
Emerging Connections between Nuclear Pore Complex Homeostasis and ALS


{kind=link}
{kind=link}
Abstract
:1. Introduction to Amyotrophic Lateral Sclerosis: Background of Genotypic and Pathological Features
2. Nuclear Transport and NE Quality Control in ALS
2.1. The Soluble Nuclear Transport Machinery Is Impacted in ALS
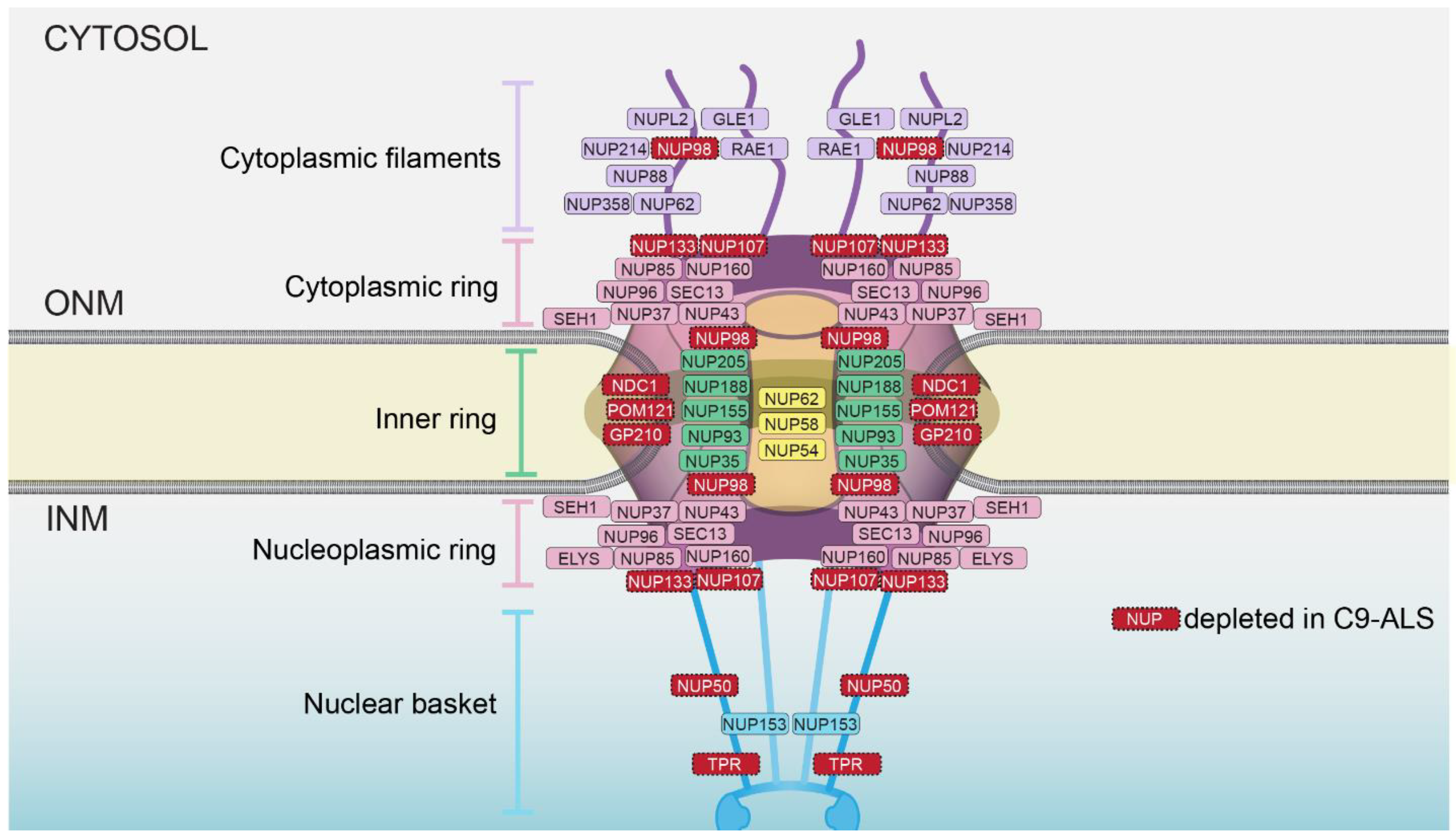
2.2. NPC Injury in C9-ALS
2.3. How Are Nups Selectively Removed from the NPC?
2.4. NE-Specific Quality Control Mechanisms
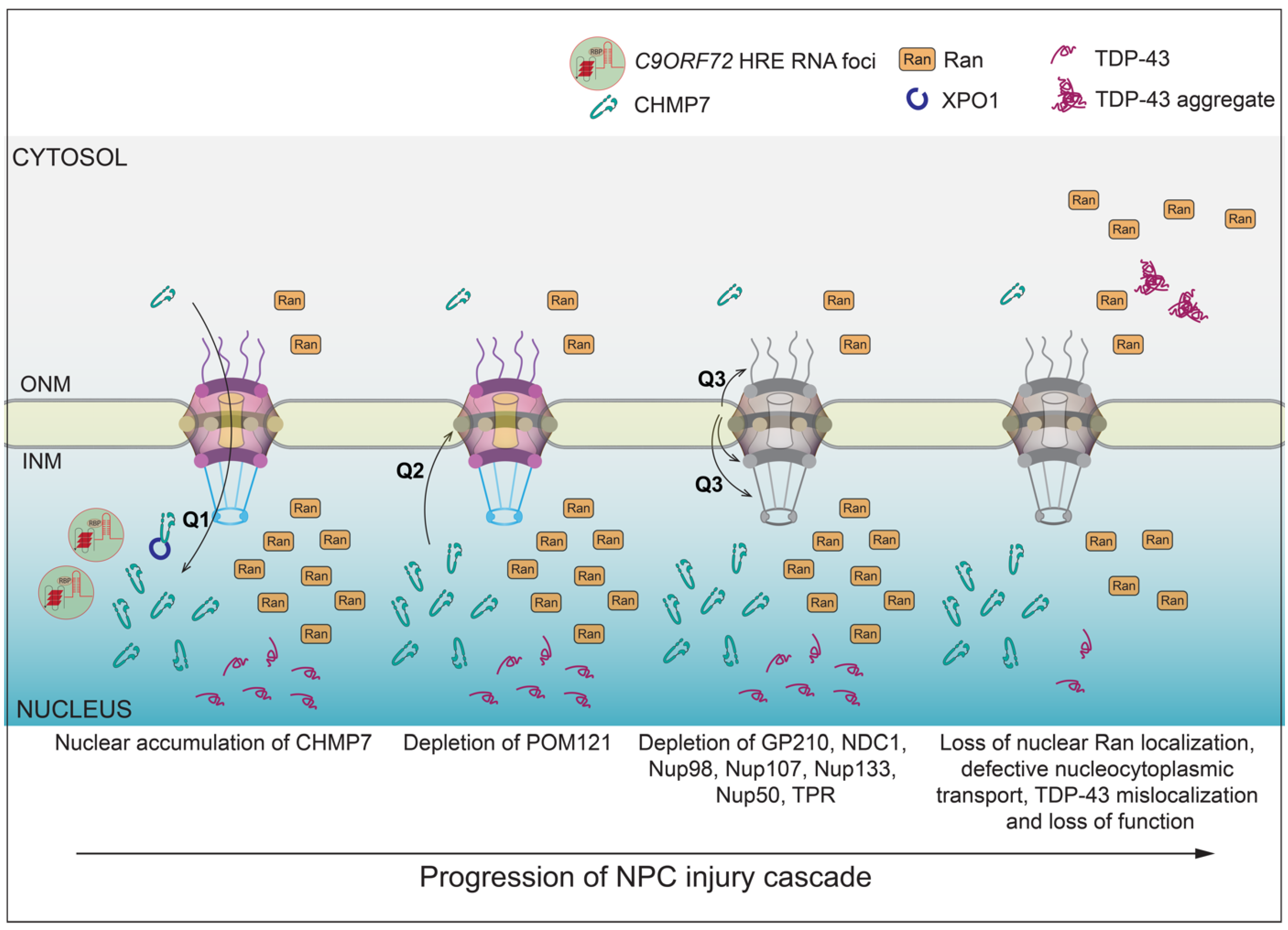
2.5. CHMP7 as a Key Player in NPC Injury in ALS
2.6. What Are the Mechanisms That Trigger CHMP7 Nuclear Accumulation and Ensuing NPC Injury?
3. Conclusions
Funding
Conflicts of Interest
References
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef] [Green Version]
- Borasio, G.D.; Voltz, R.; Miller, R.G. Palliative Care in Amyotrophic Lateral Sclerosis. Neurol. Clin. 2001, 19, 829–847. [Google Scholar] [CrossRef]
- Norris, S.P.; Likanje, M.N.; Andrews, J.A. Amyotrophic lateral sclerosis: Update on clinical management. Curr. Opin. Neurol. 2020, 33, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.; Gautier, O.; Tassoni-Tsuchida, E.; Ma, X.R.; Gitler, A.D. ALS Genetics: Gains, Losses, and Implications for Future Therapies. Neuron 2020, 108, 822–842. [Google Scholar] [CrossRef] [PubMed]
- Valdmanis, P.N.; Rouleau, G.A. Genetics of familial amyotrophic lateral sclerosis. Neurology 2008, 70, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Coyne, A.N.; Zaepfel, B.L.; Hayes, L.; Fitchman, B.; Salzberg, Y.; Luo, E.C.; Bowen, K.; Trost, H.; Aigner, S.; Rigo, F.; et al. G4C2 Repeat RNA Initiates a POM121-Mediated Reduction in Specific Nucleoporins in C9orf72 ALS/FTD. Neuron 2020, 107, 1124–1140 e1111. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Toro, A.; TG, S.; Gao, J.; Chalk, J.; Oskarsson, B.; Zhang, K. Divergence, Convergence, and Therapeutic Implications: A Cell Biology Perspective of C9ORF72-ALS/FTD. Mol. Neurodegener. 2020, 15, 34. [Google Scholar] [CrossRef]
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef]
- Frottin, F.; Perez-Berlanga, M.; Hartl, F.U.; Hipp, M.S. Multiple pathways of toxicity induced by C9orf72 dipeptide repeat aggregates and G4C2 RNA in a cellular model. eLife 2021, 10, e62718. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.; Rabichow, B.E.; Sattler, R. The Hitchhiker’s Guide to Nucleocytoplasmic Trafficking in Neurodegeneration. Neurochem. Res. 2020, 45, 1306–1327. [Google Scholar] [CrossRef] [PubMed]
