
Involvement of Fatty Acids and Their Metabolites in the Development of Inflammation in Atherosclerosis



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
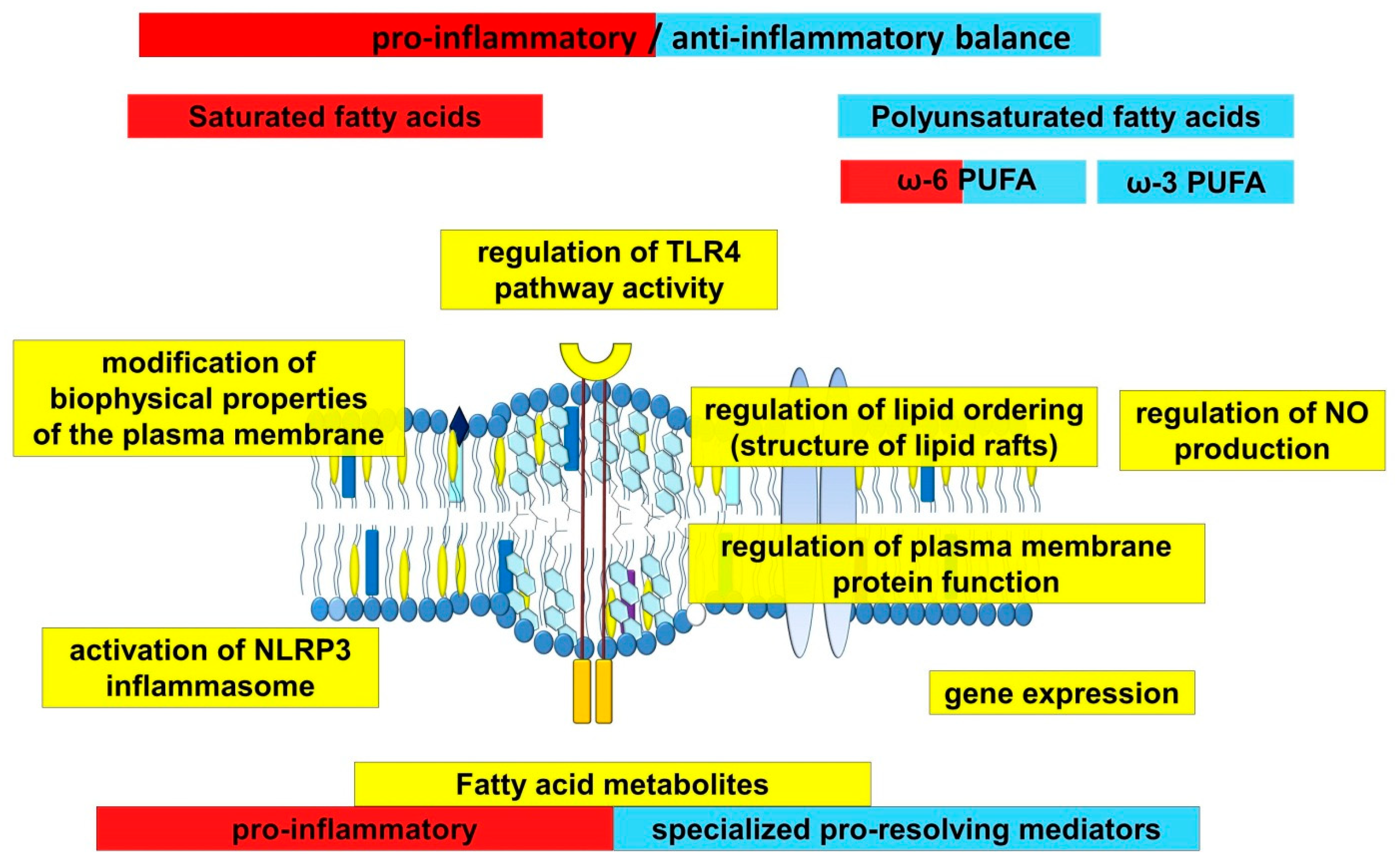
2. The Role of Fatty Acids in the Development of Inflammation in Atherosclerosis
3. The Role of Endothelial Cells in Inflammation
4. The Role of the Innate Immune System in Atherogenesis
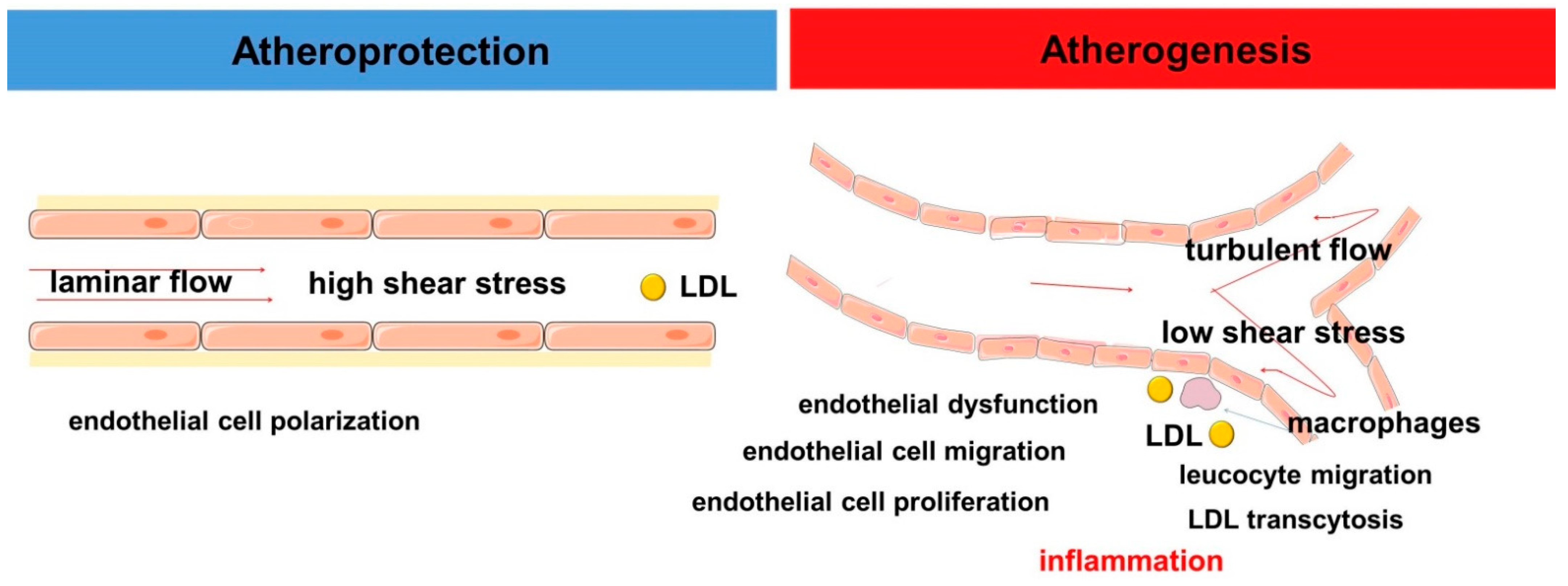
5. Hemodynamic Characteristics of Blood Flow and Inflammation
6. Participation of Fatty Acids in the Regulation of Bioactive Metabolites Related to Hemodynamics and Inflammation
6.1. Nitric Oxide
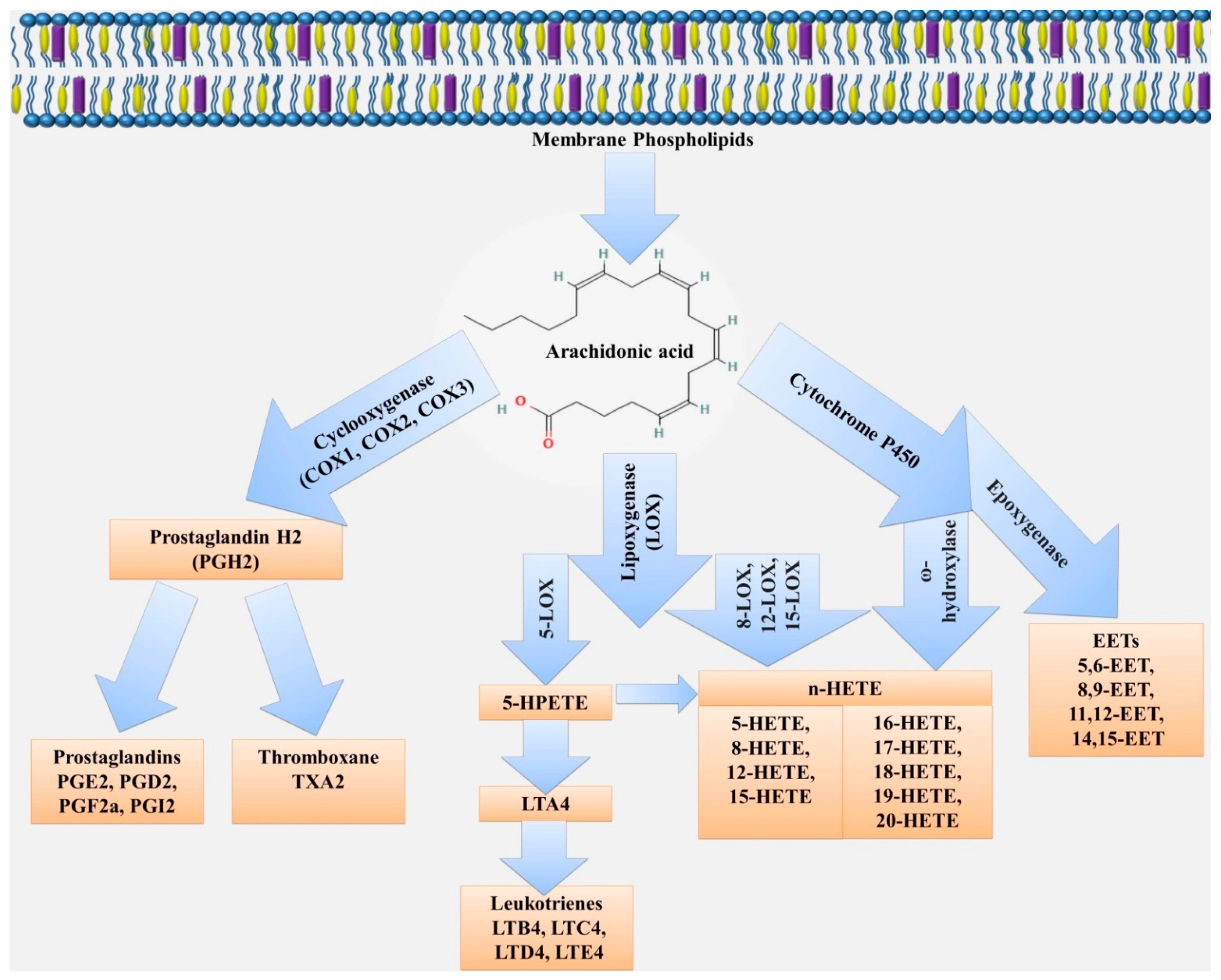
6.2. Eicosanoids
6.2.1. Cyclooxygenase Pathway of Arachidonic Acid Metabolism
6.2.2. Lipoxygenase Pathway
6.2.3. Cytochrome P450 Pathway
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Song, P.; Fang, Z.; Wang, H.; Cai, Y.; Rahimi, K.; Zhu, Y.; Fowkes, F.G.R.; Fowkes, F.J.I.; Rudan, I. Global and regional prevalence, burden, and risk factors for carotid atherosclerosis: A systematic review, meta-analysis, and modelling study. Lancet Glob. Health 2020, 8, e721–e729. [Google Scholar] [CrossRef]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, T.; Sissani, L.; Labreuche, J.; Ducrocq, G.; Lavallée, P.C.; Meseguer, E.; Guidoux, C.; Cabrejo, L.; Hobeanu, C.; Gongora-Rivera, F.; et al. Prevalence of Systemic Atherosclerosis Burdens and Overlapping Stroke Etiologies and Their Associations with Long-term Vascular Prognosis in Stroke with Intracranial Atherosclerotic Disease. JAMA Neurol. 2018, 75, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Bauersachs, R.; Zeymer, U.; Brière, J.B.; Marre, C.; Bowrin, K.; Huelsebeck, M. Burden of Coronary Artery Disease and Peripheral Artery Disease: A Literature Review. Cardiovasc. Ther. 2019, 2019, 8295054. [Google Scholar] [CrossRef] [PubMed]
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Bäck, M.; Benetos, A.; Biffi, A.; Boavida, J.M.; Capodanno, D.; et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur. Heart J. 2021, 42, 3227–3337. [Google Scholar] [CrossRef]
- Meier, R.; Rachamin, Y.; Rosemann, T.; Markun, S. The Impact of the 2019 European Guideline for Cardiovascular Risk Management: A Cross-Sectional Study in General Practice. J. Clin. Med. 2020, 9, 2140. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Ruparelia, N.; Choudhury, R. Inflammation and atherosclerosis: What is on the horizon? Heart 2020, 106, 80–85. [Google Scholar] [CrossRef]
- Insull, W., Jr. The Pathology of Atherosclerosis: Plaque Development and Plaque Responses to Medical Treatment. Am. J. Med. 2009, 122, S3–S14. [Google Scholar] [CrossRef]
- Morbiducci, U.; Kok, A.M.; Kwak, B.R.; Stone, P.H.; Steinman, D.A.; Wentzel, J.J. Atherosclerosis at arterial bifurcations: Evidence for the role of haemodynamics and geometry. Thromb. Haemost. 2016, 115, 484–492. [Google Scholar] [CrossRef]
- Jiang, P.; Chen, Z.; Hippe, D.S.; Watase, H.; Sun, B.; Lin, R.; Yang, Z.; Xue, Y.; Zhao, X.; Yuan, C. Association between Carotid Bifurcation Geometry and Atherosclerotic Plaque Vulnerability: A Chinese Atherosclerosis Risk Evaluation Study. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Bezsonov, E.E.; Sobenin, I.A.; Orekhov, A.N. Immunopathology of Atherosclerosis and Related Diseases: Focus on Molecular Biology. Int. J. Mol. Sci. 2021, 22, 4080. [Google Scholar] [CrossRef] [PubMed]
- Mezentsev, A.; Bezsonov, E.; Kashirskikh, D.; Baig, M.S.; Eid, A.H.; Orekhov, A. Proatherogenic Sialidases and Desialylated Lipoproteins: 35 Years of Research and Current State from Bench to Bedside. Biomedicines 2021, 9, 600. [Google Scholar] [CrossRef]
- Ghosh, A.; Gao, L.; Thakur, A.; Siu, P.M.; Lai, C.W.K. Role of free fatty acids in endothelial dysfunction. J. Biomed. Sci. 2017, 24, 50. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Li, H.; Bao, Y.; Zhang, X.; Yu, Y. Free fatty acids induce endothelial dysfunction and activate protein kinase C and nuclear factor-κB pathway in rat aorta. Int. J. Cardiol. 2011, 152, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Sherratt, S.C.R.; Dawoud, H.; Bhatt, D.L.; Malinski, T.; Mason, R.P. Omega-3 and omega-6 fatty acids have distinct effects on endothelial fatty acid content and nitric oxide bioavailability. Prostaglandins Leukot. Essent. Fat. Acids 2021, 173, 102337. [Google Scholar] [CrossRef] [PubMed]
- Werz, O.; Gerstmeier, J.; Libreros, S.; De la Rosa, X.; Werner, M.; Norris, P.C.; Chiang, N.; Serhan, C.N. Human macrophages differentially produce specific resolvin or leukotriene signals that depend on bacterial pathogenicity. Nat. Commun. 2018, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Haeggström, J.Z.; Funk, C.D. Lipoxygenase and leukotriene pathways: Biochemistry, biology, and roles in disease. Chem. Rev. 2011, 111, 5866–5898. [Google Scholar] [CrossRef]
- Das, U.N. “Cell Membrane Theory of Senescence” and the Role of Bioactive Lipids in Aging, and Aging Associated Diseases and Their Therapeutic Implications. Biomolecules 2021, 11, 241. [Google Scholar] [CrossRef]
- Das, U.N. Arachidonic acid and lipoxin A4 as possible endogenous anti-diabetic molecules. Prostaglandins Leukot. Essent. Fatty Acids 2013, 88, 201–210. [Google Scholar] [CrossRef]
- Gundala, N.K.V.; Naidu, V.G.M.; Das, U.N. Arachidonic acid and lipoxin A4 attenuate alloxan-induced cytotoxicity to RIN5F cells in vitro and type 1 diabetes mellitus in vivo. Biofactors 2017, 43, 251–271. [Google Scholar] [CrossRef] [PubMed]
- Gundala, N.K.V.; Naidu, V.G.M.; Das, U.N. Arachidonic acid and lipoxinA4 attenuate streptozotocin-induced cytotoxicity to RIN5 F cells in vitro and type 1 and type 2 diabetes mellitus in vivo. Nutrition 2017, 35, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Higgs, E.A. Nitric Oxide and the Vascular Endothelium. In The Vascular Endothelium I; Moncada, S., Higgs, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 213–254. [Google Scholar]
- Cines, D.B.; Pollak, E.S.; Buck, C.A.; Loscalzo, J.; Zimmerman, G.A.; McEver, R.P.; Pober, J.S.; Wick, T.M.; Konkle, B.A.; Schwartz, B.S.; et al. Endothelial Cells in Physiology and in the Pathophysiology of Vascular Disorders. Blood 1998, 91, 3527–3561. [Google Scholar] [CrossRef] [PubMed]
- Segers, V.F.M.; Brutsaert, D.L.; De Keulenaer, G.W. Cardiac Remodeling: Endothelial Cells Have More to Say than Just NO. Front. Physiol. 2018, 9, 382. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Shu, B.; Zhang, Y.; Wang, M. Endothelial Response to Pathophysiological Stress. Arterioscler. Thromb. Vasc. Biol. 2019, 39, e233–e243. [Google Scholar] [CrossRef] [PubMed]
- Mai, J.; Virtue, A.; Shen, J.; Wang, H.; Yang, X.F. An evolving new paradigm: Endothelial cells—Conditional innate immune cells. J. Hematol. Oncol. 2013, 6, 61. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Saredy, J.; Yang, W.Y.; Sun, Y.; Lu, Y.; Saaoud, F.; Drummer, C.; Johnson, C.; Xu, K.; Jiang, X.; et al. Vascular Endothelial Cells and Innate Immunity. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e138–e152. [Google Scholar] [CrossRef]
- Mallick, R.; Duttaroy, A.K. Modulation of endothelium function by fatty acids. Mol. Cell. Biochem. 2021. [Google Scholar] [CrossRef]
- Majzner, K.; Tott, S.; Roussille, L.; Deckert, V.; Chlopicki, S.; Baranska, M. Uptake of fatty acids by a single endothelial cell investigated by Raman spectroscopy supported by AFM. Analyst 2018, 143, 970–980. [Google Scholar] [CrossRef]
- Qian, X.; Yang, Z.; Mao, E.; Chen, E. Regulation of fatty acid synthesis in immune cells. Scand. J. Immunol. 2018, 88, e12713. [Google Scholar] [CrossRef]
