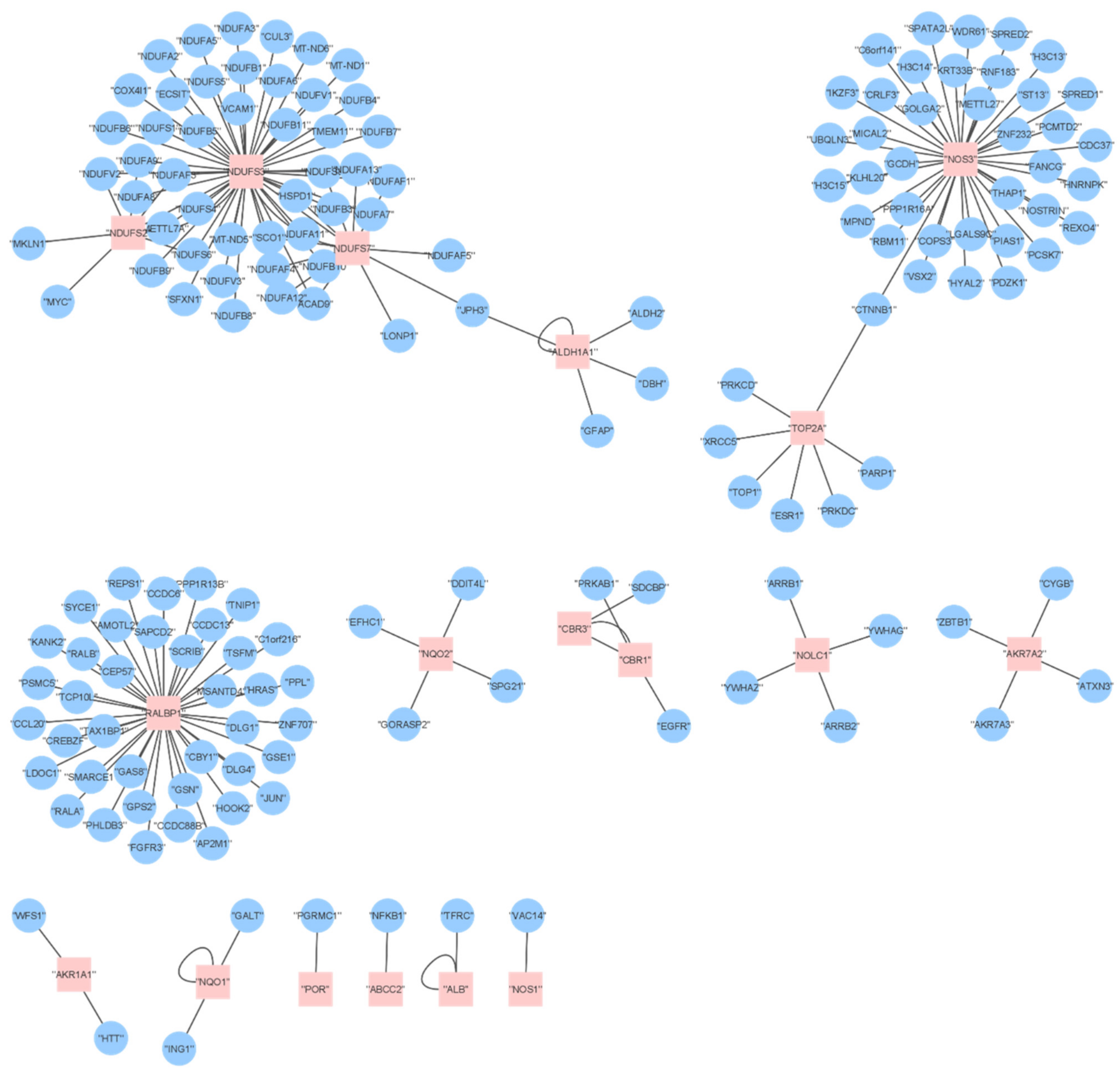
2.6. Proteome Analysis
2.7. Comparing DOX Effects on Transcriptomics and Proteomics
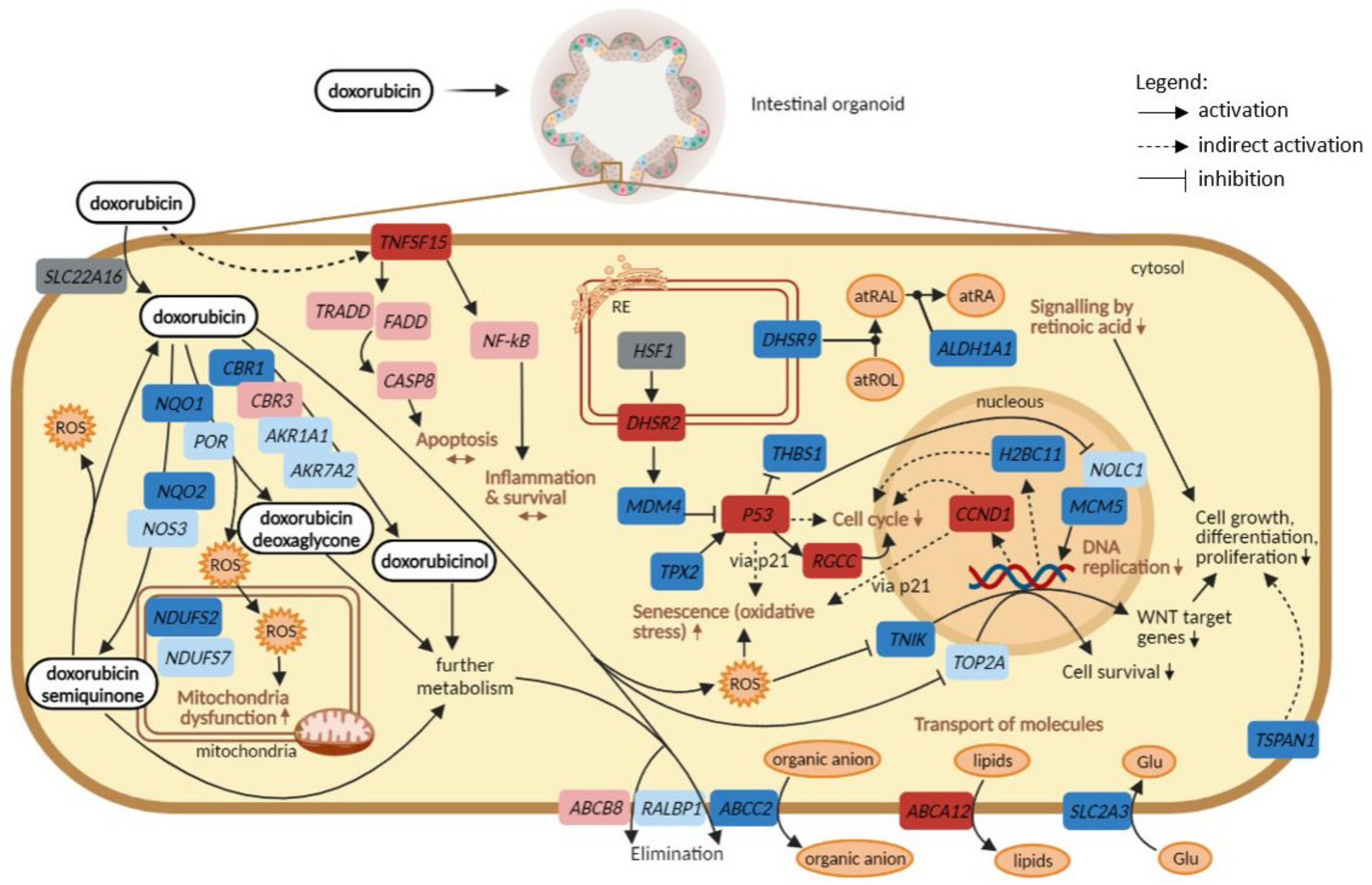
3. Discussion
4. Materials and Methods
4.1. In Vitro Culture of Healthy Intestinal Organoids
4.2. Selection of DOX In Vitro Concentrations Based on PBPK Simulation
4.3. In Vitro Exposure to DOX
4.4. Cytotoxicity Assays: ATP Measurement and Caspase 3/7 Activity
4.5. Image Analysis
4.6. RNA Isolation from Intestinal Organoids
4.7. Library Preparation and mRNA Sequencing
4.8. Pre-Processing and Data Analysis
4.9. Proteome Analysis
4.10. Pathway Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arcamone, F.; Cassinelli, G.; Fantini, G.; Grein, A.; Orezzi, P.; Pol, C.; Spalla, C. Adriamycin, 14-hydroxydaunomycin, a new antitumor antibiotic from S. peucetius var. caesius. Biotechnol. Bioeng. 1969, 11, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.; Santos, R.X.; Cardoso, S.; Correia, S.; Oliveira, P.J.; Santos, M.S.; Moreira, P.I. Doxorubicin: The good, the bad and the ugly effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef] [PubMed]
- Pugazhendhi, A.; Edison, T.; Velmurugan, B.K.; Jacob, J.A.; Karuppusamy, I. Toxicity of Doxorubicin (Dox) to different experimental organ systems. Life Sci. 2018, 200, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharmacogenet. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef]
- Damiani, R.M.; Moura, D.J.; Viau, C.M.; Caceres, R.A.; Henriques, J.A.P.; Saffi, J. Pathways of cardiac toxicity: Comparison between chemotherapeutic drugs doxorubicin and mitoxantrone. Arch. Toxicol. 2016, 90, 2063–2076. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Teves, S.S.; Kemp, C.J.; Henikoff, S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim. Biophys. Acta 2014, 1845, 84–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthiaume, J.M.; Wallace, K.B. Adriamycin-induced oxidative mitochondrial cardiotoxicity. Cell Biol. Toxicol. 2007, 23, 15–25. [Google Scholar] [CrossRef]
- Edwardson, D.W.; Narendrula, R.; Chewchuk, S.; Mispel-Beyer, K.; Mapletoft, J.P.; Parissenti, A.M. Role of Drug Metabolism in the Cytotoxicity and Clinical Efficacy of Anthracyclines. Curr. Drug Metab. 2015, 16, 412–426. [Google Scholar] [CrossRef] [Green Version]
- Kaczmarek, A.; Brinkman, B.M.; Heyndrickx, L.; Vandenabeele, P.; Krysko, D.V. Severity of doxorubicin-induced small intestinal mucositis is regulated by the TLR-2 and TLR-9 pathways. J. Pathol. 2012, 226, 598–608. [Google Scholar] [CrossRef]