- Hutten, S.; Dormann, D. Nucleocytoplasmic transport defects in neurodegeneration—Cause or consequence? Semin. Cell Dev. Biol. 2020, 99, 151–162. [Google Scholar] [CrossRef]
- Fallini, C.; Khalil, B.; Smith, C.L.; Rossoll, W. Traffic jam at the nuclear pore: All roads lead to nucleocytoplasmic transport defects in ALS/FTD. Neurobiol. Dis. 2020, 140, 104835. [Google Scholar] [CrossRef] [PubMed]
- Vanneste, J.; Van Den Bosch, L. The Role of Nucleocytoplasmic Transport Defects in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 12175. [Google Scholar] [CrossRef] [PubMed]
- Semmelink, M.F.W.; Steen, A.; Veenhoff, L.M. Measuring and Interpreting Nuclear Transport in Neurodegenerative Disease-The Example of C9orf72 ALS. Int. J. Mol. Sci. 2021, 22, 9217. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Segura, L.M.; Lafarga, M.; Berciano, M.T.; Hernandez, P.; Andres, M.A. Distribution of nuclear pores and chromatin organization in neurons and glial cells of the rat cerebellar cortex. J. Comp. Neurol. 1989, 290, 440–450. [Google Scholar] [CrossRef]
- Toyama, B.H.; Savas, J.N.; Park, S.K.; Harris, M.S.; Ingolia, N.T.; Yates, J.R., 3rd; Hetzer, M.W. Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell 2013, 154, 971–982. [Google Scholar] [CrossRef] [Green Version]
- Toda, T.; Hsu, J.Y.; Linker, S.B.; Hu, L.; Schafer, S.T.; Mertens, J.; Jacinto, F.V.; Hetzer, M.W.; Gage, F.H. Nup153 Interacts with Sox2 to Enable Bimodal Gene Regulation and Maintenance of Neural Progenitor Cells. Cell Stem Cell 2017, 21, 618–634 e617. [Google Scholar] [CrossRef] [Green Version]
- Hampoelz, B.; Andres-Pons, A.; Kastritis, P.; Beck, M. Structure and Assembly of the Nuclear Pore Complex. Ann. Rev. Biophys. 2019, 48, 515–536. [Google Scholar] [CrossRef]
- Wente, S.R.; Rout, M.P. The nuclear pore complex and nuclear transport. Cold Spring Harb. Perspect. Biol. 2010, 2, a000562. [Google Scholar] [CrossRef] [PubMed]
- Paci, G.; Caria, J.; Lemke, E.A. Cargo transport through the nuclear pore complex at a glance. J. Cell Sci. 2021, 134, jcs247874. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Taylor, J.P. Lost in Transportation: Nucleocytoplasmic Transport Defects in ALS and Other Neurodegenerative Diseases. Neuron 2017, 96, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Bitetto, G.; Di Fonzo, A. Nucleo-cytoplasmic transport defects and protein aggregates in neurodegeneration. Transl. Neurodegener. 2020, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.A.; Smikle, R.; Reid, M.J.; Mizielinska, S. Altered Phase Separation and Cellular Impact in C9orf72-Linked ALS/FTD. Front. Cell Neurosci. 2021, 15, 664151. [Google Scholar] [CrossRef]