- Van den Bossche, J.; Baardman, J.; de Winther, M.P. Metabolic Characterization of Polarized M1 and M2 Bone Marrow-derived Macrophages Using Real-time Extracellular Flux Analysis. J. Vis. Exp. 2015, 105, e53424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.; et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015, 21, 65–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.C.; Everts, B.; Ivanova, Y.; O’Sullivan, D.; Nascimento, M.; Smith, A.M.; Beatty, W.; Love-Gregory, L.; Lam, W.Y.; O’Neill, C.M.; et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol. 2014, 15, 846–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Certo, M.; Elkafrawy, H.; Pucino, V.; Cucchi, D.; Cheung, K.C.P.; Mauro, C. Endothelial cell and T-cell crosstalk: Targeting metabolism as a therapeutic approach in chronic inflammation. Br. J. Pharmacol. 2021, 178, 2041–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bock, K.; Georgiadou, M.; Carmeliet, P. Role of endothelial cell metabolism in vessel sprouting. Cell Metab. 2013, 18, 634–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Missiaen, R.; Morales-Rodriguez, F.; Eelen, G.; Carmeliet, P. Targeting endothelial metabolism for anti-angiogenesis therapy: A pharmacological perspective. Vasc. Pharmacol. 2017, 90, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Kalucka, J.; Bierhansl, L.; Conchinha, N.V.; Missiaen, R.; Elia, I.; Brüning, U.; Scheinok, S.; Treps, L.; Cantelmo, A.R.; Dubois, C.; et al. Quiescent Endothelial Cells Upregulate Fatty Acid β-Oxidation for Vasculoprotection via Redox Homeostasis. Cell Metab. 2018, 28, 881–894.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patella, F.; Schug, Z.T.; Persi, E.; Neilson, L.J.; Erami, Z.; Avanzato, D.; Maione, F.; Hernandez-Fernaud, J.R.; Mackay, G.; Zheng, L.; et al. Proteomics-Based Metabolic Modeling Reveals That Fatty Acid Oxidation (FAO) Controls Endothelial Cell (EC) Permeability. Mol. Cell. Proteom. 2015, 14, 621–634. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Schneider, J.G.; Shenouda, S.M.; Lee, A.; Towler, D.A.; Chakravarthy, M.V.; Vita, J.A.; Semenkovich, C.F. De novo lipogenesis maintains vascular homeostasis through endothelial nitric-oxide synthase (eNOS) palmitoylation. J. Biol. Chem. 2011, 286, 2933–2945. [Google Scholar] [CrossRef] [Green Version]
- Mehrotra, D.; Wu, J.; Papangeli, I.; Chun, H.J. Endothelium as a gatekeeper of fatty acid transport. Trends Endocrinol. Metab. 2014, 25, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Boden, G. Obesity and Free Fatty Acids. Endocrinol. Metab. Clin. N. Am. 2008, 37, 635–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, M.; Tay, E.; Cusi, K. Elevated plasma free fatty acids increase cardiovascular risk by inducing plasma biomarkers of endothelial activation, myeloperoxidase and PAI-1 in healthy subjects. Cardiovasc. Diabetol. 2010, 9, 9. [Google Scholar] [CrossRef] [Green Version]
- Pillon, N.J.; Azizi, P.M.; Li, Y.E.; Liu, J.; Wang, C.; Chan, K.L.; Hopperton, K.E.; Bazinet, R.P.; Heit, B.; Bilan, P.J.; et al. Palmitate-induced inflammatory pathways in human adipose microvascular endothelial cells promote monocyte adhesion and impair insulin transcytosis. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E35–E44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Akira, S. Pathogen recognition with Toll-like receptors. Curr. Opin. Immunol. 2005, 17, 338–344. [Google Scholar] [CrossRef]
- Opitz, B.; Eitel, J.; Meixenberger, K.; Suttorp, N. Role of Toll-like receptors, NOD-like receptors and RIG-I-like receptors in endothelial cells and systemic infections. Thromb. Haemost. 2009, 102, 1103–1109. [Google Scholar] [CrossRef]
- Roshan, M.H.K.; Tambo, A.; Pace, N.P. The Role of TLR2, TLR4, and TLR9 in the Pathogenesis of Atherosclerosis. Int. J. Inflamm. 2016, 2016, 1532832. [Google Scholar] [CrossRef] [Green Version]
- Dunzendorfer, S.; Lee, H.-K.; Tobias, P.S. Flow-Dependent Regulation of Endothelial Toll-Like Receptor 2 Expression through Inhibition of SP1 Activity. Circ. Res. 2004, 95, 684–691. [Google Scholar] [CrossRef] [Green Version]
- Edfeldt, K.; Swedenborg, J.; Hansson, G.K.; Yan, Z.Q. Expression of toll-like receptors in human atherosclerotic lesions: A possible pathway for plaque activation. Circulation 2002, 105, 1158–1161. [Google Scholar] [CrossRef] [Green Version]
- Zeuke, S.; Ulmer, A.J.; Kusumoto, S.; Katus, H.A.; Heine, H. TLR4-mediated inflammatory activation of human coronary artery endothelial cells by LPS. Cardiovasc. Res. 2002, 56, 126–134. [Google Scholar] [CrossRef]
- Li, H.; Sun, B. Toll-like receptor 4 in atherosclerosis. J. Cell. Mol. Med. 2007, 11, 88–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, P.K.; Galis, Z.S. Matrix metalloproteinase hypothesis of plaque rupture: Players keep piling up but questions remain. Circulation 2001, 104, 1878–1880. [Google Scholar] [CrossRef] [PubMed]
- Howell, K.W.; Meng, X.; Fullerton, D.A.; Jin, C.; Reece, T.B.; Cleveland, J.C., Jr. Toll-like receptor 4 mediates oxidized LDL-induced macrophage differentiation to foam cells. J. Surg. Res. 2011, 171, e27–e31. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.H.; Shah, P.K.; Faure, E.; Equils, O.; Thomas, L.; Fishbein, M.C.; Luthringer, D.; Xu, X.P.; Rajavashisth, T.B.; Yano, J.; et al. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation 2001, 104, 3103–3108. [Google Scholar] [CrossRef] [Green Version]
- Miller, Y.I.; Viriyakosol, S.; Worrall, D.S.; Boullier, A.; Butler, S.; Witztum, J.L. Toll-like receptor 4-dependent and -independent cytokine secretion induced by minimally oxidized low-density lipoprotein in macrophages. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1213–1219. [Google Scholar] [CrossRef] [Green Version]
- Castrillo, A.; Joseph, S.B.; Vaidya, S.A.; Haberland, M.; Fogelman, A.M.; Cheng, G.; Tontonoz, P. Crosstalk between LXR and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol. Cell 2003, 12, 805–816. [Google Scholar] [CrossRef]
- Ruysschaert, J.-M.; Lonez, C. Role of lipid microdomains in TLR-mediated signalling. Biochim. Biophys. Acta (BBA)-Biomembr. 2015, 1848, 1860–1867. [Google Scholar] [CrossRef]
- Suzuki, K.; Kawakami, Y.; Yamauchi, K. Impact of TLR 2, TLR 4-activation on the Expression of ABCA1 and ABCG1 in Raw Cells. Ann. Clin. Lab. Sci. 2017, 47, 436–446. [Google Scholar]
- Hoshino, K.; Takeuchi, O.; Kawai, T.; Sanjo, H.; Ogawa, T.; Takeda, Y.; Takeda, K.; Akira, S. Pillars Article: Cutting Edge: Toll-Like Receptor 4 (TLR4)-Deficient Mice Are Hyporesponsive to Lipopolysaccharide: Evidence for TLR4 as the Lps Gene Product. J. Immunol. 1999, 162, 3749–3752. [Google Scholar]
- Rocha, D.M.; Caldas, A.P.; Oliveira, L.L.; Bressan, J.; Hermsdorff, H.H. Saturated fatty acids trigger TLR4-mediated inflammatory response. Atherosclerosis 2016, 244, 211–215. [Google Scholar] [CrossRef]
- Kawahara, K. Variation, Modification and Engineering of Lipid A in Endotoxin of Gram-Negative Bacteria. Int. J. Mol. Sci. 2021, 22, 2281. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.W.; Kwon, M.-J.; Choi, A.M.K.; Kim, H.-P.; Nakahira, K.; Hwang, D.H. Fatty acids modulate Toll-like receptor 4 activation through regulation of receptor dimerization and recruitment into lipid rafts in a reactive oxygen species-dependent manner. J. Biol. Chem. 2009, 284, 27384–27392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, G.I.; Langley, K.G.; Berglund, N.A.; Kammoun, H.L.; Reibe, S.; Estevez, E.; Weir, J.; Mellett, N.A.; Pernes, G.; Conway, J.R.W.; et al. Evidence that TLR4 Is Not a Receptor for Saturated Fatty Acids but Mediates Lipid-Induced Inflammation by Reprogramming Macrophage Metabolism. Cell Metab. 2018, 27, 1096–1110.e1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Qian, Y.; Fang, Q.; Zhong, P.; Li, W.; Wang, L.; Fu, W.; Zhang, Y.; Xu, Z.; Li, X.; et al. Saturated palmitic acid induces myocardial inflammatory injuries through direct binding to TLR4 accessory protein MD2. Nat. Commun. 2017, 8, 13997. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, E.A.; Zhang, W.Y.; Karnik, S.K.; Borwege, S.; Anand, V.R.; Laine, P.S.; Su, Y.; Reaven, P.D. Nutrient modification of the innate immune response: A novel mechanism by which saturated fatty acids greatly amplify monocyte inflammation. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 802–808. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Li, Y.; Jin, J.; Zhang, X.; Hannun, Y.A.; Huang, Y. GPR40/FFA1 and neutral sphingomyelinase are involved in palmitate-boosted inflammatory response of microvascular endothelial cells to LPS. Atherosclerosis 2015, 240, 163–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, R.; Al-Roub, A.; Kochumon, S.; Akther, N.; Thomas, R.; Kumari, M.; Koshy, M.S.; Tiss, A.; Hannun, Y.A.; Tuomilehto, J.; et al. The Synergy between Palmitate and TNF-α for CCL2 Production Is Dependent on the TRIF/IRF3 Pathway: Implications for Metabolic Inflammation. J. Immunol. 2018, 200, 3599–3611. [Google Scholar] [CrossRef]
- Huang, S.; Rutkowsky, J.M.; Snodgrass, R.G.; Ono-Moore, K.D.; Schneider, D.A.; Newman, J.W.; Adams, S.H.; Hwang, D.H. Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J. Lipid Res. 2012, 53, 2002–2013. [Google Scholar] [CrossRef] [Green Version]
- Korbecki, J.; Bajdak-Rusinek, K. The effect of palmitic acid on inflammatory response in macrophages: An overview of molecular mechanisms. Inflamm. Res. 2019, 68, 915–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, M.T.; Favelyukis, S.; Nguyen, A.K.; Reichart, D.; Scott, P.A.; Jenn, A.; Liu-Bryan, R.; Glass, C.K.; Neels, J.G.; Olefsky, J.M. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J. Biol. Chem. 2007, 282, 35279–35292. [Google Scholar] [CrossRef] [Green Version]