- Rodrigues, D.; de Souza, T.; Coyle, L.; Di Piazza, M.; Herpers, B.; Ferreira, S.; Zhang, M.; Vappiani, J.; Sevin, D.C.; Gabor, A.; et al. New insights into the mechanisms underlying 5-fluorouracil-induced intestinal toxicity based on transcriptomic and metabolomic responses in human intestinal organoids. Arch. Toxicol. 2021, 95, 2691–2718. [Google Scholar] [CrossRef]
- Chaicharoenaudomrung, N.; Kunhorm, P.; Noisa, P. Three-dimensional cell culture systems as an in vitro platform for cancer and stem cell modeling. World J. Stem Cells 2019, 11, 1065–1083. [Google Scholar] [CrossRef]
- Nii, T.; Makino, K.; Tabata, Y. Three-Dimensional Culture System of Cancer Cells Combined with Biomaterials for Drug Screening. Cancers 2020, 12, 2754. [Google Scholar] [CrossRef]
- Benton, G.; Arnaoutova, I.; George, J.; Kleinman, H.K.; Koblinski, J. Matrigel: From discovery and ECM mimicry to assays and models for cancer research. Adv. Drug Deliv. Rev. 2014, 79, 3–18. [Google Scholar] [CrossRef]
- Barros, A.S.; Costa, E.C.; Nunes, A.S.; de Melo-Diogo, D.; Correia, I.J. Comparative study of the therapeutic effect of Doxorubicin and Resveratrol combination on 2D and 3D (spheroids) cell culture models. Int. J. Pharm. 2018, 551, 76–83. [Google Scholar] [CrossRef]
- Senkowski, W.; Jarvius, M.; Rubin, J.; Lengqvist, J.; Gustafsson, M.G.; Nygren, P.; Kultima, K.; Larsson, R.; Fryknas, M. Large-Scale Gene Expression Profiling Platform for Identification of Context-Dependent Drug Responses in Multicellular Tumor Spheroids. Cell Chem. Biol. 2016, 23, 1428–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, A.S.; Costa, E.C.; Barros, A.S.; de Melo-Diogo, D.; Correia, I.J. Establishment of 2D Cell Cultures Derived From 3D MCF-7 Spheroids Displaying a Doxorubicin Resistant Profile. Biotechnol. J. 2019, 14, e1800268. [Google Scholar] [CrossRef]
- Devarasetty, M.; Skardal, A.; Cowdrick, K.; Marini, F.; Soker, S. Bioengineered Submucosal Organoids for In Vitro Modeling of Colorectal Cancer. Tissue Eng. Part A 2017, 23, 1026–1041. [Google Scholar] [CrossRef]
- Darling, N.J.; Mobbs, C.L.; Gonzalez-Hau, A.L.; Freer, M.; Przyborski, S. Bioengineering Novel in vitro Co-culture Models That Represent the Human Intestinal Mucosa With Improved Caco-2 Structure and Barrier Function. Front. Bioeng. Biotechnol. 2020, 8, 992. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Zachos, N.C.; Kovbasnjuk, O.; Foulke-Abel, J.; In, J.; Blutt, S.E.; de Jonge, H.R.; Estes, M.K.; Donowitz, M. Human Enteroids/Colonoids and Intestinal Organoids Functionally Recapitulate Normal Intestinal Physiology and Pathophysiology. J. Biol. Chem. 2016, 291, 3759–3766. [Google Scholar] [CrossRef] [Green Version]
