
Identification of New KRAS G12D Inhibitors through Computer-Aided Drug Discovery Methods


 ,
,  ,
,
Abstract
:1. Introduction
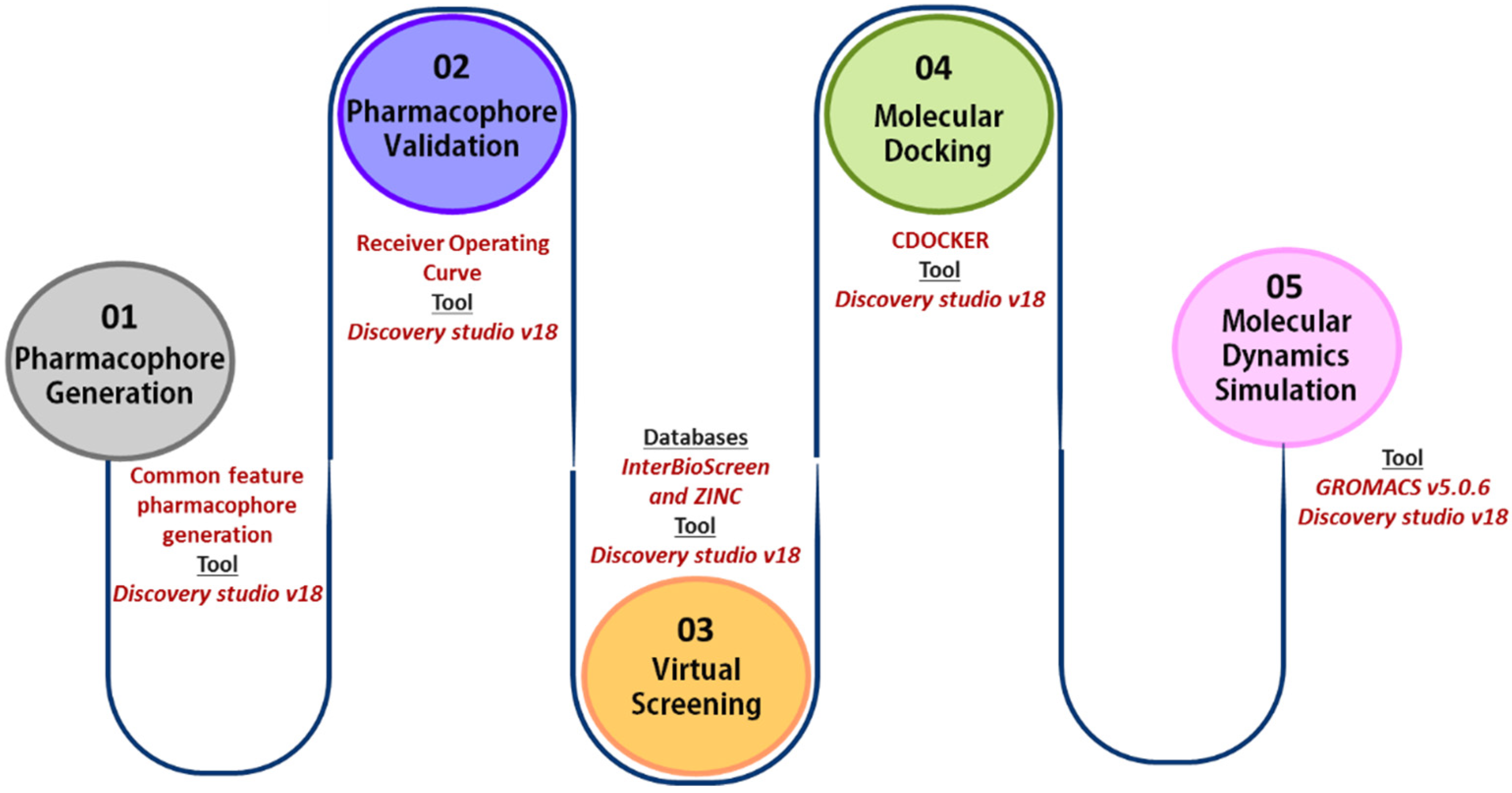
2. Results
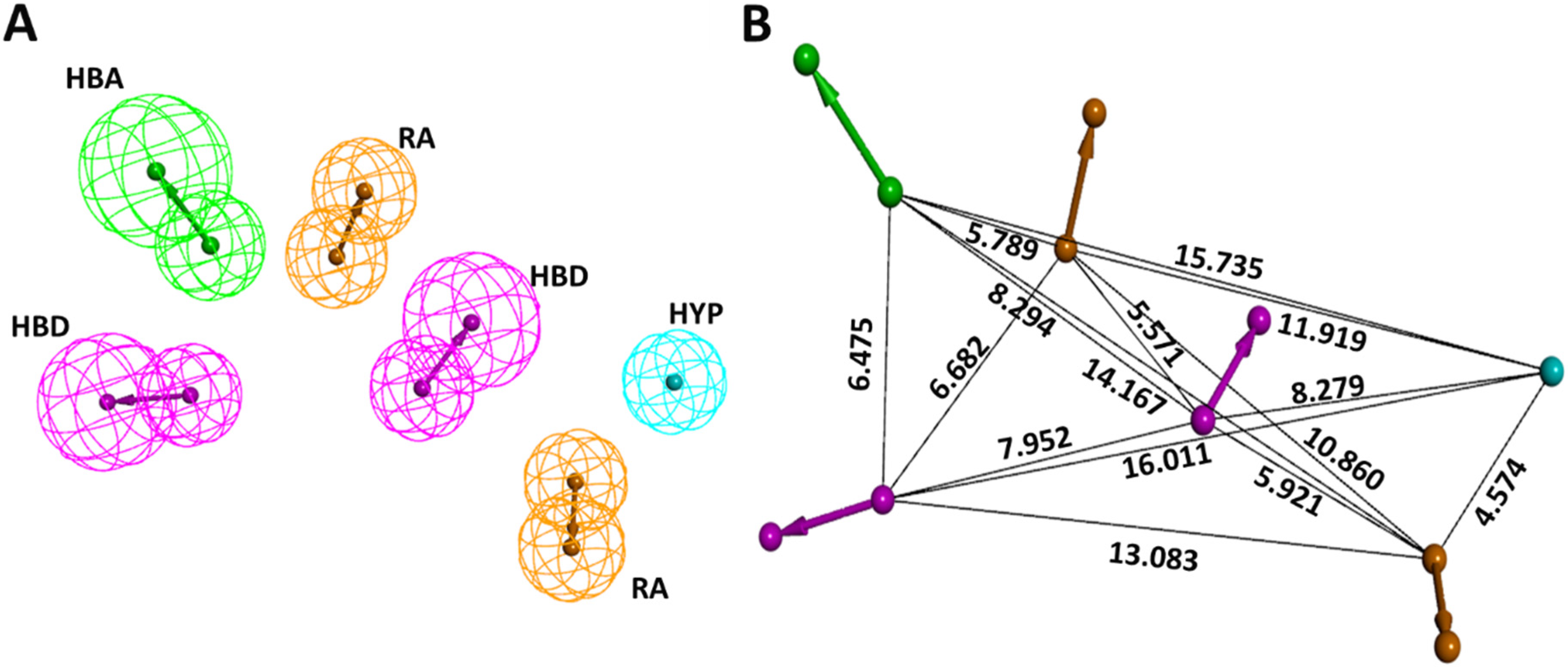
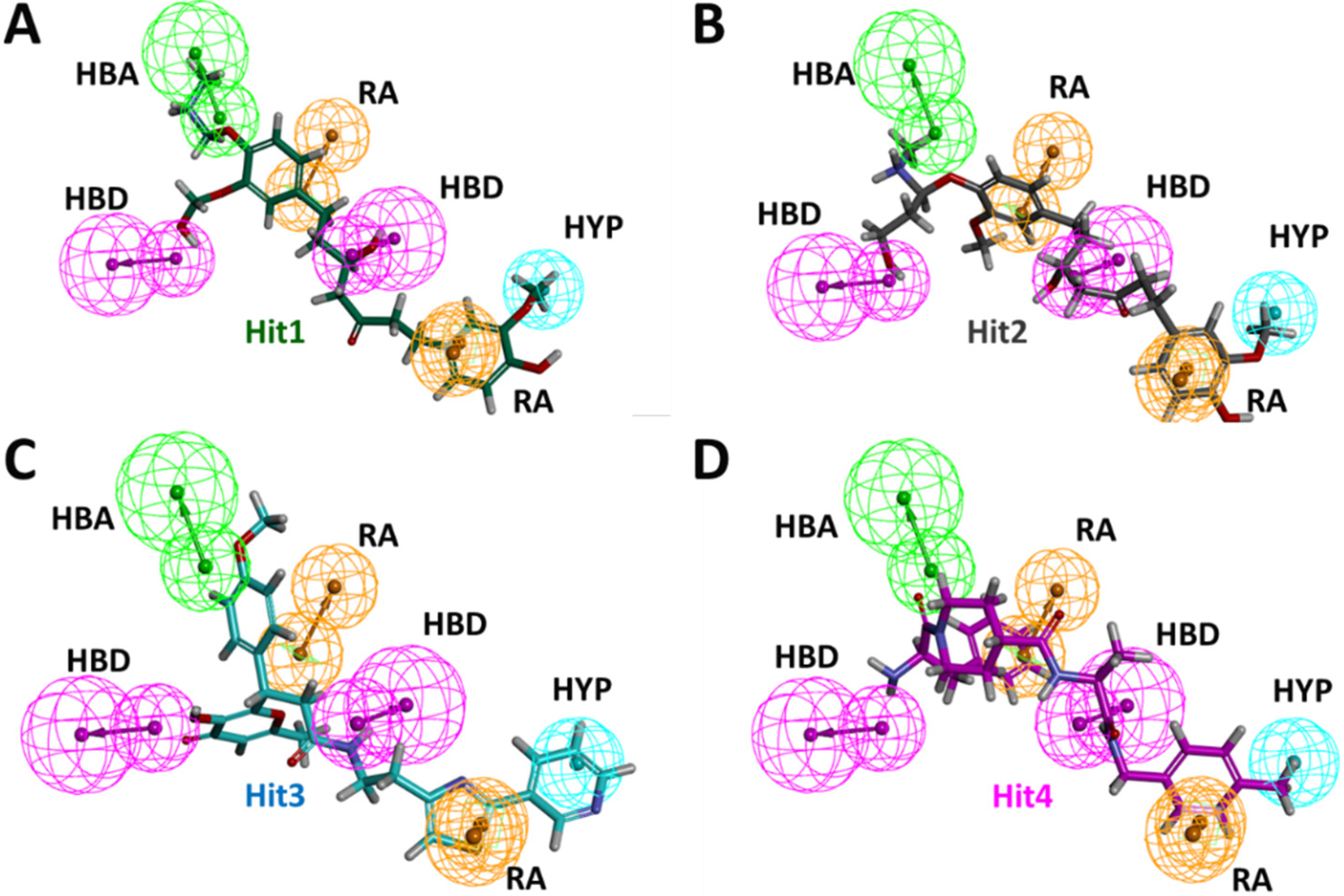
2.1. Common Feature Pharmacophore Generation
2.2. Pharmacophore Validation
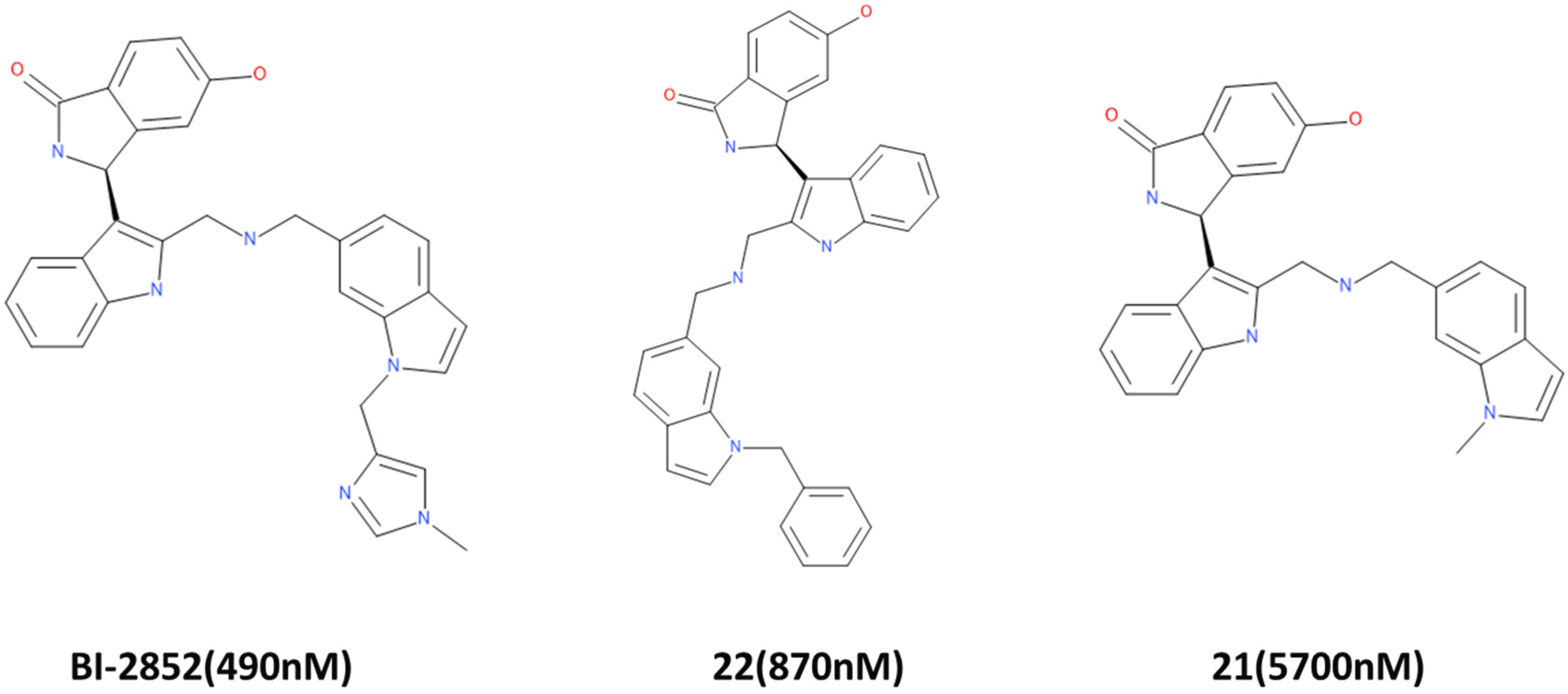
2.3. Virtual Screening
2.4. Molecular Docking
2.5. Molecular Dynamics Simulations
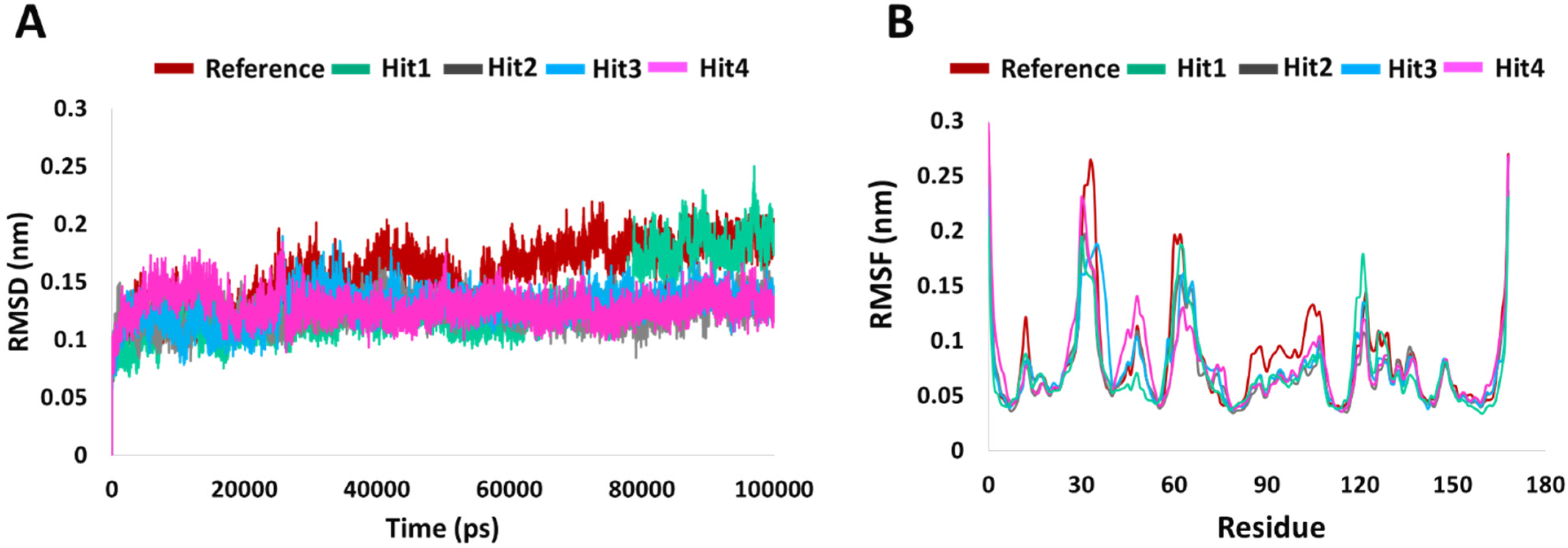
2.5.1. RMSD and RMSF Assessment
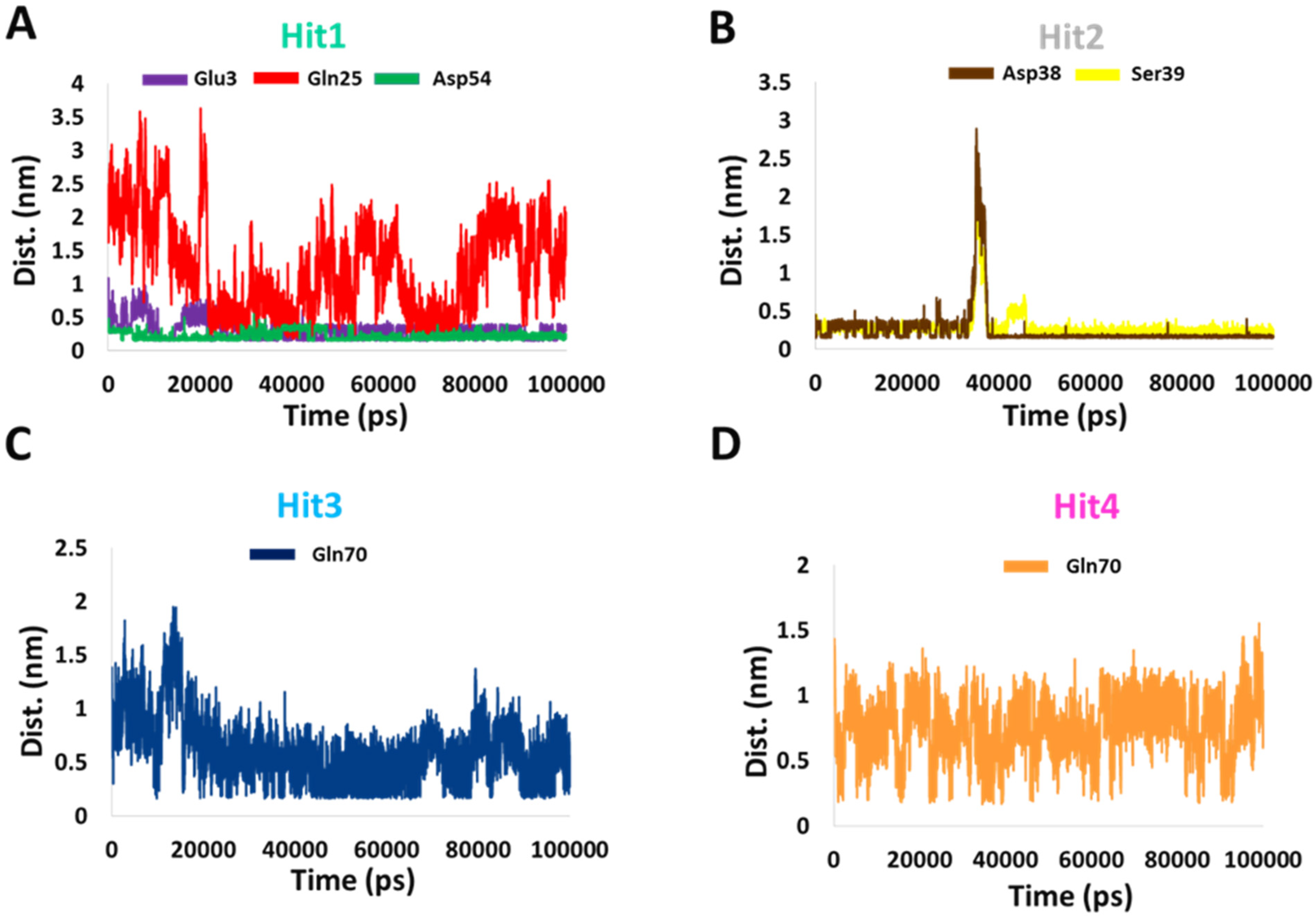