- Tashiro, H.; Takahashi, K.; Sadamatsu, H.; Kato, G.; Kurata, K.; Kimura, S.; Sueoka-Aragane, N. Saturated Fatty Acid Increases Lung Macrophages and Augments House Dust Mite-Induced Airway Inflammation in Mice Fed with High-Fat Diet. Inflammation 2017, 40, 1072–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suganami, T.; Tanimoto-Koyama, K.; Nishida, J.; Itoh, M.; Yuan, X.; Mizuarai, S.; Kotani, H.; Yamaoka, S.; Miyake, K.; Aoe, S.; et al. Role of the Toll-like receptor 4/NF-kappaB pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 84–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullberg, K.B.; Larsen, J.Ø.; Pedersen, S.B.; Richelsen, B. Effects of LPS and dietary free fatty acids on MCP-1 in 3T3-L1 adipocytes and macrophages in vitro. Nutr. Diabetes 2014, 4, e113. [Google Scholar] [CrossRef] [PubMed]
- Kochumon, S.; Wilson, A.; Chandy, B.; Shenouda, S.; Tuomilehto, J.; Sindhu, S.; Ahmad, R. Palmitate Activates CCL4 Expression in Human Monocytic Cells via TLR4/MyD88 Dependent Activation of NF-κB/MAPK/ PI3K Signaling Systems. Cell. Physiol. Biochem. 2018, 46, 953–964. [Google Scholar] [CrossRef]
- Lee, J.Y.; Sohn, K.H.; Rhee, S.H.; Hwang, D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J. Biol. Chem. 2001, 276, 16683–16689. [Google Scholar] [CrossRef] [Green Version]
- Sindhu, S.; Al-Roub, A.; Koshy, M.; Thomas, R.; Ahmad, R. Palmitate-Induced MMP-9 Expression in the Human Monocytic Cells is Mediated through the TLR4-MyD88 Dependent Mechanism. Cell. Physiol. Biochem. 2016, 39, 889–900. [Google Scholar] [CrossRef]
- Das, T.; Yount, J.S.; Hang, H.C. Protein S-palmitoylation in immunity. Open Biol. 2021, 11, 200411. [Google Scholar] [CrossRef]
- Chesarino, N.M.; Hach, J.C.; Chen, J.L.; Zaro, B.W.; Rajaram, M.V.; Turner, J.; Schlesinger, L.S.; Pratt, M.R.; Hang, H.C.; Yount, J.S. Chemoproteomics reveals Toll-like receptor fatty acylation. BMC Biol. 2014, 12, 91. [Google Scholar] [CrossRef]
- Kim, Y.C.; Lee, S.E.; Kim, S.K.; Jang, H.D.; Hwang, I.; Jin, S.; Hong, E.B.; Jang, K.S.; Kim, H.S. Toll-like receptor mediated inflammation requires FASN-dependent MYD88 palmitoylation. Nat. Chem. Biol. 2019, 15, 907–916. [Google Scholar] [CrossRef]
- Qi, H.Y.; Shelhamer, J.H. Toll-like receptor 4 signaling regulates cytosolic phospholipase A2 activation and lipid generation in lipopolysaccharide-stimulated macrophages. J. Biol. Chem. 2005, 280, 38969–38975. [Google Scholar] [CrossRef] [Green Version]
- Olona, A.; Hateley, C.; Muralidharan, S.; Wenk, M.R.; Torta, F.; Behmoaras, J. Sphingolipid metabolism during Toll-like receptor 4 (TLR4)-mediated macrophage activation. Br. J. Pharmacol. 2021, 178, 4575–4587. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, H.; Zhang, W.; Cai, Y.; Shan, P.; Wu, D.; Zhang, B.; Liu, H.; Khan, Z.A.; Liang, G. Arachidonic acid inhibits inflammatory responses by binding to myeloid differentiation factor-2 (MD2) and preventing MD2/toll-like receptor 4 signaling activation. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165683. [Google Scholar] [CrossRef] [PubMed]
- Rogero, M.M.; Calder, P.C. Obesity, Inflammation, Toll-Like Receptor 4 and Fatty Acids. Nutrients 2018, 10, 432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Ye, J.; Gao, Z.; Youn, H.S.; Lee, W.H.; Zhao, L.; Sizemore, N.; Hwang, D.H. Reciprocal modulation of Toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J. Biol. Chem. 2003, 278, 37041–37051. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, M.A.; Carretta, M.D.; Burgos, R.A. Long Chain Fatty Acids as Modulators of Immune Cells Function: Contribution of FFA1 and FFA4 Receptors. Front. Physiol. 2021, 12. [Google Scholar] [CrossRef]
- Pavillard, L.E.; Marín-Aguilar, F.; Bullon, P.; Cordero, M.D. Cardiovascular diseases, NLRP3 inflammasome, and western dietary patterns. Pharmacol. Res. 2018, 131, 44–50. [Google Scholar] [CrossRef]
- Robblee, M.M.; Kim, C.C.; Porter Abate, J.; Valdearcos, M.; Sandlund, K.L.M.; Shenoy, M.K.; Volmer, R.; Iwawaki, T.; Koliwad, S.K. Saturated Fatty Acids Engage an IRE1α-Dependent Pathway to Activate the NLRP3 Inflammasome in Myeloid Cells. Cell Rep. 2016, 14, 2611–2623. [Google Scholar] [CrossRef] [Green Version]
- Ralston, J.C.; Lyons, C.L.; Kennedy, E.B.; Kirwan, A.M.; Roche, H.M. Fatty Acids and NLRP3 Inflammasome-Mediated Inflammation in Metabolic Tissues. Annu. Rev. Nutr. 2017, 37, 77–102. [Google Scholar] [CrossRef]
- Karasawa, T.; Takahashi, M. Role of NLRP3 Inflammasomes in Atherosclerosis. J. Atheroscler. Thromb. 2017, 24, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Gijsen, F.; Katagiri, Y.; Barlis, P.; Bourantas, C.; Collet, C.; Coskun, U.; Daemen, J.; Dijkstra, J.; Edelman, E.; Evans, P.; et al. Expert recommendations on the assessment of wall shear stress in human coronary arteries: Existing methodologies, technical considerations, and clinical applications. Eur. Heart J. 2019, 40, 3421–3433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, P.F. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 2009, 6, 16–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malek, A.M.; Alper, S.L.; Izumo, S. Hemodynamic shear stress and its role in atherosclerosis. JAMA 1999, 282, 2035–2042. [Google Scholar] [CrossRef] [PubMed]
- Campinho, P.; Vilfan, A.; Vermot, J. Blood Flow Forces in Shaping the Vascular System: A Focus on Endothelial Cell Behavior. Front. Physiol. 2020, 11, 552. [Google Scholar] [CrossRef]
- Kotlyarov, S. Diversity of Lipid Function in Atherogenesis: A Focus on Endothelial Mechanobiology. Int. J. Mol. Sci. 2021, 22, 11545. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.F.; Remuzzi, A.; Gordon, E.J.; Dewey, C.F.; Gimbrone, M.A. Turbulent fluid shear stress induces vascular endothelial cell turnover in vitro. Proc. Natl. Acad. Sci. USA 1986, 83, 2114–2117. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Li, Y.-S.; Chien, S. Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2191–2198. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Cardeña, G.; Comander, J.; Anderson, K.R.; Blackman, B.R.; Gimbrone, M.A., Jr. Biomechanical activation of vascular endothelium as a determinant of its functional phenotype. Proc. Natl. Acad. Sci. USA 2001, 98, 4478–4485. [Google Scholar] [CrossRef] [Green Version]
- Haidekker, M.A.; L’Heureux, N.; Frangos, J.A. Fluid shear stress increases membrane fluidity in endothelial cells: A study with DCVJ fluorescence. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1401–H1406. [Google Scholar] [CrossRef]
- Butler, P.J.; Norwich, G.; Weinbaum, S.; Chien, S. Shear stress induces a time- and position-dependent increase in endothelial cell membrane fluidity. Am. J. Physiol. Cell Physiol. 2001, 280, C962–C969. [Google Scholar] [CrossRef]
- Sriram, K.; Laughlin, J.G.; Rangamani, P.; Tartakovsky, D.M. Shear-Induced Nitric Oxide Production by Endothelial Cells. Biophys. J. 2016, 111, 208–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frangos, J.A.; Eskin, S.G.; McIntire, L.V.; Ives, C.L. Flow effects on prostacyclin production by cultured human endothelial cells. Science 1985, 227, 1477–1479. [Google Scholar] [CrossRef] [PubMed]
- Joannides, R.; Haefeli, W.E.; Linder, L.; Richard, V.; Bakkali, E.H.; Thuillez, C.; Luscher, T.F. Nitric oxide is responsible for flow-dependent dilatation of human peripheral conduit arteries in vivo. Circulation 1995, 91, 1314–1319. [Google Scholar] [CrossRef]
- Loot, A.E.; Popp, R.; Fisslthaler, B.; Vriens, J.; Nilius, B.; Fleming, I. Role of cytochrome P450-dependent transient receptor potential V4 activation in flow-induced vasodilatation. Cardiovasc. Res. 2008, 80, 445–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vriens, J.; Owsianik, G.; Fisslthaler, B.; Suzuki, M.; Janssens, A.; Voets, T.; Morisseau, C.; Hammock, B.D.; Fleming, I.; Busse, R. Modulation of the Ca2 permeable cation channel TRPV4 by cytochrome P450 epoxygenases in vascular endothelium. Circ. Res. 2005, 97, 908–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Félétou, M.; Vanhoutte, P.M. Endothelium-derived hyperpolarizing factor: Where are we now? Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1215–1225. [Google Scholar] [CrossRef]
- Qin, X.; Tian, J.; Zhang, P.; Fan, Y.; Chen, L.; Guan, Y.; Fu, Y.; Zhu, Y.; Chien, S.; Wang, N. Laminar shear stress up-regulates the expression of stearoyl-CoA desaturase-1 in vascular endothelial cells. Cardiovas. Res. 2007, 74, 506–514. [Google Scholar] [CrossRef] [Green Version]