- Perez-Blanco, J.S.; Santos-Buelga, D.; de Gatta, M.D.F.; Hernandez-Rivas, J.M.; Martin, A.; Garcia, M.J. Population pharmacokinetics of doxorubicin and doxorubicinol in patients diagnosed with non-Hodgkin’s lymphoma. Br. J. Clin. Pharmacol. 2016, 82, 1517–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanke, N.; Teifel, M.; Moj, D.; Wojtyniak, J.G.; Britz, H.; Aicher, B.; Sindermann, H.; Ammer, N.; Lehr, T. A physiologically based pharmacokinetic (PBPK) parent-metabolite model of the chemotherapeutic zoptarelin doxorubicin-integration of in vitro results, Phase I and Phase II data and model application for drug-drug interaction potential analysis. Cancer Chemother. Pharm. 2018, 81, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Fuzi, B.; Gurinova, J.; Hermjakob, H.; Ecker, G.F.; Sheriff, R. Path4Drug: Data Science Workflow for Identification of Tissue-Specific Biological Pathways Modulated by Toxic Drugs. Front. Pharmacol. 2021, 12, 708296. [Google Scholar] [CrossRef] [PubMed]
- Whirl-Carrillo, M.; McDonagh, E.M.; Hebert, J.M.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Altman, R.B.; Klein, T.E. Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 2012, 92, 414–417. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Felix, E.; Magarinos, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, S.; Li, F.; Zhou, Y.; Zhang, Y.; Wang, Z.; Zhang, R.; Zhu, J.; Ren, Y.; Tan, Y.; et al. Therapeutic target database 2020: Enriched resource for facilitating research and early development of targeted therapeutics. Nucleic Acids Res. 2020, 48, D1031–D1041. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Southan, C.; Sharman, J.L.; Campo, B.; Cavanagh, D.R.; Alexander, S.P.H.; Davenport, A.P.; et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2020: Extending immunopharmacology content and introducing the IUPHAR/MMV Guide to MALARIA PHARMACOLOGY. Nucleic Acids Res. 2020, 48, D1006–D1021. [Google Scholar] [CrossRef]
- Illumina Considerations for RNA-Seq Read Length and Coverage. Available online: https://support.illumina.com/bulletins/2017/04/considerations-for-rna-seq-read-length-and-coverage-.html (accessed on 10 January 2022).