2.5.2. Binding Dynamics and Molecular Interactions
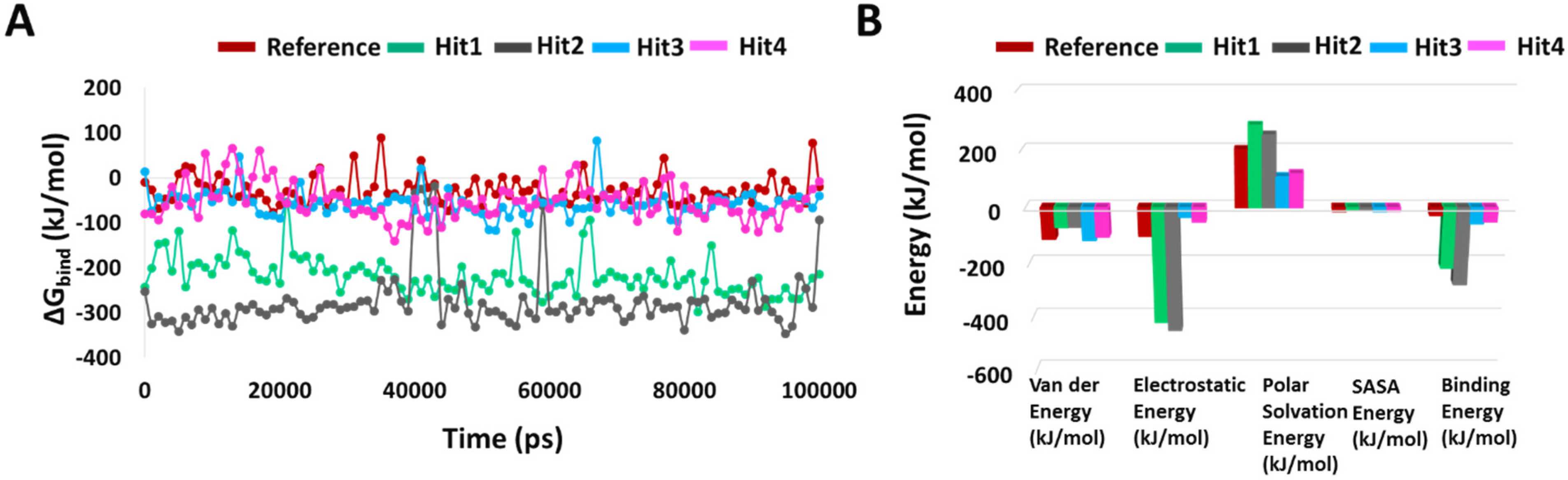
2.5.3. Binding Free Energy
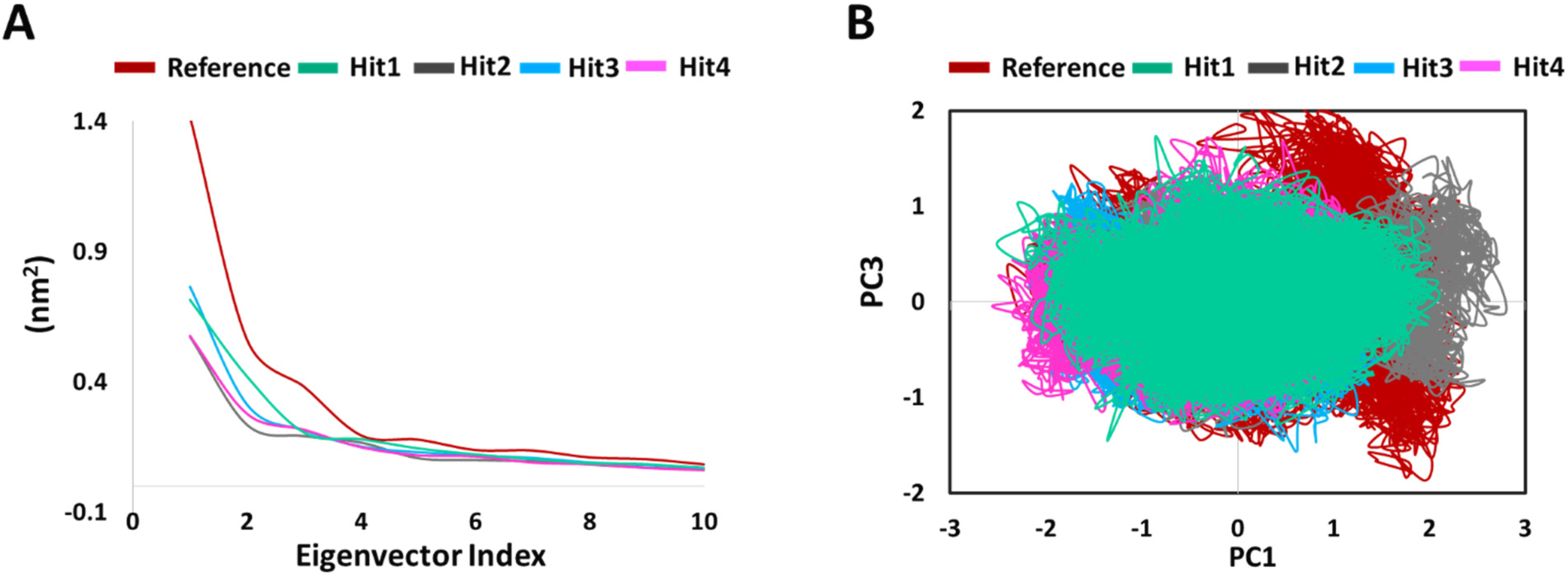
2.5.4. PCA
3. Discussion
4. Material and Methods
4.1. Common Feature Pharmacophore Generation
4.2. Common Feature Pharmacophore Validation
4.3. Virtual Screening
4.4. Molecular Docking
4.5. Molecular Dynamics Simulation
4.5.1. Root Mean Square Deviation (RMSD) and Root Mean Square Fluctuation (RMSF) Analysis
4.5.2. Binding Dynamics and Molecular Interactions
4.5.3. Binding Free Energy