- Ntambi, J.M.; Miyazaki, M. Recent insights into stearoyl-CoA desaturase-1. Curr. Opin. Lipidol. 2003, 14, 255–261. [Google Scholar] [CrossRef]
- Ntambi, J.M.; Miyazaki, M. Regulation of stearoyl-CoA desaturases and role in metabolism. Prog. Lipid Res. 2004, 43, 91–104. [Google Scholar] [CrossRef]
- Carluccio, M.A.; Massaro, M.; Bonfrate, C.; Siculella, L.; Maffia, M.; Nicolardi, G.; Distante, A.; Storelli, C.; De Caterina, R. Oleic acid inhibits endothelial activation: A direct vascular antiatherogenic mechanism of a nutritional component in the mediterranean diet. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 220–228. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chen, B.P.; Lu, M.; Zhu, Y.; Stemerman, M.B.; Chien, S.; Shyy, J.Y. Shear stress activation of SREBP1 in endothelial cells is mediated by integrins. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 76–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhu, Y.; Rannou, F.; Lee, T.S.; Formentin, K.; Zeng, L.; Yuan, X.; Wang, N.; Chien, S.; Forman, B.M.; et al. Laminar flow activates peroxisome proliferator-activated receptor-gamma in vascular endothelial cells. Circulation 2004, 110, 1128–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Fang, Y.; Wu, D. Mechanical forces and metabolic changes cooperate to drive cellular memory and endothelial phenotypes. Curr. Top. Membr. 2021, 87, 199–253. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Lu, M.; Lin, T.Y.; Chen, Z.; Chen, G.; Wang, W.C.; Marin, T.; Shentu, T.P.; Wen, L.; Gongol, B.; et al. Sterol regulatory element binding protein 2 activation of NLRP3 inflammasome in endothelium mediates hemodynamic-induced atherosclerosis susceptibility. Circulation 2013, 128, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Mullick, A.E.; Soldau, K.; Kiosses, W.B.; Bell, T.A., 3rd; Tobias, P.S.; Curtiss, L.K. Increased endothelial expression of Toll-like receptor 2 at sites of disturbed blood flow exacerbates early atherogenic events. J. Exp. Med. 2008, 205, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.L.; Chang, Y.W.; Wu, J.Y.; Wu, M.L.; Wu, K.K.; Yet, S.F.; Kuo, C.C. TLR 2 induces vascular smooth muscle cell migration through cAMP response element-binding protein-mediated interleukin-6 production. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2751–2760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surapisitchat, J.; Hoefen, R.J.; Pi, X.; Yoshizumi, M.; Yan, C.; Berk, B.C. Fluid shear stress inhibits TNF-α activation of JNK but not ERK1/2 or p38 in human umbilical vein endothelial cells: Inhibitory crosstalk among MAPK family members. Proc. Natl. Acad. Sci. USA 2001, 98, 6476–6481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boon, R.A.; Leyen, T.A.; Fontijn, R.D.; Fledderus, J.O.; Baggen, J.M.; Volger, O.L.; van Nieuw Amerongen, G.P.; Horrevoets, A.J. KLF2-induced actin shear fibers control both alignment to flow and JNK signaling in vascular endothelium. Blood 2010, 115, 2533–2542. [Google Scholar] [CrossRef] [Green Version]
- Sweet, D.R.; Vasudevan, N.T.; Fan, L.; Booth, C.E.; Keerthy, K.S.; Liao, X.; Vinayachandran, V.; Takami, Y.; Tugal, D.; Sharma, N.; et al. Myeloid Krüppel-like factor 2 is a critical regulator of metabolic inflammation. Nat. Commun. 2020, 11, 5872. [Google Scholar] [CrossRef]
- Szmitko, P.E.; Wang, C.-H.; Weisel, R.D.; de Almeida, J.R.; Anderson, T.J.; Verma, S. New markers of inflammation and endothelial cell activation: Part I. Circulation 2003, 108, 1917–1923. [Google Scholar] [CrossRef]
- Nishizaki, Y.; Shimada, K.; Tani, S.; Ogawa, T.; Ando, J.; Takahashi, M.; Yamamoto, M.; Shinozaki, T.; Miyauchi, K.; Nagao, K. Significance of imbalance in the ratio of serum n-3 to n-6 polyunsaturated fatty acids in patients with acute coronary syndrome. Am. J. Cardiol. 2014, 113, 441–445. [Google Scholar] [CrossRef]
- Huang, P.L.; Huang, Z.; Mashimo, H.; Bloch, K.D.; Moskowitz, M.A.; Bevan, J.A.; Fishman, M.C. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 1995, 377, 239–242. [Google Scholar] [CrossRef] [PubMed]
- van Haperen, R.; de Waard, M.; van Deel, E.; Mees, B.; Kutryk, M.; van Aken, T.; Hamming, J.; Grosveld, F.; Duncker, D.J.; de Crom, R. Reduction of Blood Pressure, Plasma Cholesterol, and Atherosclerosis by Elevated Endothelial Nitric Oxide. J. Biol. Chem. 2002, 277, 48803–48807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.L.; Zhang, L.; Youker, K.; Zhang, M.X.; Wang, J.; LeMaire, S.A.; Coselli, J.S.; Shen, Y.H. Free fatty acids inhibit insulin signaling-stimulated endothelial nitric oxide synthase activation through upregulating PTEN or inhibiting Akt kinase. Diabetes 2006, 55, 2301–2310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.R.; Li, H.L.; Zhang, X.X. [Effects of free fatty acids on nitric oxide synthase activity and mRNA expression in endothelial cell of SD rat aorta]. Sichuan Da Xue Xue Bao Yi Xue Ban 2008, 39, 193–196. [Google Scholar] [PubMed]
- Steinberg, H.O.; Tarshoby, M.; Monestel, R.; Hook, G.; Cronin, J.; Johnson, A.; Bayazeed, B.; Baron, A.D. Elevated circulating free fatty acid levels impair endothelium-dependent vasodilation. J. Clin. Investig. 1997, 100, 1230–1239. [Google Scholar] [CrossRef]
- Steinberg, H.O.; Paradisi, G.; Hook, G.; Crowder, K.; Cronin, J.; Baron, A.D. Free fatty acid elevation impairs insulin-mediated vasodilation and nitric oxide production. Diabetes 2000, 49, 1231–1238. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; García-Cardeña, G.; Sessa, W.C. Palmitoylation of endothelial nitric oxide synthase is necessary for optimal stimulated release of nitric oxide: Implications for caveolae localization. Biochemistry 1996, 35, 13277–13281. [Google Scholar] [CrossRef]
- Connelly, L.; Jacobs, A.T.; Palacios-Callender, M.; Moncada, S.; Hobbs, A.J. Macrophage endothelial nitric-oxide synthase autoregulates cellular activation and pro-inflammatory protein expression. J. Biol. Chem. 2003, 278, 26480–26487. [Google Scholar] [CrossRef] [Green Version]
- Bucci, M.; Roviezzo, F.; Posadas, I.; Yu, J.; Parente, L.; Sessa, W.C.; Ignarro, L.J.; Cirino, G. Endothelial nitric oxide synthase activation is critical for vascular leakage during acute inflammation in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 904–908. [Google Scholar] [CrossRef] [Green Version]
- Fritzsche, C.; Schleicher, U.; Bogdan, C. Endothelial nitric oxide synthase limits the inflammatory response in mouse cutaneous leishmaniasis. Immunobiology 2010, 215, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Aktan, F. iNOS-mediated nitric oxide production and its regulation. Life Sci. 2004, 75, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Depre, C.; Havaux, X.; Renkin, J.; Vanoverschelde, J.L.J.; Wijns, W. Expression of inducible nitric oxide synthase in human coronary atherosclerotic plaque. Cardiovasc. Res. 1999, 41, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, J.N.; Subramanian, R.R.; Sundell, C.L.; Tracey, W.R.; Pollock, J.S.; Harrison, D.G.; Marsden, P.A. Expression of Multiple Isoforms of Nitric Oxide Synthase in Normal and Atherosclerotic Vessels. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2479–2488. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S. Nitric oxide: Physiology, pathophysiology and pharmacology. Pharmacol. Rev. 1991, 43, 109–142. [Google Scholar] [PubMed]
- Lamoke, F.; Mazzone, V.; Persichini, T.; Maraschi, A.; Harris, M.B.; Venema, R.C.; Colasanti, M.; Gliozzi, M.; Muscoli, C.; Bartoli, M. Amyloid β peptide-induced inhibition of endothelial nitric oxide production involves oxidative stress-mediated constitutive eNOS/HSP90 interaction and disruption of agonist-mediated Akt activation. J. Neuroinflamm. 2015, 12, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Salvemini, D.; Mollace, V.; Pistelli, A.; Anggård, E.; Vane, J. Cultured astrocytoma cells generate a nitric oxide-like factor from endogenous L-arginine and glyceryl trinitrate: Effect of E. coli lipopolysaccharide. Br. J. Pharmacol. 1992, 106, 931–936. [Google Scholar] [CrossRef] [Green Version]