- Robles, J.A.; Qureshi, S.E.; Stephen, S.J.; Wilson, S.R.; Burden, C.J.; Taylor, J.M. Efficient experimental design and analysis strategies for the detection of differential expression using RNA-Sequencing. BMC Genom. 2012, 13, 484. [Google Scholar] [CrossRef] [Green Version]
- Sundaram, P.; Hultine, S.; Smith, L.M.; Dews, M.; Fox, J.L.; Biyashev, D.; Schelter, J.M.; Huang, Q.; Cleary, M.A.; Volpert, O.V.; et al. p53-responsive miR-194 inhibits thrombospondin-1 and promotes angiogenesis in colon cancers. Cancer Res. 2011, 71, 7490–7501. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.; Truong, D.; Shangari, N.; O’Brien, P.J. Drug-induced mitochondrial toxicity. Expert Opin Drug Metab. Toxicol. 2005, 1, 655–669. [Google Scholar] [CrossRef]
- Mourelle, M.; Casellas, F.; Guarner, F.; Salas, A.; Riveros-Moreno, V.; Moncada, S.; Malagelada, J.R. Induction of nitric oxide synthase in colonic smooth muscle from patients with toxic megacolon. Gastroenterology 1995, 109, 1497–1502. [Google Scholar] [CrossRef]
- Fan, L.; Goh, B.C.; Wong, C.I.; Sukri, N.; Lim, S.E.; Tan, S.H.; Guo, J.Y.; Lim, R.; Yap, H.L.; Khoo, Y.M.; et al. Genotype of human carbonyl reductase CBR3 correlates with doxorubicin disposition and toxicity. Pharmacogenet. Genom. 2008, 18, 621–631. [Google Scholar] [CrossRef]
- Mross, K.; Maessen, P.; van der Vijgh, W.J.; Gall, H.; Boven, E.; Pinedo, H.M. Pharmacokinetics and metabolism of epidoxorubicin and doxorubicin in humans. J. Clin. Oncol. 1988, 6, 517–526. [Google Scholar] [CrossRef]
- Pritchard, M.D.; Potten, C.S.; Hitknian, J.A. The relationships between p53-dependent apoptosis, inhibition of proliferation, and 5-fluorouracil-induced histopathology in murine intestinal epithelia. Cancer Res. 1998, 58, 5453–5465. [Google Scholar]
- Chang, C.T.; Ho, T.Y.; Lin, H.; Liang, J.A.; Huang, H.C.; Li, C.C.; Lo, H.Y.; Wu, S.L.; Huang, Y.F.; Hsiang, C.Y. 5-Fluorouracil induced intestinal mucositis via nuclear factor-kappaB activation by transcriptomic analysis and in vivo bioluminescence imaging. PLoS ONE 2012, 7, e31808. [Google Scholar]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, D.; Pinto, J.; Araujo, A.M.; Jeronimo, C.; Henrique, R.; Bastos, M.L.; Guedes de Pinho, P.; Carvalho, M. GC-MS Metabolomics Reveals Distinct Profiles of Low- and High-Grade Bladder Cancer Cultured Cells. Metabolites 2019, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- Derave, W.; Everaert, I.; Beeckman, S.; Baguet, A. Muscle carnosine metabolism and beta-alanine supplementation in relation to exercise and training. Sports Med. 2010, 40, 247–263. [Google Scholar] [CrossRef] [Green Version]
- Holecek, M. Histidine in Health and Disease: Metabolism, Physiological Importance, and Use as a Supplement. Nutrients 2020, 12, 848. [Google Scholar] [CrossRef] [Green Version]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Lyon, P.; Strippoli, V.; Fang, B.; Cimmino, L. B Vitamins and One-Carbon Metabolism: Implications in Human Health and Disease. Nutrients 2020, 12, 2867. [Google Scholar] [CrossRef]
- Bartke, N.; Hannun, Y.A. Bioactive sphingolipids: Metabolism and function. J. Lipid Res. 2009, 50, S91–S96. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Hwang, J.T.; Lee, H.J.; Jung, S.N.; Kang, I.; Chi, S.G.; Kim, S.S.; Ha, J. AMP-activated protein kinase activity is critical for hypoxia-inducible factor-1 transcriptional activity and its target gene expression under hypoxic conditions in DU145 cells. J. Biol. Chem. 2003, 278, 39653–39661. [Google Scholar] [CrossRef] [Green Version]
- Roden, D.M.; Wilke, R.A.; Kroemer, H.K.; Stein, C.M. Pharmacogenomics: The genetics of variable drug responses. Circulation 2011, 123, 1661–1670. [Google Scholar] [CrossRef] [Green Version]
- Verma, M.; Khan, M.I.K.; Kadumuri, R.V.; Chakrapani, B.; Awasthi, S.; Mahesh, A.; Govindaraju, G.; Chavali, P.L.; Rajavelu, A.; Chavali, S.; et al. PRMT3 interacts with ALDH1A1 and regulates gene-expression by inhibiting retinoic acid signaling. Commun. Biol. 2021, 4, 109. [Google Scholar] [CrossRef]
- Yuan, F.W.; Zhang, Y.; Ma, L.W.; Cheng, Q.; Li, G.D.; Tong, T.J. Enhanced NOLC1 promotes cell senescence and represses hepatocellular carcinoma cell proliferation by disturbing the organization of nucleolus. Aging Cell 2017, 16, 726–737. [Google Scholar] [CrossRef]