4.5.4. Principal Component Analysis (PCA)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vetter, I.R.; Wittinghofer, A. The Guanine Nucleotide-Binding Switch in Three Dimensions. Science 2001, 294, 1299–1304. [Google Scholar] [CrossRef] [Green Version]
- Liceras-Boillos, P.; García-Navas, R.; Ginel-Picardo, A.; Anta, B.; Perez-Andres, M.; Lillo, C.; Gómez, C.; Jimeno, D.; Fernández-Medarde, A.; Baltanas, F.C.; et al. Sos1 disruption impairs cellular proliferation and viability through an increase in mitochondrial oxidative stress in primary MEFs. Oncogene 2016, 35, 6389–6402. [Google Scholar] [CrossRef] [PubMed]
- Kessler, D.; Gmachl, M.; Mantoulidis, A.; Martin, L.J.; Zoephel, A.; Mayer, M.; Gollner, A.; Covini, D.; Fischer, S.; Gerstberger, T.; et al. Drugging an undruggable pocket on KRAS. Proc. Natl. Acad. Sci. USA 2019, 116, 15823–15829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Maldonado, C.; Zimmer, Y.; Medová, M. A Comparative Analysis of Individual RAS Mutations in Cancer Biology. Front. Oncol. 2019, 9, 1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina-Arcas, M.; Samani, A.; Downward, J. Drugging the Undruggable: Advances on RAS Targeting in Cancer. Genes 2021, 12, 899. [Google Scholar] [CrossRef]
- Papke, B.; Der, C.J. Drugging RAS: Know the enemy. Science 2017, 355, 1158–1163. [Google Scholar] [CrossRef] [Green Version]
- Tisi, R.; Gaponenko, V.; Vanoni, M.; Sacco, E. Natural Products Attenuating Biosynthesis, Processing, and Activity of Ras Oncoproteins: State of the Art and Future Perspectives. Biomolecules 2020, 10, 1535. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotorasib, F. FDA Grants Accelerated Approval to Sotorasib for KRAS G12C Mutated NSCLC. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-sotorasib-kras-g12c-mutated-nsclc (accessed on 22 June 2021).