- Fan, A.C.; Leung, A.K.L. RNA Granules and Diseases: A Case Study of Stress Granules in ALS and FTLD. In RNA Processing: Disease and Genome-Wide Probing; Yeo, G.W., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 263–296. [Google Scholar]
- Yoshizawa, T.; Ali, R.; Jiou, J.; Fung, H.Y.J.; Burke, K.A.; Kim, S.J.; Lin, Y.; Peeples, W.B.; Saltzberg, D.; Soniat, M.; et al. Nuclear Import Receptor Inhibits Phase Separation of FUS through Binding to Multiple Sites. Cell 2018, 173, 693–705 e622. [Google Scholar] [CrossRef] [Green Version]
- Springhower, C.E.; Rosen, M.K.; Chook, Y.M. Karyopherins and condensates. Curr. Opin. Cell Biol. 2020, 64, 112–123. [Google Scholar] [CrossRef]
- Lin, Y.C.; Kumar, M.S.; Ramesh, N.; Anderson, E.N.; Nguyen, A.T.; Kim, B.; Cheung, S.; McDonough, J.A.; Skarnes, W.C.; Lopez-Gonzalez, R.; et al. Interactions between ALS-linked FUS and nucleoporins are associated with defects in the nucleocytoplasmic transport pathway. Nat. Neurosci. 2021, 24, 1077–1088. [Google Scholar] [CrossRef]
- D’Angelo, M.A.; Raices, M.; Panowski, S.H.; Hetzer, M.W. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell 2009, 136, 284–295. [Google Scholar] [CrossRef] [Green Version]
- Savas, J.N.; Toyama, B.H.; Xu, T.; Yates, J.R., 3rd; Hetzer, M.W. Extremely long-lived nuclear pore proteins in the rat brain. Science 2012, 335, 942. [Google Scholar] [CrossRef] [Green Version]
- Sydor, A.M.; Czymmek, K.J.; Puchner, E.M.; Mennella, V. Super-Resolution Microscopy: From Single Molecules to Supramolecular Assemblies. Trends Cell Biol. 2015, 25, 730–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schermelleh, L.; Carlton, P.M.; Haase, S.; Shao, L.; Winoto, L.; Kner, P.; Burke, B.; Cardoso, M.C.; Agard, D.A.; Gustafsson, M.G.L.; et al. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science 2008, 320, 1332–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coyne, A.N.V.; Zaepfel, B.L.; Dickson, D.W.; Rigo, F.; Bennett, F.; Lusk, C.P.; Rothstein, J.D. Nuclear accumulation of CHMP7 initiates nuclear pore complex injury and subsequent TDP-43 dysfunction in sporadic and familial ALS. Sci. Transl. Med. 2021, 13, eabe1923. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Grima, J.C.; Rothstein, J.D.; Lloyd, T.E. Nucleocytoplasmic transport in C9orf72-mediated ALS/FTD. Nucleus 2016, 7, 132–137. [Google Scholar] [CrossRef] [Green Version]