- Jin, R.C.; Loscalzo, J. Vascular Nitric Oxide: Formation and Function. J. Blood Med. 2010, 2010, 147–162. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Lucke-Wold, B.P.; Li, X.; Logsdon, A.F.; Xu, L.-C.; Xu, S.; LaPenna, K.B.; Wang, H.; Talukder, M.A.H.; Siedlecki, C.A.; et al. Reduction of Endothelial Nitric Oxide Increases the Adhesiveness of Constitutive Endothelial Membrane ICAM-1 through Src-Mediated Phosphorylation. Front. Physiol. 2018, 8, 1124. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, M.; Konopinski, R.; Krishnan, M.; Roman, L.; Bera, A.; Hongying, Z.; Habib, S.L.; Mohan, S. Inhibitor-κB kinase attenuates Hsp90-dependent endothelial nitric oxide synthase function in vascular endothelial cells. Am. J. Physiol. Cell Physiol. 2015, 308, C673–C683. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, K.A., Jr.; Ackerman, A.W.; Gross, E.R.; Stepp, D.W.; Shi, Y.; Fontana, J.T.; Baker, J.E.; Sessa, W.C. Heat shock protein 90 mediates the balance of nitric oxide and superoxide anion from endothelial nitric-oxide synthase. J. Biol. Chem. 2001, 276, 17621–17624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollace, R.; Gliozzi, M.; Tavernese, A.; Musolino, V.; Carresi, C.; Scicchitano, M.; Palma, E.; Nucera, S.; Bosco, F.; Scarano, F. Bergamot Polyphenolic Fraction supplementation improves metabolic balance, endothelial function and maximal oxygen uptake in athletes. J. Sports Med. 2018, 3, 53–61. [Google Scholar]
- Gliozzi, M.; Scicchitano, M.; Bosco, F.; Musolino, V.; Carresi, C.; Scarano, F.; Maiuolo, J.; Nucera, S.; Maretta, A.; Paone, S.; et al. Modulation of Nitric Oxide Synthases by Oxidized LDLs: Role in Vascular Inflammation and Atherosclerosis Development. Int. J. Mol. Sci. 2019, 20, 3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eligini, S.; Colli, S.; Habib, A.; Aldini, G.; Altomare, A.; Banfi, C. Cyclooxygenase-2 Glycosylation Is Affected by Peroxynitrite in Endothelial Cells: Impact on Enzyme Activity and Degradation. Antioxidants 2021, 10, 496. [Google Scholar] [CrossRef] [PubMed]
- Sadekuzzaman, M.; Stanley, D.; Kim, Y. Nitric Oxide Mediates Insect Cellular Immunity via Phospholipase A2 Activation. J. Innate Immun. 2018, 10, 70–81. [Google Scholar] [CrossRef]
- Foley, E.; O’Farrell, P.H. Nitric oxide contributes to induction of innate immune responses to gram-negative bacteria in Drosophila. Genes Dev. 2003, 17, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Stanley, D. Eicosanoid Signaling in Insect Immunology: New Genes and Unresolved Issues. Genes 2021, 12, 211. [Google Scholar] [CrossRef]
- Salvemini, D.; Misko, T.P.; Masferrer, J.L.; Seibert, K.; Currie, M.G.; Needleman, P. Nitric oxide activates cyclooxygenase enzymes. Proc. Natl. Acad. Sci. USA 1993, 90, 7240–7244. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.F. The role of nitric oxide in prostaglandin biology; update. Nitric Oxide 2011, 25, 255–264. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Borchert, G.L.; Phang, J.M. Polyoma Enhancer Activator 3, an Ets Transcription Factor, Mediates the Induction of Cyclooxygenase-2 by Nitric Oxide in Colorectal Cancer Cells. J. Biol. Chem. 2004, 279, 18694–18700. [Google Scholar] [CrossRef] [Green Version]
- Park, S.W.; Sung, M.W.; Heo, D.S.; Inoue, H.; Shim, S.H.; Kim, K.H. Nitric oxide upregulates the cyclooxygenase-2 expression through the cAMP-response element in its promoter in several cancer cell lines. Oncogene 2005, 24, 6689–6698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, Y.; Uno, E.; Sakuma, S. Effects of reactive oxygen and nitrogen species on cyclooxygenase-1 and -2 activities. Prostaglandins Leukot. Essent. Fat. Acids (PLEFA) 2004, 71, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Holowatz, L.A.; Kenney, W.L. Acute localized administration of tetrahydrobiopterin and chronic systemic atorvastatin treatment restore cutaneous microvascular function in hypercholesterolaemic humans. J. Physiol. 2011, 589, 4787–4797. [Google Scholar] [CrossRef] [PubMed]
- Maiuolo, J.; Gliozzi, M.; Musolino, V.; Carresi, C.; Nucera, S.; Macrì, R.; Scicchitano, M.; Bosco, F.; Scarano, F.; Ruga, S.; et al. The Role of Endothelial Dysfunction in Peripheral Blood Nerve Barrier: Molecular Mechanisms and Pathophysiological Implications. Int. J. Mol. Sci. 2019, 20, 3022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Çiftçi, G.A.; Ertorun, İ.; Akalin, A.; Alataş, İ.Ö.; Musmul, A. The effects of atorvastatin on antioxidant/antiinflammatory properties of HDLs in hypercholesterolemics. Turk. J. Med. Sci. 2015, 45, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.; Cioni, E.; Nuzzo, A.; Origliani, G.; Modena, M.G. Endothelial-dependent vasodilation and incidence of type 2 diabetes in a population of healthy postmenopausal women. Diabetes Care 2005, 28, 702–707. [Google Scholar] [CrossRef] [Green Version]
- O’Neal, W.T.; Efird, J.T.; Yeboah, J.; Nazarian, S.; Alonso, A.; Heckbert, S.R.; Soliman, E.Z. Brachial flow-mediated dilation and incident atrial fibrillation: The multi-ethnic study of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2717–2720. [Google Scholar] [CrossRef] [Green Version]
- Kenney, W.L.; Cannon, J.G.; Alexander, L.M. Cutaneous microvascular dysfunction correlates with serum LDL and sLOX-1 receptor concentrations. Microvasc. Res. 2013, 85, 112–117. [Google Scholar] [CrossRef] [Green Version]
- Schopfer, F.J.; Cipollina, C.; Freeman, B.A. Formation and signaling actions of electrophilic lipids. Chem. Rev. 2011, 111, 5997–6021. [Google Scholar] [CrossRef] [Green Version]
- Kansanen, E.; Kuosmanen, S.M.; Ruotsalainen, A.-K.; Hynynen, H.; Levonen, A.-L. Nitro-Oleic Acid Regulates Endothelin Signaling in Human Endothelial Cells. Mol. Pharmacol. 2017, 92, 481–490. [Google Scholar] [CrossRef]
- Khoo, N.K.H.; Rudolph, V.; Cole, M.P.; Golin-Bisello, F.; Schopfer, F.J.; Woodcock, S.R.; Batthyany, C.; Freeman, B.A. Activation of vascular endothelial nitric oxide synthase and heme oxygenase-1 expression by electrophilic nitro-fatty acids. Free Radic. Biol. Med. 2010, 48, 230–239. [Google Scholar] [CrossRef] [Green Version]
- Lima, E.S.; Di Mascio, P.; Abdalla, D.S.P. Cholesteryl nitrolinoleate, a nitrated lipid present in human blood plasma and lipoproteins. J. Lipid Res. 2003, 44, 1660–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, P.R.S.; Schopfer, F.J.; O’Donnell, V.B.; Freeman, B.A. Convergence of nitric oxide and lipid signaling: Anti-inflammatory nitro-fatty acids. Free Radic. Biol. Med. 2009, 46, 989–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, M.J.; McIntyre, T.M.; Prescott, S.M.; Zimmerman, G.A.; Whatley, R.E. Shear stress activates cytosolic phospholipase A2 (cPLA2) and MAP kinase in human endothelial cells. Biochem. Biophys. Res. Commun. 1996, 218, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V. Phospholipase A2 and signal transduction. J. Am. Soc. Nephrol. 1992, 3, 128–150. [Google Scholar] [CrossRef]
- Harizi, H.; Corcuff, J.B.; Gualde, N. Arachidonic-acid-derived eicosanoids: Roles in biology and immunopathology. Trends Mol. Med. 2008, 14, 461–469. [Google Scholar] [CrossRef]
- Moncada, S.; Higgs, E.A. Arachidonate metabolism in blood cells and the vessel wall. Clin. Haematol. 1986, 15, 273–292. [Google Scholar] [CrossRef]
- Van Diest, M.J.; Verbeuren, T.J.; Herman, A.G. 15-lipoxygenase metabolites of arachidonic acid evoke contractions and relaxations in isolated canine arteries: Role of thromboxane receptors, endothelial cells and cyclooxygenase. J. Pharmacol. Exp. Ther. 1991, 256, 194–203. [Google Scholar]
- Kuhn, H.; Banthiya, S.; van Leyen, K. Mammalian lipoxygenases and their biological relevance. Biochim. Biophys. Acta 2015, 1851, 308–330. [Google Scholar] [CrossRef] [Green Version]
- Horn, T.; Adel, S.; Schumann, R.; Sur, S.; Kakularam, K.R.; Polamarasetty, A.; Redanna, P.; Kuhn, H.; Heydeck, D. Evolutionary aspects of lipoxygenases and genetic diversity of human leukotriene signaling. Prog. Lipid Res. 2015, 57, 13–39. [Google Scholar] [CrossRef]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.J. Paracetamol (Acetaminophen): Mechanisms of action. Paediatr. Anaesth. 2008, 18, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Wlodawer, P.; Samuelsson, B. On the organization and mechanism of prostaglandin synthetase. J. Biol. Chem. 1973, 248, 5673–5678. [Google Scholar] [CrossRef]
- Smith, W.L.; DeWitt, D.L.; Garavito, R.M. Cyclooxygenases: Structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000, 69, 145–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FitzGerald, G.A.; Patrono, C. The coxibs, selective inhibitors of cyclooxygenase-2. N. Engl. J. Med. 2001, 345, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Dozier, B.L.; Watanabe, K.; Duffy, D.M. Two pathways for prostaglandin F2 alpha synthesis by the primate periovulatory follicle. Reproduction 2008, 136, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Belton, O.; Byrne, D.; Kearney, D.; Leahy, A.; Fitzgerald, D.J. Cyclooxygenase-1 and -2—Dependent Prostacyclin Formation in Patients with Atherosclerosis. Circulation 2000, 102, 840–845. [Google Scholar] [CrossRef] [Green Version]