- MRTX1133 n.d. KRASG12D Inhibitor. Available online: https://www.mirati.com/science/programs/kras-inhibitors/kras-g12d-inhibitor/ (accessed on 22 June 2021).
- Zhang, Z.; Gao, R.; Hu, Q.; Peacock, H.; Peacock, D.M.; Dai, S.; Shokat, K.M.; Suga, H. GTP-State-Selective Cyclic Peptide Ligands of K-Ras(G12D) Block Its Interaction with Raf. ACS Cent. Sci. 2020, 6, 1753–1761. [Google Scholar] [CrossRef]
- Sakamoto, K.; Masutani, T.; Hirokawa, T. Generation of KS-58 as the first K-Ras(G12D)-inhibitory peptide presenting anti-cancer activity in vivo. Sci. Rep. 2020, 10, 21671. [Google Scholar] [CrossRef] [PubMed]
- Osborne, J.K.; Minna, J.D. Defining the First Part of the Oncogenic KRAS Journey. Cell Stem Cell 2020, 27, 499–500. [Google Scholar] [CrossRef] [PubMed]
- Menyhárd, D.K.; Pálfy, G.; Orgován, Z.; Vida, I.; Keserű, G.M.; Perczel, A. Structural impact of GTP binding on downstream KRAS signaling. Chem. Sci. 2020, 11, 9272–9289. [Google Scholar] [CrossRef]
- Yin, G.; Kistler, S.; George, S.D.; Kuhlmann, N.; Garvey, L.; Huynh, M.; Bagni, R.K.; Lammers, M.; Der, C.; Campbell, S.L. A KRAS GTPase K104Q Mutant Retains Downstream Signaling by Offsetting Defects in Regulation. J. Biol. Chem. 2017, 292, 4446–4456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, B.E.; Bar-Sagi, D.; Nassar, N. The structural basis for the transition from Ras-GTP to Ras-GDP. Proc. Natl. Acad. Sci. USA 2002, 99, 12138–12142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrem, J.M.L.; Shokat, K.M. Direct small-molecule inhibitors of KRAS: From structural insights to mechanism-based design. Nat. Rev. Drug Discov. 2016, 15, 771–785. [Google Scholar] [CrossRef]
- Pantsar, T. The current understanding of KRAS protein structure and dynamics. Comput. Struct. Biotechnol. J. 2020, 18, 189–198. [Google Scholar] [CrossRef]
- Patricelli, M.P.; Janes, M.R.; Li, L.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T.; et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016, 6, 316–329. [Google Scholar] [CrossRef] [Green Version]
- Fell, J.B.; Fischer, J.P.; Baer, B.R.; Ballard, J.; Blake, J.F.; Bouhana, K.; Brandhuber, B.J.; Briere, D.M.; Burgess, L.E.; Burkard, M.R.; et al. Discovery of Tetrahydropyridopyrimidines as Irreversible Covalent Inhibitors of KRAS-G12C with In Vivo Activity. ACS Med. Chem. Lett. 2018, 9, 1230–1234. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Ou, S.-H.I.; Johnson, M.L.; Christensen, J.; Velastegui, K.; Potvin, D.; Faltaos, D.; Chao, R.C. A phase I/II multiple expansion cohort trial of MRTX849 in patients with advanced solid tumors with KRAS G12C mutation. J. Clin. Oncol. 2019, 37, TPS3161. [Google Scholar] [CrossRef]
- Fakih, M.; O’Neil, B.; Price, T.J.; Falchook, G.S.; Desai, J.; Kuo, J.; Govindan, R.; Rasmussen, E.; Morrow, P.K.H.; Ngang, J.; et al. Phase 1 study evaluating the safety, tolerability, pharmacokinetics (PK), and efficacy of AMG 510, a novel small molecule KRASG12C inhibitor, in advanced solid tumors. J. Clin. Oncol. 2019, 37, 3003. [Google Scholar] [CrossRef]
- Chen, H.; Smaill, J.B.; Liu, T.; Ding, K.; Lu, X. Small-Molecule Inhibitors Directly Targeting KRAS as Anticancer Therapeutics. J. Med. Chem. 2020, 63, 14404–14424. [Google Scholar] [CrossRef] [PubMed]
- Opo, F.A.D.M.; Rahman, M.M.; Ahammad, F.; Ahmed, I.; Bhuiyan, M.A.; Asiri, A.M. Structure based pharmacophore modeling, virtual screening, molecular docking and ADMET approaches for identification of natural anti-cancer agents targeting XIAP protein. Sci. Rep. 2021, 11, 4049. [Google Scholar] [CrossRef] [PubMed]
- Pantsar, T. KRAS(G12C)–AMG 510 interaction dynamics revealed by all-atom molecular dynamics simulations. Sci. Rep. 2020, 10, 11992. [Google Scholar] [CrossRef]
- Vatansever, S.; Erman, B.; Gümüş, Z.H. Oncogenic G12D mutation alters local conformations and dynamics of K-Ras. Sci. Rep. 2019, 9, 11730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Patel, D.; Athar, M.; Jha, P.C. Exploring Ruthenium-Based Organometallic Inhibitors against Plasmodium falciparum Calcium Dependent Kinase 2 (PfCDPK2): A Combined Ensemble Docking, QM/MM and Molecular Dynamics Study. ChemistrySelect 2021, 6, 8189–8199. [Google Scholar] [CrossRef]
- Dhasmana, D.; Singh, A.; Shukla, R.; Tripathi, T.; Garg, N. Targeting Nucleotide Binding Domain of Multidrug Resistance-associated Protein-1 (MRP1) for the Reversal of Multi Drug Resistance in Cancer. Sci. Rep. 2018, 8, 11973. [Google Scholar] [CrossRef] [PubMed]