- McGowan, E.M.; Tran, N.; Alling, N.; Yagoub, D.; Sedger, L.M.; Martiniello-Wilks, R. p14ARF post-transcriptional regulation of nuclear cyclin D1 in MCF-7 breast cancer cells: Discrimination between a good and bad prognosis? PLoS ONE 2012, 7, e42246. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, T.; Li, V.S.W.; Ng, S.S.; Taouatas, N.; Vries, R.G.J.; Mohammed, S.; Heck, A.J.; Clevers, H. The kinase TNIK is an essential activator of Wnt target genes. EMBO J. 2009, 28, 3329–3340. [Google Scholar] [CrossRef]
- Woodfield, S.E.; Shi, Y.; Patel, R.H.; Chen, Z.; Shah, A.P.; Whitlock, R.S.; Ibarra, A.M.; Larson, S.R.; Sarabia, S.F.; Badachhape, A.; et al. MDM4 inhibition: A novel therapeutic strategy to reactivate p53 in hepatoblastoma. Sci. Rep. 2021, 11, 2967. [Google Scholar] [CrossRef]
- Dewaele, M.; Tabaglio, T.; Willekens, K.; Bezzi, M.; Teo, S.X.; Low, D.H.; Koh, C.M.; Rambow, F.; Fiers, M.; Rogiers, A.; et al. Antisense oligonucleotide-mediated MDM4 exon 6 skipping impairs tumor growth. J. Clin. Investig. 2016, 126, 68–84. [Google Scholar] [CrossRef] [PubMed]
- Pascreau, G.; Eckerdt, F.; Lewellyn, A.L.; Prigent, C.; Maller, J.L. Phosphorylation of p53 Is Regulated by TPX2-Aurora A in Xenopus Oocytes. J. Biol. Chem. 2009, 284, 5497–5505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Counts, S.E.; Mufson, E.J. Regulator of Cell Cycle (RGCC) Expression During the Progression of Alzheimer’s Disease. Cell Transplant. 2017, 26, 693–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhinn, M.; Dolle, P. Retinoic acid signalling during development. Development 2012, 139, 843–858. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef]
- Holmgren, G.; Synnergren, J.; Bogestal, Y.; Ameen, C.; Akesson, K.; Holmgren, S.; Lindahl, A.; Sartipy, P. Identification of novel biomarkers for doxorubicin-induced toxicity in human cardiomyocytes derived from pluripotent stem cells. Toxicology 2015, 328, 102–111. [Google Scholar] [CrossRef]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J. Temporary importation of Doxorubicin Hydrochloride 50 mg Powder for Injection (50 mg/Vial) to Address Drug Shortage Issue; Hospira, Inc., Pfizer Company: New York, NY, USA, 2016. [Google Scholar]
- Fisher, C.; Simeon, S.; Jamei, M.; Gardner, I.; Bois, Y.F. VIVD: Virtual in vitro distribution model for the mechanistic prediction of intracellular concentrations of chemicals in in vitro toxicity assays. Toxicol In Vitro 2019, 58, 42–50. [Google Scholar] [CrossRef]
- Di, Z.; Klop, M.J.; Rogkoti, V.M.; Le Devedec, S.E.; van de Water, B.; Verbeek, F.J.; Price, L.S.; Meerman, J.H. Ultra high content image analysis and phenotype profiling of 3D cultured micro-tissues. PLoS ONE 2014, 9, e109688. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC. A Quality Control Tool for High Throughput Sequence Data. 2014. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 21 April 2021).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aickin, M.; Gensler, H. Adjusting for multiple testing when reporting research results: The Bonferroni vs Holm methods. Am. J. Public Health 1996, 86, 726–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Licata, L.; Briganti, L.; Peluso, D.; Perfetto, L.; Iannuccelli, M.; Galeota, E.; Sacco, F.; Palma, A.; Nardozza, A.P.; Santonico, E.; et al. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012, 40, D857–D861. [Google Scholar] [CrossRef] [PubMed]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; del-Toro, N.; et al. The MIntAct project--IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef] [Green Version]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Kamburov, A.; Stelzl, U.; Lehrach, H.; Herwig, R. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 2013, 41, D793–D800. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Du, J.; Yuan, Z.; Ma, Z.; Song, J.; Xie, X.; Chen, Y. KEGG-PATH: Kyoto encyclopedia of genes and genomes-based pathway analysis using a path analysis model. Mol. Biosyst. 2014, 10, 2441–2447. [Google Scholar] [CrossRef]
- Available online: https://bioinfogp.cnb.csic.es/tools/venny (accessed on 11 November 2021).