- Dorris, S.L.; Peebles, R.S., Jr. PGI2 as a regulator of inflammatory diseases. Mediat. Inflamm. 2012, 2012, 926968. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, G.A. Coxibs and cardiovascular disease. N. Engl. J. Med. 2004, 351, 1709–1711. [Google Scholar] [CrossRef]
- Mitchell, J.A.; Kirkby, N.S. Eicosanoids, prostacyclin and cyclooxygenase in the cardiovascular system. Br. J. Pharmacol. 2019, 176, 1038–1050. [Google Scholar] [CrossRef]
- Bouros, D.; Steiropoulos, P.; Trakada, G. Current pharmacological treatment of pulmonary arterial hypertension. Curr. Clin. Pharmacol. 2008, 3, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Tahara, Y.; Matsumoto, M.; Iguchi, M.; Sano, H.; Murayama, T.; Arai, H.; Oida, H.; Yurugi-Kobayashi, T.; Yamashita, J.K.; et al. Roles of thromboxane A(2) and prostacyclin in the development of atherosclerosis in apoE-deficient mice. J. Clin. Investig. 2004, 114, 784–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouaziz, A.; de Ficquelmont-Loïzos, M.M.; Richert, A.; Caprani, A. Direct Physical Factors and PGI2 and TXA2 Secretions by a Human Endothelial Cell Line: In Vitro Investigation of Pressure and Shear Stress Applied Independently or in Synergy. Thromb. Res. 1998, 90, 279–289. [Google Scholar] [CrossRef]
- Morita, I. Distinct functions of COX-1 and COX-2. Prostaglandins Other Lipid Mediat. 2002, 68–69, 165–175. [Google Scholar] [CrossRef]
- Kis, B.; Snipes, J.A.; Simandle, S.A.; Busija, D.W. Acetaminophen-sensitive prostaglandin production in rat cerebral endothelial cells. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2005, 288, R897–R902. [Google Scholar] [CrossRef]
- Schwab, J.M.; Schluesener, H.J.; Meyermann, R.; Serhan, C.N. COX-3 the enzyme and the concept: Steps towards highly specialized pathways and precision therapeutics? Prostaglandins Leukot. Essent. Fat. Acids 2003, 69, 339–343. [Google Scholar] [CrossRef]
- Bishop-Bailey, D.; Pepper, J.R.; Larkin, S.W.; Mitchell, J.A. Differential induction of cyclooxygenase-2 in human arterial and venous smooth muscle: Role of endogenous prostanoids. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1655–1661. [Google Scholar] [CrossRef] [Green Version]
- Bishop-Bailey, D.; Larkin, S.W.; Warner, T.D.; Chen, G.; Mitchell, J.A. Characterization of the induction of nitric oxide synthase and cyclo-oxygenase in rat aorta in organ culture. Br. J. Pharmacol. 1997, 121, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Gomez, I.; Foudi, N.; Longrois, D.; Norel, X. The role of prostaglandin E2 in human vascular inflammation. Prostaglandins Leukot. Essent. Fat. Acids 2013, 89, 55–63. [Google Scholar] [CrossRef]
- Schönbeck, U.; Sukhova, G.K.; Graber, P.; Coulter, S.; Libby, P. Augmented expression of cyclooxygenase-2 in human atherosclerotic lesions. Am. J. Pathol. 1999, 155, 1281–1291. [Google Scholar] [CrossRef] [Green Version]
- Nasrallah, R.; Zimpelmann, J.; Robertson, S.J.; Ghossein, J.; Thibodeau, J.-F.; Kennedy, C.R.J.; Gutsol, A.; Xiao, F.; Burger, D.; Burns, K.D.; et al. Prostaglandin E2 receptor EP1 (PGE2/EP1) deletion promotes glomerular podocyte and endothelial cell injury in hypertensive TTRhRen mice. Lab. Investig. 2020, 100, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Breyer, R.M.; Bagdassarian, C.K.; Myers, S.A.; Breyer, M.D. Prostanoid receptors: Subtypes and signaling. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 661–690. [Google Scholar] [CrossRef] [PubMed]
- Rutkai, I.; Feher, A.; Erdei, N.; Henrion, D.; Papp, Z.; Edes, I.; Koller, A.; Kaley, G.; Bagi, Z. Activation of prostaglandin E2 EP1 receptor increases arteriolar tone and blood pressure in mice with type 2 diabetes. Cardiovasc. Res. 2009, 83, 148–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Y.; Zhang, Y.; Wu, J.; Qi, Z.; Yang, G.; Dou, D.; Gao, Y.; Chen, L.; Zhang, X.; Davis, L.S.; et al. Antihypertensive effects of selective prostaglandin E2 receptor subtype 1 targeting. J. Clin. Investig. 2007, 117, 2496–2505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suganami, T.; Mori, K.; Tanaka, I.; Mukoyama, M.; Sugawara, A.; Makino, H.; Muro, S.; Yahata, K.; Ohuchida, S.; Maruyama, T.; et al. Role of prostaglandin E receptor EP1 subtype in the development of renal injury in genetically hypertensive rats. Hypertension 2003, 42, 1183–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef]
- Aoki, T.; Narumiya, S. Prostaglandins and chronic inflammation. Trends Pharmacol. Sci. 2012, 33, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Yang, G.; Grosser, T. Prostanoids and inflammatory pain. Prostaglandins Other Lipid Mediat. 2013, 104, 58–66. [Google Scholar] [CrossRef]
- Linton, M.F.; Fazio, S. Cyclooxygenase-2 and inflammation in atherosclerosis. Curr. Opin. Pharmacol. 2004, 4, 116–123. [Google Scholar] [CrossRef]
- Arnett, D.K.; McClelland, R.L.; Bank, A.; Bluemke, D.A.; Cushman, M.; Szalai, A.J.; Jain, N.; Gomes, A.S.; Heckbert, S.R.; Hundley, W.G. Biomarkers of inflammation and hemostasis associated with left ventricular mass: The Multiethnic Study of Atherosclerosis (MESA). Int. J. Mol. Epidemiol. Genet. 2011, 2, 391. [Google Scholar]
- Raval, M.; Frank, P.G.; Laury-Kleintop, L.; Yan, G.; Lanza-Jacoby, S. Celecoxib combined with atorvastatin prevents progression of atherosclerosis. J. Surg. Res. 2010, 163, e113–e122. [Google Scholar] [CrossRef] [PubMed]
- Schober, L.J.; Khandoga, A.L.; Dwivedi, S.; Penz, S.M.; Maruyama, T.; Brandl, R.; Siess, W. The role of PGE(2) in human atherosclerotic plaque on platelet EP(3) and EP(4) receptor activation and platelet function in whole blood. J. Thromb. Thrombolysis 2011, 32, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Narumiya, S.; Sugimoto, Y.; Ushikubi, F. Prostanoid Receptors: Structures, Properties, and Functions. Physiol. Rev. 1999, 79, 1193–1226. [Google Scholar] [CrossRef] [PubMed]
- Di Francesco, L.; Totani, L.; Dovizio, M.; Piccoli, A.; Di Francesco, A.; Salvatore, T.; Pandolfi, A.; Evangelista, V.; Dercho, R.A.; Seta, F.; et al. Induction of prostacyclin by steady laminar shear stress suppresses tumor necrosis factor-alpha biosynthesis via heme oxygenase-1 in human endothelial cells. Circ. Res. 2009, 104, 506–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Gong, Y.; Yu, Y. PG F2α Receptor: A Promising Therapeutic Target for Cardiovascular Disease. Front. Pharmacol. 2010, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Lucitt, M.B.; Stubbe, J.; Cheng, Y.; Friis, U.G.; Hansen, P.B.; Jensen, B.L.; Smyth, E.M.; FitzGerald, G.A. Prostaglandin F2α elevates blood pressure and promotes atherosclerosis. Proc. Natl. Acad. Sci. USA 2009, 106, 7985–7990. [Google Scholar] [CrossRef] [Green Version]
- Clarke, R.J.; Mayo, G.; Price, P.; FitzGerald, G.A. Suppression of thromboxane A2 but not of systemic prostacyclin by controlled-release aspirin. N. Engl. J. Med. 1991, 325, 1137–1141. [Google Scholar] [CrossRef]
- Fu, J.Y.; Masferrer, J.L.; Seibert, K.; Raz, A.; Needleman, P. The induction and suppression of prostaglandin H2 synthase (cyclooxygenase) in human monocytes. J. Biol. Chem. 1990, 265, 16737–16740. [Google Scholar] [CrossRef]
- FitzGerald, G.A.; Pedersen, A.K.; Patrono, C. Analysis of prostacyclin and thromboxane biosynthesis in cardiovascular disease. Circulation 1983, 67, 1174–1177. [Google Scholar] [CrossRef] [Green Version]
- Hamberg, M.; Svensson, J.; Samuelsson, B. Thromboxanes: A new group of biologically active compounds derived from prostaglandin endoperoxides. Proc. Natl. Acad. Sci. USA 1975, 72, 2994–2998. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Liu, P.; Zhou, X.; Li, M.T.; Li, F.L.; Wang, Z.; Meng, Z.; Sun, Y.P.; Yu, Y.; Xiong, Y.; et al. Thromboxane A2 Activates YAP/TAZ Protein to Induce Vascular Smooth Muscle Cell Proliferation and Migration. J. Biol. Chem. 2016, 291, 18947–18958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, K.M.; Wang, M.; Lucitt, M.B.; Zukas, A.M.; Puré, E.; Lawson, J.A.; FitzGerald, G.A. Cyclooxygenases, Thromboxane, and Atherosclerosis. Circulation 2005, 111, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Hanasaki, K.; Nakano, T.; Arita, H. Receptor-mediated mitogenic effect of thromboxane A2 in vascular smooth muscle cells. Biochem. Pharmacol. 1990, 40, 2535–2542. [Google Scholar] [CrossRef]
- Praticò, D.; Iuliano, L.; Mauriello, A.; Spagnoli, L.; Lawson, J.A.; Rokach, J.; Maclouf, J.; Violi, F.; FitzGerald, G.A. Localization of distinct F2-isoprostanes in human atherosclerotic lesions. J. Clin. Investig. 1997, 100, 2028–2034. [Google Scholar] [CrossRef] [PubMed]