- Regmi, S.G.; Lee, H.; Kaufhold, R.; Fichtman, B.; Chen, S.; Aksenova, V.; Turcotte, E.; Harel, A.; Arnaoutov, A.; Dasso, M. The nuclear pore complex consists of two independent scaffolds. BioRixv 2020. [Google Scholar] [CrossRef]
- Aksenova, V.; Smith, A.; Lee, H.; Bhat, P.; Esnault, C.; Chen, S.; Iben, J.; Kaufhold, R.; Yau, K.C.; Echeverria, C.; et al. Nucleoporin TPR is an integral component of the TREX-2 mRNA export pathway. Nat. Commun. 2020, 11, 4577. [Google Scholar] [CrossRef]
- Schuller, A.P.; Wojtynek, M.; Mankus, D.; Tatli, M.; Kronenberg-Tenga, R.; Regmi, S.G.; Dip, P.V.; Lytton-Jean, A.K.R.; Brignole, E.J.; Dasso, M.; et al. The cellular environment shapes the nuclear pore complex architecture. Nature 2021, 598, 667–671. [Google Scholar] [CrossRef]
- Mitchell, J.M.; Mansfeld, J.; Capitanio, J.; Kutay, U.; Wozniak, R.W. Pom121 links two essential subcomplexes of the nuclear pore complex core to the membrane. J. Cell Biol. 2010, 191, 505–521. [Google Scholar] [CrossRef] [Green Version]
- Doucet, C.M.; Talamas, J.A.; Hetzer, M.W. Cell cycle-dependent differences in nuclear pore complex assembly in metazoa. Cell 2010, 141, 1030–1041. [Google Scholar] [CrossRef] [Green Version]
- Pohl, C.; Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 2019, 366, 818–822. [Google Scholar] [CrossRef]
- Mehrtash, A.B.; Hochstrasser, M. Ubiquitin-dependent protein degradation at the endoplasmic reticulum and nuclear envelope. Semin. Cell Dev. Biol. 2019, 93, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.J.; Gendron, T.F.; Grima, J.C.; Sasaguri, H.; Jansen-West, K.; Xu, Y.F.; Katzman, R.B.; Gass, J.; Murray, M.E.; Shinohara, M.; et al. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat. Neurosci. 2016, 19, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Ruz, C.; Alcantud, J.L.; Vives Montero, F.; Duran, R.; Bandres-Ciga, S. Proteotoxicity and Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 5646. [Google Scholar] [CrossRef] [PubMed]
- Mandrioli, J.; D’Amico, R.; Zucchi, E.; Gessani, A.; Fini, N.; Fasano, A.; Caponnetto, C.; Chio, A.; Dalla Bella, E.; Lunetta, C.; et al. Rapamycin treatment for amyotrophic lateral sclerosis: Protocol for a phase II randomized, double-blind, placebo-controlled, multicenter, clinical trial (RAP-ALS trial). Medicine 2018, 97, e11119. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Hochstrasser, M. Spatially regulated ubiquitin ligation by an ER/nuclear membrane ligase. Nature 2006, 443, 827–831. [Google Scholar] [CrossRef]