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef] [Green Version]
- Available online: BioRender.com (accessed on 23 April 2021).
- Lombardo, F.; Berellini, G.; Obach, R.S. Trend Analysis of a Database of Intravenous Pharmacokinetic Parameters in Humans for 1352 Drug Compounds. Drug Metab. Dispos. 2018, 46, 1466–1477. [Google Scholar] [CrossRef] [Green Version]
- Yee, S. In vitro permeability across Caco-2 cells (colonic) can predict in vivo (small intestinal) absorption in man—Fact or myth. Pharm. Res. 1997, 14, 763–766. [Google Scholar] [CrossRef]
- Rodgers, T.; Leahy, D.; Rowland, M. Physiologically based pharmacokinetic modeling 1: Predicting the tissue distribution of moderate-to-strong bases. J. Pharm. Sci. 2005, 94, 1259–1276. [Google Scholar] [CrossRef]
- Rodgers, T.; Rowland, M. Physiologically based pharmacokinetic modelling 2: Predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 2006, 95, 1238–1257. [Google Scholar] [CrossRef]
- Speth, P.A.; van Hoesel, Q.G.; Haanen, C. Clinical pharmacokinetics of doxorubicin. Clin. Pharmacokinet. 1988, 15, 15–31. [Google Scholar] [CrossRef]
- Meylan, W.M.; Howard, P. User’s Guide for HENRYWIN Version 3.10: Henry’s Law Constant Program; Syracuse Research Corporation: Syracuse, NY, USA, 2000. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IV Dose Infused Over 20 min in Human (mg/m2) | In Vivo Gut Tissue Total Cmax (µM) | In Vitro Nominal Concentration (µM) |
|---|---|---|
| 2.50 | 1.06 | 0.96 |
| 15.00 | 6.36 | 5.76 |
| 40.00 | 17.00 | 15.40 |
| Name of the Pathway | Pathway Source | Time of Exposure (h) | DOX Conc. (µM) | q Value | |
|---|---|---|---|---|---|
| Colon | SI | ||||
| Cell Cycle | Reactome | 24 | 1 | 2.74 × 10−20 | NA |
| 10 | 1.64 × 10–13 | 3.04 × 10–4 | |||
| 30 | 2.57 × 10–8 | 0.14 | |||
| 60 | 2.04 × 10–6 | NA | |||
| 48 | 1 | NA | NA | ||
| 10 | 4.19 × 10–3 | 9.96 × 10–3 | |||
| 30 | 1.08 × 10–3 | 0.16 | |||
| 60 | NA | 0.06 | |||
| 72 | 1 | NA | NA | ||
| 10 | 0.01 | 0.06 | |||
| 30 | 0.04 | 0.24 | |||
| 60 | 0.09 | NA | |||
| Cell cycle—DNA repair | Reactome | 24 | 1 | 0.027 | NA |
| 10 | 2.81 × 10–8 | 2.16 × 10–3 | |||
| 30 | 1.04 × 10–3 | NA | |||
| 60 | 5.19 × 10–3 | NA | |||
| 48 | 1 | NA | NA | ||
| 10 | 0.04 | 1.08 × 10–3 | |||
| 30 | 0.01 | NA | |||
| 60 | 0.06 | NA | |||
| 72 | 1 | NA | NA | ||
| 10 | NA | NA | |||
| 30 | NA | NA | |||
| 60 | 0.25 | NA | |||
| Gene expression—the p53 signalling | KEGG | 24 | 1 | 1.32 × 10–5 | NA |
| 10 | 7.84 × 10–13 | 0.02 | |||
| 30 | 2.79 × 10–7 | 7.77 × 10–3 | |||
| 60 | 4.99 × 10–5 | 0.11 | |||
| 48 | 1 | 0.03 | NA | ||
| 10 | 6.60 × 10–5 | 0.05 | |||
| 30 | 3.92 × 10–5 | 0.12 | |||
| 60 | 8.63 × 10–4 | NA | |||
| 72 | 1 | 0.06 | NA | ||
| 10 | 2.65 × 10–6 | 0.17 | |||
| 30 | 8.25 × 10–7 | NA | |||
| 60 | 4.56 × 10–6 | NA | |||
| Epigenetic regulation of gene expression—DNA methylation | Reactome | 24 | 1 | NA | 0.04 |
| 10 | 6.63 x 10–14 | 4.95 × 10–11 | |||
| 30 | 3.98 × 10–14 | 8.08 × 10–6 | |||
| 60 | 4.44 × 10–11 | 1.03 × 10–3 | |||
| 48 | 1 | NA | NA | ||
| 10 | 2.60 × 10–7 | 2.58 × 10–6 | |||
| 30 | 3.12 × 10–8 | 7.41 × 10–5 | |||
| 60 | 5.58 × 10–7 | 7.49 × 10–4 | |||