- Kessler, D.; Bergner, A.; Böttcher, J.; Fischer, G.; Döbel, S.; Hinkel, M.; Müllauer, B.; Weiss-Puxbaum, A.; McConnell, D.B. Drugging all RAS isoforms with one pocket. Futur. Med. Chem. 2020, 12, 1911–1923. [Google Scholar] [CrossRef]
- Kaserer, T.; Beck, K.R.; Akram, M.; Odermatt, A.; Schuster, D. Pharmacophore Models and Pharmacophore-Based Virtual Screening: Concepts and Applications Exemplified on Hydroxysteroid Dehydrogenases. Molecules 2015, 20, 22799–22832. [Google Scholar] [CrossRef] [Green Version]
- Parasrampuria, D.; Benet, L.Z.; Sharma, A. Why Drugs Fail in Late Stages of Development: Case Study Analyses from the Last Decade and Recommendations. AAPS J. 2018, 20, 46. [Google Scholar] [CrossRef] [PubMed]
- Rampogu, S.; Kim, S.M.; Son, M.; Baek, A.; Park, C.; Lee, G.; Kim, Y.; Kim, G.S.; Kim, J.H.; Lee, K.W. A Computational Approach with Biological Evaluation: Combinatorial Treatment of Curcumin and Exemestane Synergistically Regulates DDX3 Expression in Cancer Cell Lines. Biomolecules 2020, 10, 857. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Sawney, S.; Saini, V.; Steffi, C.; Tiwari, M.; Saluja, D. Esculetin induces antiproliferative and apoptotic response in pancreatic cancer cells by directly binding to KEAP1. Mol. Cancer 2016. [CrossRef] [PubMed] [Green Version]
- Maurer, T.; Garrenton, L.S.; Oh, A.; Pitts, K.; Anderson, D.J.; Skelton, N.J.; Fauber, B.P.; Pan, B.; Malek, S.; Stokoe, D.; et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc. Natl. Acad. Sci. USA 2012, 109, 5299–5304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fetics, S.K.; Guterres, H.; Kearney, B.M.; Buhrman, G.; Ma, B.; Nussinov, R.; Mattos, C. Allosteric Effects of the Oncogenic RasQ61L Mutant on Raf-RBD. Structure 2015, 23, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Pacold, M.E.; Suire, S.; Perisic, O.; Lara-Gonzalez, S.; Davis, C.T.; Walker, E.H.; Hawkins, P.; Stephens, L.R.; Eccleston, J.F.; Williams, R.L. Crystal Structure and Functional Analysis of Ras Binding to Its Effector Phosphoinositide 3-Kinase γ. Cell 2000, 103, 931–944. [Google Scholar] [CrossRef] [Green Version]
- Welsch, M.E.; Kaplan, A.; Chambers, J.M.; Stokes, M.E.; Bos, P.; Zask, A.; Zhang, Y.; Sanchez-Martin, M.; Badgley, M.A.; Huang, C.; et al. Multivalent Small-Molecule Pan-RAS Inhibitors. Cell 2017, 168, 878–889.e29. [Google Scholar] [CrossRef] [Green Version]
- Bhati, A.P.; Wan, S.; Wright, D.W.; Coveney, P.V. Rapid, Accurate, Precise, and Reliable Relative Free Energy Prediction Using Ensemble Based Thermodynamic Integration. J. Chem. Theory Comput. 2017, 13, 210–222. [Google Scholar] [CrossRef]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.; Willett, P.; Glen, R.C. A genetic algorithm for flexible molecular overlay and pharmacophore elucidation. J. Comput. Mol. Des. 1995, 9, 532–549. [Google Scholar] [CrossRef]
- BIOVIA DS. Discovery Studio Modeling Environment, Release 2017; Dassault Systèmes: San Diego, CA, USA, 2016. [Google Scholar]
- Pascual, R.; Almansa, C.; Plata-Salamán, C.; Vela, J.M. A New Pharmacophore Model for the Design of Sigma-1 Ligands Validated on a Large Experimental Dataset. Front. Pharmacol. 2019, 10, 519. [Google Scholar] [CrossRef] [PubMed]
- Florkowski, C.M. Sensitivity, specificity, receiver-operating characteristic (ROC) curves and likelihood ratios: Communicating the performance of diagnostic tests. Clin. Biochem. Rev. 2008, 29, S83–S87. [Google Scholar] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and Permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Wu, G.; Robertson, D.H.; Brooks, C.L.; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER-A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef]
- Zhou, X.; Yu, S.; Su, J.; Sun, L. Computational Study on New Natural Compound Inhibitors of Pyruvate Dehydrogenase Kinases. Int. J. Mol. Sci. 2016, 17, 340. [Google Scholar] [CrossRef] [Green Version]
- Rampogu, S.; Lemuel, M.R. Network Based Approach in the Establishment of the Relationship between Type 2 Diabetes Mellitus and Its Complications at the Molecular Level Coupled with Molecular Docking Mechanism. BioMed Res. Int. 2016, 2016, 6068437. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.; Hess, B.; van der Spoel, D.; Lindahl, E. Gromacs 5.0.7. WwwGromacsOrg 2015. Available online: https://ftp.gromacs.org/pub/manual/manual-5.0.7.pdf (accessed on 14 December 2021).