- Foresti, O.; Rodriguez-Vaello, V.; Funaya, C.; Carvalho, P. Quality control of inner nuclear membrane proteins by the Asi complex. Science 2014, 346, 751–755. [Google Scholar] [CrossRef]
- Khmelinskii, A.; Blaszczak, E.; Pantazopoulou, M.; Fischer, B.; Omnus, D.J.; Le Dez, G.; Brossard, A.; Gunnarsson, A.; Barry, J.D.; Meurer, M.; et al. Protein quality control at the inner nuclear membrane. Nature 2014, 516, 410–413. [Google Scholar] [CrossRef] [Green Version]
- Koch, B.A.; Jin, H.; Tomko, R.J., Jr.; Yu, H.G. The anaphase-promoting complex regulates the degradation of the inner nuclear membrane protein Mps3. J. Cell Biol. 2019, 218, 839–854. [Google Scholar] [CrossRef] [Green Version]
- Tsai, P.L.; Zhao, C.; Turner, E.; Schlieker, C. The Lamin B receptor is essential for cholesterol synthesis and perturbed by disease-causing mutations. eLife 2016, 5, e16011. [Google Scholar] [CrossRef]
- Albert, S.; Schaffer, M.; Beck, F.; Mosalaganti, S.; Asano, S.; Thomas, H.F.; Plitzko, J.M.; Beck, M.; Baumeister, W.; Engel, B.D. Proteasomes tether to two distinct sites at the nuclear pore complex. Proc. Natl. Acad. Sci. USA 2017, 114, 13726–13731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, K.; Yanagida, M. Regulation of nuclear proteasome by Rhp6/Ubc2 through ubiquitination and destruction of the sensor and anchor Cut8. Cell 2005, 122, 393–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, K.; Tonthat, N.K.; Glover, T.; Xu, W.; Koonin, E.V.; Yanagida, M.; Schumacher, M.A. Implications for proteasome nuclear localization revealed by the structure of the nuclear proteasome tether protein Cut8. Proc. Natl. Acad. Sci. USA 2011, 108, 16950–16955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Romero, L.; Chuang, S.M.; Tournier, V.; Joshi, K.K.; Lee, J.A.; Kovvali, G.; Madura, K. Sts1 plays a key role in targeting proteasomes to the nucleus. J. Biol. Chem. 2011, 286, 3104–3118. [Google Scholar] [CrossRef] [Green Version]
- Niepel, M.; Molloy, K.R.; Williams, R.; Farr, J.C.; Meinema, A.C.; Vecchietti, N.; Cristea, I.M.; Chait, B.T.; Rout, M.P.; Strambio-De-Castillia, C. The nuclear basket proteins Mlp1p and Mlp2p are part of a dynamic interactome including Esc1p and the proteasome. Mol. Biol. Cell 2013, 24, 3920–3938. [Google Scholar] [CrossRef]
- de Almeida, M.; Hinterndorfer, M.; Brunner, H.; Grishkovskaya, I.; Singh, K.; Schleiffer, A.; Jude, J.; Deswal, S.; Kalis, R.; Vunjak, M.; et al. AKIRIN2 controls the nuclear import of proteasomes in vertebrates. Nature 2021, 599, 491–496. [Google Scholar] [CrossRef]