| 72 | 1 | NA | NA | ||
| 10 | 0.02 | NA | |||
| 30 | 2.96 × 10–3 | NA | |||
| 60 | 9.08 × 10–5 | NA | |||
| Metabolism of carbohydrates—Glycolysis/Gluconeogenesis | KEGG | 24 | 1 | NA | NA |
| 10 | NA | NA | |||
| 30 | NA | 0.15 | |||
| 60 | NA | NA | |||
| 48 | 1 | NA | NA | ||
| 10 | 6.81 × 10–3 | NA | |||
| 30 | 0.02 | 0.11 | |||
| 60 | 0.04 | 0.14 | |||
| 72 | 1 | NA | NA | ||
| 10 | 6.43 × 10–4 | NA | |||
| 30 | 6.84 × 10–5 | NA | |||
| 60 | 7.95 × 10–6 | NA | |||
| Metabolism—Respiratory electron transport, ATP synthesis by chemiosmotic coupling, and eat production by uncoupling proteins | Reactome | 24 | 1 | NA | NA |
| 10 | NA | NA | |||
| 30 | NA | NA | |||
| 60 | 6.24 × 10–3 | NA | |||
| 48 | 1 | NA | NA | ||
| 10 | NA | NA | |||
| 30 | NA | NA | |||
| 60 | 0.05 | NA | |||
| 72 | 1 | NA | NA | ||
| 10 | NA | NA | |||
| 30 | NA | NA | |||
| 60 | 0.20 | NA | |||
| Metabolism of lipids | Reactome | 24 | 1 | NA | NA |
| 10 | 0.15 | 0.09 | |||
| 30 | 7.01 × 10–3 | 0.15 | |||
| 60 | 0.07 | NA | |||
| 48 | 1 | NA | NA | ||
| 10 | 8.49 × 10–5 | NA | |||
| 30 | 5.41 × 10–5 | NA | |||
| 60 | 9.11 × 10–6 | NA | |||
| 72 | 1 | NA | NA | ||
| 10 | 1.00 × 10–3 | NA | |||
| 30 | 5.19 × 10–4 | NA | |||
| 60 | 3.91 × 10–3 | NA | |||
| Metabolism of amino acids and derivatives | Reactome | 24 | 1 | NA | NA |
| 10 | 0.08 | 0.03 | |||
| 30 | 0.15 | 4.46 × 10–17 | |||
| 60 | 0.07 | 1.29 × 10–36 | |||
| 48 | 1 | 0.13 | NA | ||
| 10 | 1.00 × 10–3 | NA | |||
| 30 | 0.05 | 6.77 × 10–27 | |||
| 60 | NA | 1.99× 10–4 | |||
| 72 | 1 | NA | NA | ||
| 10 | 3.12 × 10–4 | NA | |||
| 30 | 2.13 × 10–10 | NA | |||
| 60 | 6.34 × 10–7 | NA | |||
| Cellular responses to external stimuli—oxidative stress induced senescence | Reactome | 24 | 1 | NA | 0.05 |
| 10 | 2.12 × 10–9 | 9.19 × 10–9 | |||
| 30 | 2.78 × 10–11 | 3.28 × 10–6 | |||
| 60 | 8.00 × 10–10 | 9.02 × 10–5 | |||
| 48 | 1 | NA | NA | ||
| 10 | 1.45 × 10–5 | 1.88 × 10–5 | |||
| 30 | 3.09 × 10–7 | 1.82 × 10–4 | |||
| 60 | 4.63 × 10–6 | 2.00 × 10–3 | |||
| 72 | 1 | NA | NA | ||
| 10 | 0.05 | NA | |||
| 30 | 4.82 × 10–3 | NA | |||
| 60 | 6.23 × 10–4 | NA | |||
| Concentration (µM) | Gene Symbol | Name | Direction of Expression (Control vs. DOX) | Main Pathway(s) Involved |
|---|---|---|---|---|
| Colon | ||||
| 30 | DHRS2 | Dehydrogenase/reductase SDR family member 2 | ↑ | Metabolism of several compounds |
| RGCC | Regulator of cell cycle | ↑ | Regulation of cell cycle progression via p53 | |
| LAMP3 | Lysosome-associated membrane glycoprotein 3 | ↑ | Gene expression; adaptive immunity | |
| TP53I3 | Tumour Protein P53 Inducible Protein 3 | ↑ | Cellular responses to oxidative stress | |
| TNFSF15 | TNF Superfamily Member 15 | ↑ | Apoptosis modulation and signalling | |
| 60 | ABCA12 | ATP Binding Cassette Subfamily A Member 12 | ↑ | Transport of molecules |
| RGCC | Regulator of cell cycle | ↑ | Regulation of cell cycle progression via p53 | |
| DHRS2 | Dehydrogenase/reductase SDR family member 2 | ↑ | Metabolism of several compounds | |
| MFAP3L | Microfibril Associated Protein 3 Like | ↑ | Nuclear signalling pathways (EGFR and MAPK) | |