- Udhaya, K.S.; Bithia, R.; Thirumal Kumar, D.; Priya Dos, C.G.; Zayed, H. Mutational landscape of K-Ras substitutions at 12th position—A systematic molecular dynamics approach. J. Biomol. Struct. Dyn. 2020, 1–15. [Google Scholar] [CrossRef]
- Prakash, P.; Sayyed-Ahmad, A.; Gorfe, A.A. The Role of Conserved Waters in Conformational Transitions of Q61H K-ras. PLOS Comput. Biol. 2012, 8, e1002394. [Google Scholar] [CrossRef]
- Sayyed-Ahmad, A.; Prakash, P.; Gorfe, A.A. Distinct dynamics and interaction patterns in H- and K-Ras oncogenic P-loop mutants. Proteins Struct. Funct. Bioinform. 2017, 85, 1618–1632. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Lopes, P.E.M.; Mackerell, A.D., Jr. Recent developments and applications of the CHARMM force fields. WIREs Comput. Mol. Sci. 2011, 2, 167–185. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model No. | Features | Score | Direct Hit | Partial Hit | Max Fit |
|---|---|---|---|---|---|
| 1 | RRHDDA | 36.17 | 11 | 00 | 6 |
| 2 | RRHDDA | 36.17 | 11 | 00 | 6 |
| 3 | RRHDDA | 36.17 | 11 | 00 | 6 |
| 4 | RRHDDA | 36.17 | 11 | 00 | 6 |
| 5 | RRHDDA | 36.17 | 11 | 00 | 6 |
| 6 | RRHDDA | 36.17 | 11 | 00 | 6 |
| 7 | RRHDDA | 36.17 | 11 | 00 | 6 |
| 8 | RRHDDA | 36.17 | 11 | 00 | 6 |
| 9 | RRHDDA | 35.96 | 11 | 00 | 6 |
| 10 | RRHDDA | 35.96 | 11 | 00 | 6 |
| Model No. | Total Actives | Total Inactives | True Positives | True Negatives | False Positives | False Negatives | Sensitivity | Specificity |
|---|---|---|---|---|---|---|---|---|
| 1 | 3 | 8 | 3 | 5 | 3 | 0 | 1 | 0.62 |
| 2 | 3 | 8 | 3 | 5 | 3 | 0 | 1 | 0.62 |
| 3 | 3 | 8 | 3 | 7 | 1 | 0 | 1 | 0.87 |
| 4 | 3 | 8 | 3 | 5 | 3 | 0 | 1 | 0.62 |
| 5 | 3 | 8 | 3 | 5 | 3 | 0 | 1 | 0.62 |
| 6 | 3 | 8 | 3 | 6 | 2 | 0 | 1 | 0.75 |
| 7 | 3 | 8 | 3 | 4 | 4 | 0 | 1 | 0.50 |
| 8 | 3 | 8 | 3 | 6 | 2 | 0 | 1 | 0.75 |
| 9 | 3 | 8 | 3 | 4 | 4 | 0 | 1 | 0.50 |
| 10 | 3 | 8 | 3 | 7 | 1 | 0 | 1 | 0.87 |
| Compound | -CDOCKER Energy (kcal/mol) | -CDOCKER Interaction Energy (kcal/mol) |
|---|---|---|
| Hit1 | 45.6694 | 53.1082 |
| Hit2 | 35.3224 | 51.3697 |
| Hit3 | 22.8951 | 49.2084 |
| Hit4 | 43.3661 | 49.1899 |
| Reference (BI-2852) | 25.0164 | 46.9 |
| Compound | Hydrogen Bond | Occupancy (%) |
|---|---|---|
| Hit1 | Glu3 | 88.4 |
| Gln25 | 5.8 | |
| Asp54 | 119 | |
| Hit2 | Asp38 | 94.9 |
| Ser39 | 96.6 | |
| Hit3 | Gln70 | 14.6 |
| Hit4 | Gln70 | 8.6 |
| Compound | Hydrogen Bonds (Molecular Docking) | Hydrogen Bonds (MD Simulations) |
|---|---|---|
| Hit1 | Leu6 (0.30) Glu37 (0.19) Ser39 (0.26) Asp54 * (0.27) | Glu3 * (0.17) Gln25 (0.30) Asp54 * (0.18) |
| Hit2 | Glu37 (0.18) Asp54 (0.24) Asp54 (0.26) | Asp38 * (0.15) Ser39 (0.19) |
| Hit3 | Lys5 (0.28) Ser39 (0.30) Arg41 (0.33) | Gln70 (0.19) |
| Hit4 | Glu37 (0.22) Glu37 (0.24) | Gln70 (0.21) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulkarni, A.M.; Kumar, V.; Parate, S.; Lee, G.; Yoon, S.; Lee, K.W. Identification of New KRAS G12D Inhibitors through Computer-Aided Drug Discovery Methods. Int. J. Mol. Sci. 2022, 23, 1309. https://doi.org/10.3390/ijms23031309
Kulkarni AM, Kumar V, Parate S, Lee G, Yoon S, Lee KW. Identification of New KRAS G12D Inhibitors through Computer-Aided Drug Discovery Methods. International Journal of Molecular Sciences. 2022; 23(3):1309. https://doi.org/10.3390/ijms23031309
Chicago/Turabian StyleKulkarni, Apoorva M., Vikas Kumar, Shraddha Parate, Gihwan Lee, Sanghwa Yoon, and Keun Woo Lee. 2022. "Identification of New KRAS G12D Inhibitors through Computer-Aided Drug Discovery Methods" International Journal of Molecular Sciences 23, no. 3: 1309. https://doi.org/10.3390/ijms23031309
APA StyleKulkarni, A. M., Kumar, V., Parate, S., Lee, G., Yoon, S., & Lee, K. W. (2022). Identification of New KRAS G12D Inhibitors through Computer-Aided Drug Discovery Methods. International Journal of Molecular Sciences, 23(3), 1309. https://doi.org/10.3390/ijms23031309