- Davi, G.; Alessandrini, P.; Mezzetti, A.; Minotti, G.; Bucciarelli, T.; Costantini, F.; Cipollone, F.; Bon, G.B.; Ciabattoni, G.; Patrono, C. In vivo formation of 8-Epi-prostaglandin F2 alpha is increased in hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3230–3235. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.S.; Roberts, L.J., 2nd. F2-isoprostanes as an indicator and risk factor for coronary heart disease. Free Radic. Biol. Med. 2011, 50, 559–566. [Google Scholar] [CrossRef] [Green Version]
- Morrow, J.D.; Hill, K.E.; Burk, R.F.; Nammour, T.M.; Badr, K.F.; Roberts, L.J., 2nd. A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc. Natl. Acad. Sci. USA 1990, 87, 9383–9387. [Google Scholar] [CrossRef] [Green Version]
- Kadiiska, M.B.; Gladen, B.C.; Baird, D.D.; Germolec, D.; Graham, L.B.; Parker, C.E.; Nyska, A.; Wachsman, J.T.; Ames, B.N.; Basu, S.; et al. Biomarkers of oxidative stress study II: Are oxidation products of lipids, proteins, and DNA markers of CCl4 poisoning? Free Radic. Biol. Med. 2005, 38, 698–710. [Google Scholar] [CrossRef]
- Milne, G.L.; Musiek, E.S.; Morrow, J.D. F2-isoprostanes as markers of oxidative stress in vivo: An overview. Biomarkers 2005, 10 (Suppl. 1), S10–S23. [Google Scholar] [CrossRef]
- Janicka, M.; Kot-Wasik, A.; Kot, J.; Namieśnik, J. Isoprostanes-biomarkers of lipid peroxidation: Their utility in evaluating oxidative stress and analysis. Int. J. Mol. Sci. 2010, 11, 4631–4659. [Google Scholar] [CrossRef] [Green Version]
- van ‘t Erve, T.J.; Kadiiska, M.B.; London, S.J.; Mason, R.P. Classifying oxidative stress by F(2)-isoprostane levels across human diseases: A meta-analysis. Redox Biol. 2017, 12, 582–599. [Google Scholar] [CrossRef] [PubMed]
- Castro-Diehl, C.; Ehrbar, R.; Obas, V.; Oh, A.; Vasan, R.S.; Xanthakis, V. Biomarkers representing key aging-related biological pathways are associated with subclinical atherosclerosis and all-cause mortality: The Framingham Study. PLoS ONE 2021, 16, e0251308. [Google Scholar] [CrossRef] [PubMed]
- Biernacki, W.A.; Kharitonov, S.A.; Barnes, P.J. Increased leukotriene B4 and 8-isoprostane in exhaled breath condensate of patients with exacerbations of COPD. Thorax 2003, 58, 294–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antczak, A.; Montuschi, P.; Kharitonov, S.; Gorski, P.; Barnes, P.J. Increased exhaled cysteinyl-leukotrienes and 8-isoprostane in aspirin-induced asthma. Am. J. Respir. Crit. Care Med. 2002, 166, 301–306. [Google Scholar] [CrossRef]
- Baraldi, E.; Ghiro, L.; Piovan, V.; Carraro, S.; Ciabattoni, G.; Barnes, P.J.; Montuschi, P. Increased exhaled 8-isoprostane in childhood asthma. Chest 2003, 124, 25–31. [Google Scholar] [CrossRef]
- Baraldi, E.; Carraro, S.; Alinovi, R.; Pesci, A.; Ghiro, L.; Bodini, A.; Piacentini, G.; Zacchello, F.; Zanconato, S. Cysteinyl leukotrienes and 8-isoprostane in exhaled breath condensate of children with asthma exacerbations. Thorax 2003, 58, 505–509. [Google Scholar] [CrossRef] [Green Version]
- Zanconato, S.; Carraro, S.; Corradi, M.; Alinovi, R.; Pasquale, M.F.; Piacentini, G.; Zacchello, F.; Baraldi, E. Leukotrienes and 8-isoprostane in exhaled breath condensate of children with stable and unstable asthma. J. Allergy Clin. Immunol. 2004, 113, 257–263. [Google Scholar] [CrossRef]
- Montuschi, P.; Ciabattoni, G.; Paredi, P.; Pantelidis, P.; du Bois, R.M.; Kharitonov, S.A.; Barnes, P.J. 8-Isoprostane as a biomarker of oxidative stress in interstitial lung diseases. Am. J. Respir. Crit. Care Med. 1998, 158, 1524–1527. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, C.T.; Price, P.V.; Christman, B.W. Exhaled breath condensate isoprostanes are elevated in patients with acute lung injury or ARDS. Chest 1998, 114, 1653–1659. [Google Scholar] [CrossRef] [Green Version]
- Psathakis, K.; Papatheodorou, G.; Plataki, M.; Panagou, P.; Loukides, S.; Siafakas, N.M.; Bouros, D. 8-Isoprostane, a marker of oxidative stress, is increased in the expired breath condensate of patients with pulmonary sarcoidosis. Chest 2004, 125, 1005–1011. [Google Scholar] [CrossRef] [Green Version]
- Carpagnano, G.E.; Kharitonov, S.A.; Resta, O.; Foschino-Barbaro, M.P.; Gramiccioni, E.; Barnes, P.J. Increased 8-isoprostane and interleukin-6 in breath condensate of obstructive sleep apnea patients. Chest 2002, 122, 1162–1167. [Google Scholar] [CrossRef] [PubMed]
- Bunting, S.; Gryglewski, R.; Moncada, S.; Vane, J.R. Arterial walls generate from prostaglandin endoperoxides a substance (prostaglandin X) which relaxes strips of mesenteric and coeliac ateries and inhibits platelet aggregation. Prostaglandins 1976, 12, 897–913. [Google Scholar] [CrossRef]
- Miyata, N.; Roman, R.J. Role of 20-hydroxyeicosatetraenoic acid (20-HETE) in vascular system. J. Smooth Muscle Res. 2005, 41, 175–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roman, R.J. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol. Rev. 2002, 82, 131–185. [Google Scholar] [CrossRef] [Green Version]
- Mollace, V.; Muscoli, C.; Masini, E.; Cuzzocrea, S.; Salvemini, D. Modulation of prostaglandin biosynthesis by nitric oxide and nitric oxide donors. Pharmacol. Rev. 2005, 57, 217–252. [Google Scholar] [CrossRef] [Green Version]
- Cuzzocrea, S.; Salvemini, D. Molecular mechanisms involved in the reciprocal regulation of cyclooxygenase and nitric oxide synthase enzymes. Kidney Int. 2007, 71, 290–297. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, I.; Kuhn, H.; Heydeck, D. Structural and functional biology of arachidonic acid 15-lipoxygenase-1 (ALOX15). Gene 2015, 573, 1–32. [Google Scholar] [CrossRef]
- Ivanov, I.; Heydeck, D.; Hofheinz, K.; Roffeis, J.; O’Donnell, V.B.; Kuhn, H.; Walther, M. Molecular enzymology of lipoxygenases. Arch. Biochem. Biophys. 2010, 503, 161–174. [Google Scholar] [CrossRef]
- Morgan, E.L.; Maskrey, B.H.; Rowley, A.F. At what stage in metazoan evolution did leukotriene generation first appear?—Key insights from cartilaginous fish. Dev. Comp. Immunol. 2005, 29, 53–59. [Google Scholar] [CrossRef]
- Spanbroek, R.; Gräbner, R.; Lötzer, K.; Hildner, M.; Urbach, A.; Rühling, K.; Moos, M.P.W.; Kaiser, B.; Cohnert, T.U.; Wahlers, T.; et al. Expanding expression of the 5-lipoxygenase pathway within the arterial wall during human atherogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 1238. [Google Scholar] [CrossRef] [Green Version]
- Subbarao, K.; Jala, V.R.; Mathis, S.; Suttles, J.; Zacharias, W.; Ahamed, J.; Ali, H.; Tseng, M.T.; Haribabu, B. Role of leukotriene B4 receptors in the development of atherosclerosis: Potential mechanisms. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 369–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Borne, P.; van der Laan, S.W.; Bovens, S.M.; Koole, D.; Kowala, M.C.; Michael, L.F.; Schoneveld, A.H.; van de Weg, S.M.; Velema, E.; de Vries, J.-P.; et al. Leukotriene B4 levels in human atherosclerotic plaques and abdominal aortic aneurysms. PLoS ONE 2014, 9, e86522. [Google Scholar] [CrossRef] [PubMed]
- Fredman, G.; Hellmann, J.; Proto, J.D.; Kuriakose, G.; Colas, R.A.; Dorweiler, B.; Connolly, E.S.; Solomon, R.; Jones, D.M.; Heyer, E.J.; et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat. Commun. 2016, 7, 12859. [Google Scholar] [CrossRef] [PubMed]
- Snodgrass, R.G.; Brüne, B. Regulation and Functions of 15-Lipoxygenases in Human Macrophages. Front. Pharmacol. 2019, 10, 719. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Ran, Y. Role of 15-lipoxygenase/15-hydroxyeicosatetraenoic acid in hypoxia-induced pulmonary hypertension. J. Physiol. Sci. 2012, 62, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.C.; Makheja, A.N.; Bailey, J.M. Formation of 15-hydroxyeicosatetraenoic acid (15-HETE) as the predominant eicosanoid in aortas from Watanabe Heritable Hyperlipidemic and cholesterol-fed rabbits. Atherosclerosis 1989, 75, 31–38. [Google Scholar] [CrossRef]
- Wittwer, J.; Hersberger, M. The two faces of the 15-lipoxygenase in atherosclerosis. Prostaglandins Leukot. Essent. Fat. Acids 2007, 77, 67–77. [Google Scholar] [CrossRef]
- Hutchins, P.M.; Murphy, R.C. Cholesteryl ester acyl oxidation and remodeling in murine macrophages: Formation of oxidized phosphatidylcholine. J. Lipid Res. 2012, 53, 1588–1597. [Google Scholar] [CrossRef] [Green Version]
- Bender, G.; Schexnaydre, E.E.; Murphy, R.C.; Uhlson, C.; Newcomer, M.E. Membrane-dependent Activities of Human 15-LOX-2 and Its Murine Counterpart: Implications for Murine Models of Atherosclerosis. J. Biol. Chem. 2016, 291, 19413–19424. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Sodhi, K.; Puri, N.; Gotlinger, K.H.; Cao, J.; Rezzani, R.; Falck, J.R.; Abraham, N.G.; Laniado-Schwartzman, M. Endothelial-specific CYP4A2 overexpression leads to renal injury and hypertension via increased production of 20-HETE. Am. J. Physiol. Renal Physiol. 2009, 297, F875–F884. [Google Scholar] [CrossRef]