| LAMP3 | Lysosome-associated membrane glycoprotein 3 | ↑ | Gene expression; adaptive immunity | |
| SI | ||||
| 30 | CAPN8 | Calpain 8 | ↓ | Degradation of the extracellular matrix |
| CTNND1 | Catenin Delta 1 | ↓ | Cell adhesion and signal transduction | |
| MPRIP | Myosin Phosphatase Rho Interacting Protein | ↓ | Signalling by BRAF and RAF fusions | |
| TSPAN1 | Tetraspanin 1 | ↓ | Regulation of cell development, activation, growth and motility | |
| TPX2 | Microtubule Nucleation Factor | ↓ | Cell cycle and Regulation of p53 activity | |
| 60 | MCM5 | Minichromosome Maintenance Complex Component 5 | ↓ | DNA replication |
| DHRS9 | Dehydrogenase/Reductase 9 | ↓ | Metabolism | |
| SLC2A3 | Solute Carrier Family 2 Member 3 | ↓ | Transport of glucose | |
| PPP1R3C | Protein Phosphatase 1 Regulatory Subunit 3C | ↓ | Glycogen synthesis | |
| MT1X | Metallothionein 1X | ↓ | Metallothioneins bind metals | |
| UniProt Accession | Gene Name | Protein Name |
|---|---|---|
| O43488 | AKR7A2 | Aflatoxin B1 aldehyde reductase member 2 |
| O75251 | NDUFS7 | NADH dehydrogenase [ubiquinone] iron–sulphur protein 7, mitochondrial |
| O75306 | NDUFS2 | NADH dehydrogenase [ubiquinone] iron–sulphur protein 2, mitochondrial |
| O75489 | NDUFS3 | NADH dehydrogenase [ubiquinone] iron–sulphur protein 3, mitochondrial |
| O75828 | CBR3 | Carbonyl reductase [NADPH] 3 |
| P00352 | ALDH1A1 | Retinal dehydrogenase 1 |
| P02768 | ALB | Albumin |
| P11388 | TOP2A | DNA topoisomerase 2-alpha |
| P14550 | AKR1A1 | Aldo-keto reductase family 1 member A1 |
| P15559 | NQO1 | NAD(P)H dehydrogenase [quinone] 1 |
| P16083 | NQO2 | Ribosyldihydronicotinamide dehydrogenase [quinone] |
| P16152 | CBR1 | Carbonyl reductase [NADPH] 1 |
| P16435 | POR | NADPH--cytochrome P450 reductase |
| P29474 | NOS3 | Nitric oxide synthase, endothelial |
| P29475 | NOS1 | Nitric oxide synthase, brain |
| Q14978 | NOLC1 | Nucleolar and coiled-body phosphoprotein 1 |
| Q15311 | RALBP1 | RalA-binding protein 1 |
| Q92887 | ABCC2 | Canalicular multispecific organic anion transporter 1 |
| Q9NUT2 | ABCB8 | Mitochondrial potassium channel ATP-binding subunit |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, D.; Coyle, L.; Füzi, B.; Ferreira, S.; Jo, H.; Herpers, B.; Chung, S.-W.; Fisher, C.; Kleinjans, J.C.S.; Jennen, D.; et al. Unravelling Mechanisms of Doxorubicin-Induced Toxicity in 3D Human Intestinal Organoids. Int. J. Mol. Sci. 2022, 23, 1286. https://doi.org/10.3390/ijms23031286
Rodrigues D, Coyle L, Füzi B, Ferreira S, Jo H, Herpers B, Chung S-W, Fisher C, Kleinjans JCS, Jennen D, et al. Unravelling Mechanisms of Doxorubicin-Induced Toxicity in 3D Human Intestinal Organoids. International Journal of Molecular Sciences. 2022; 23(3):1286. https://doi.org/10.3390/ijms23031286
Chicago/Turabian StyleRodrigues, Daniela, Luke Coyle, Barbara Füzi, Sofia Ferreira, Heeseung Jo, Bram Herpers, Seung-Wook Chung, Ciarán Fisher, Jos C. S. Kleinjans, Danyel Jennen, and et al. 2022. "Unravelling Mechanisms of Doxorubicin-Induced Toxicity in 3D Human Intestinal Organoids" International Journal of Molecular Sciences 23, no. 3: 1286. https://doi.org/10.3390/ijms23031286