- Cheng, J.; Ou, J.S.; Singh, H.; Falck, J.R.; Narsimhaswamy, D.; Pritchard, K.A., Jr.; Schwartzman, M.L. 20-hydroxyeicosatetraenoic acid causes endothelial dysfunction via eNOS uncoupling. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1018–H1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.S.; Singh, H.; Zhang, F.; Ishizuka, T.; Deng, H.; Kemp, R.; Wolin, M.S.; Hintze, T.H.; Abraham, N.G.; Nasjletti, A.; et al. Endothelial dysfunction and hypertension in rats transduced with CYP4A2 adenovirus. Circ. Res. 2006, 98, 962–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuez, T.; Korkmaz, B.; Buharalioglu, C.K.; Sahan-Firat, S.; Falck, J.; Malik, K.U.; Tunctan, B. A synthetic analogue of 20-HETE, 5,14-HEDGE, reverses endotoxin-induced hypotension via increased 20-HETE levels associated with decreased iNOS protein expression and vasodilator prostanoid production in rats. Basic Clin. Pharmacol. Toxicol. 2010, 106, 378–388. [Google Scholar] [CrossRef] [Green Version]
- Ishizuka, T.; Cheng, J.; Singh, H.; Vitto, M.D.; Manthati, V.L.; Falck, J.R.; Laniado-Schwartzman, M. 20-Hydroxyeicosatetraenoic acid stimulates nuclear factor-kappaB activation and the production of inflammatory cytokines in human endothelial cells. J. Pharmacol. Exp. Ther. 2008, 324, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Hoopes, S.L.; Garcia, V.; Edin, M.L.; Schwartzman, M.L.; Zeldin, D.C. Vascular actions of 20-HETE. Prostaglandins Other Lipid Mediat. 2015, 120, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Wu, C.C.; Gotlinger, K.H.; Zhang, F.; Falck, J.R.; Narsimhaswamy, D.; Schwartzman, M.L. 20-hydroxy-5,8,11,14-eicosatetraenoic acid mediates endothelial dysfunction via IkappaB kinase-dependent endothelial nitric-oxide synthase uncoupling. J. Pharmacol. Exp. Ther. 2010, 332, 57–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, V.; Gilani, A.; Shkolnik, B.; Pandey, V.; Zhang, F.F.; Dakarapu, R.; Gandham, S.K.; Reddy, N.R.; Graves, J.P.; Gruzdev, A.; et al. 20-HETE Signals through G-Protein-Coupled Receptor GPR75 (G(q)) to Affect Vascular Function and Trigger Hypertension. Circ. Res. 2017, 120, 1776–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascale, J.V.; Park, E.J.; Adebesin, A.M.; Falck, J.R.; Schwartzman, M.L.; Garcia, V. Uncovering the signalling, structure and function of the 20-HETE-GPR75 pairing: Identifying the chemokine CCL5 as a negative regulator of GPR75. Br. J. Pharmacol. 2021, 178, 3813–3828. [Google Scholar] [CrossRef]
- Gebremedhin, D.; Lange, A.R.; Narayanan, J.; Aebly, M.R.; Jacobs, E.R.; Harder, D.R. Cat cerebral arterial smooth muscle cells express cytochrome P450 4A2 enzyme and produce the vasoconstrictor 20-HETE which enhances L-type Ca2+ current. J. Physiol. 1998, 507 Pt 3, 771–781. [Google Scholar] [CrossRef]
- Hill, E.; Murphy, R.C. Quantitation of 20-hydroxy-5,8,11,14-eicosatetraenoic acid (20-HETE) produced by human polymorphonuclear leukocytes using electron capture ionization gas chromatography/mass spectrometry. Biol. Mass Spectrom. 1992, 21, 249–253. [Google Scholar] [CrossRef]
- Tsai, I.J.; Croft, K.D.; Puddey, I.B.; Beilin, L.J.; Barden, A. 20-Hydroxyeicosatetraenoic acid synthesis is increased in human neutrophils and platelets by angiotensin II and endothelin-1. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1194–H1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, J.; Chen, C. The Role of Epoxyeicosatrienoic Acids in Cardiac Remodeling. Front. Physiol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Mäki-Petäjä, K.; Cheriyan, J.; McEniery, C.; Wilkinson, I.B. The role of epoxyeicosatrienoic acids in the cardiovascular system. Br. J. Clin. Pharmacol. 2015, 80, 28–44. [Google Scholar] [CrossRef] [Green Version]
- Rand, A.A.; Rajamani, A.; Kodani, S.D.; Harris, T.R.; Schlatt, L.; Barnych, B.; Passerini, A.G.; Hammock, B.D. Epoxyeicosatrienoic acid (EET)-stimulated angiogenesis is mediated by epoxy hydroxyeicosatrienoic acids (EHETs) formed from COX-2. J. Lipid Res. 2019, 60, 1996–2005. [Google Scholar] [CrossRef]
- Sahebkar, A.; Simental-Mendía, L.E.; Pedone, C.; Ferretti, G.; Nachtigal, P.; Bo, S.; Derosa, G.; Maffioli, P.; Watts, G.F. Statin therapy and plasma free fatty acids: A systematic review and meta-analysis of controlled clinical trials. Br. J. Clin. Pharmacol. 2016, 81, 807–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciucanu, C.I.; Olariu, S.; Vlad, D.C.; Dumitraşcu, V. Effect of rosuvastatin on the concentration of each fatty acid in the fraction of free fatty acids and total lipids in human plasma: The role of cholesterol homeostasis. Biochem. Biophys. Rep. 2020, 24, 100822. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Essential fatty acids and their metabolites could function as endogenous HMG-CoA reductase and ACE enzyme inhibitors, anti-arrhythmic, anti-hypertensive, anti-atherosclerotic, anti-inflammatory, cytoprotective, and cardioprotective molecules. Lipids Health Dis. 2008, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- Das, U.N. Essential fatty acids: Biochemistry, physiology and pathology. Biotechnol. J. 2006, 1, 420–439. [Google Scholar] [CrossRef] [PubMed]
- Das, U. Essential fatty acids as possible mediators of the actions of statins. Prostaglandins Leukot. Essent. Fat. Acids (PLEFA) 2001, 65, 37–40. [Google Scholar] [CrossRef]
- Dobrucki, L.W.; Kalinowski, L.; Dobrucki, I.T.; Malinski, T. Statin-stimulated nitric oxide release from endothelium. Med. Sci. Monit. 2001, 7, 622–627. [Google Scholar]
- McGown, C.C.; Brookes, Z.L. Beneficial effects of statins on the microcirculation during sepsis: The role of nitric oxide. Br. J. Anaesth. 2007, 98, 163–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuda, Y.; Kawashima, K.; Sawada, T.; Tsurumaru, K.; Asano, M.; Suzuki, S.; Soma, M.; Nakajima, T.; Yamashita, K. Eicosapentaenoic acid enhances nitric oxide production by cultured human endothelial cells. Biochem. Biophys. Res. Commun. 1997, 232, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Azekoshi, Y.; Yasu, T.; Watanabe, S.; Tagawa, T.; Abe, S.; Yamakawa, K.; Uehara, Y.; Momomura, S.; Urata, H.; Ueda, S. Free Fatty Acid Causes Leukocyte Activation and Resultant Endothelial Dysfunction through Enhanced Angiotensin II Production in Mononuclear and Polymorphonuclear Cells. Hypertension 2010, 56, 136–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, S.; Tagawa, T.; Yamakawa, K.; Shimabukuro, M.; Ueda, S. Inhibition of the renin-angiotensin system prevents free fatty acid-induced acute endothelial dysfunction in humans. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2376–2380. [Google Scholar] [CrossRef] [Green Version]
- Das, U.N. Long-chain polyunsaturated fatty acids interact with nitric oxide, superoxide anion, and transforming growth factor-β to prevent human essential hypertension. Eur. J. Clin. Nutr. 2004, 58, 195–203. [Google Scholar] [CrossRef] [Green Version]
- Petri, M.H.; Laguna-Fernandez, A.; Arnardottir, H.; Wheelock, C.E.; Perretti, M.; Hansson, G.K.; Bäck, M. Aspirin-triggered lipoxin A4 inhibits atherosclerosis progression in apolipoprotein E(-/-) mice. Br. J. Pharmacol. 2017, 174, 4043–4054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, K.J.; Spite, M.; Owens, C.D.; Lancero, H.; Kroemer, A.H.K.; Pande, R.; Creager, M.A.; Serhan, C.N.; Conte, M.S. Aspirin-Triggered Lipoxin and Resolvin E1 Modulate Vascular Smooth Muscle Phenotype and Correlate with Peripheral Atherosclerosis. Am. J. Pathol. 2010, 177, 2116–2123. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotlyarov, S.; Kotlyarova, A. Involvement of Fatty Acids and Their Metabolites in the Development of Inflammation in Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 1308. https://doi.org/10.3390/ijms23031308
Kotlyarov S, Kotlyarova A. Involvement of Fatty Acids and Their Metabolites in the Development of Inflammation in Atherosclerosis. International Journal of Molecular Sciences. 2022; 23(3):1308. https://doi.org/10.3390/ijms23031308
Chicago/Turabian StyleKotlyarov, Stanislav, and Anna Kotlyarova. 2022. "Involvement of Fatty Acids and Their Metabolites in the Development of Inflammation in Atherosclerosis" International Journal of Molecular Sciences 23, no. 3: 1308. https://doi.org/10.3390/ijms23031308
APA StyleKotlyarov, S., & Kotlyarova, A. (2022). Involvement of Fatty Acids and Their Metabolites in the Development of Inflammation in Atherosclerosis. International Journal of Molecular Sciences, 23(3), 1308. https://doi.org/10.3390/ijms23031308