- Chandra, S.; Mannino, P.J.; Thaller, D.J.; Ader, N.R.; King, M.C.; Melia, T.J.; Lusk, C.P. Atg39 selectively captures inner nuclear membrane into lumenal vesicles for delivery to the autophagosome. J. Cell Biol. 2021, 220, e202103030. [Google Scholar] [CrossRef]
- Mochida, K.; Otani, T.; Katsumata, Y.; Kirisako, H.; Kakuta, C.; Kotani, T.; Nakatogawa, H. Atg39 links and deforms the outer and inner nuclear membranes in selective autophagy of the nucleus. BioRixv 2021. [Google Scholar] [CrossRef]
- Lee, C.W.; Wilfling, F.; Ronchi, P.; Allegretti, M.; Mosalaganti, S.; Jentsch, S.; Beck, M.; Pfander, B. Selective autophagy degrades nuclear pore complexes. Nat. Cell Biol. 2020, 22, 159–166. [Google Scholar] [CrossRef]
- Tomioka, Y.; Kotani, T.; Kirisako, H.; Oikawa, Y.; Kimura, Y.; Hirano, H.; Ohsumi, Y.; Nakatogawa, H. TORC1 inactivation stimulates autophagy of nucleoporin and nuclear pore complexes. J. Cell Biol. 2020, 219, e201910063. [Google Scholar] [CrossRef]
- Allegretti, M.; Zimmerli, C.E.; Rantos, V.; Wilfling, F.; Ronchi, P.; Fung, H.K.H.; Lee, C.W.; Hagen, W.; Turonova, B.; Karius, K.; et al. In-cell architecture of the nuclear pore and snapshots of its turnover. Nature 2020, 586, 796–800. [Google Scholar] [CrossRef] [PubMed]
- Toyama, B.H.; Arrojo, E.D.R.; Lev-Ram, V.; Ramachandra, R.; Deerinck, T.J.; Lechene, C.; Ellisman, M.H.; Hetzer, M.W. Visualization of long-lived proteins reveals age mosaicism within nuclei of postmitotic cells. J. Cell Biol. 2019, 218, 433–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCullough, J.; Frost, A.; Sundquist, W.I. Structures, functions, and dynamics of ESCRT-III/Vps4 membrane remodeling and fission complexes. Ann. Rev. Cell Dev. Biol. 2018, 34, 85–109. [Google Scholar] [CrossRef] [PubMed]
- Webster, B.M.; Colombi, P.; Jager, J.; Lusk, C.P. Surveillance of nuclear pore complex assembly by ESCRT-III/Vps4. Cell 2014, 159, 388–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, B.M.; Thaller, D.J.; Jager, J.; Ochmann, S.E.; Borah, S.; Lusk, C.P. Chm7 and Heh1 collaborate to link nuclear pore complex quality control with nuclear envelope sealing. EMBO J. 2016, 35, 2447–2467. [Google Scholar] [CrossRef]
- Thaller, D.J.; Allegretti, M.; Borah, S.; Ronchi, P.; Beck, M.; Lusk, C.P. An ESCRT-LEM protein surveillance system is poised to directly monitor the nuclear envelope and nuclear transport system. eLife 2019, 8, e45284. [Google Scholar] [CrossRef]
- van der Zee, J.; Urwin, H.; Engelborghs, S.; Bruyland, M.; Vandenberghe, R.; Dermaut, B.; De Pooter, T.; Peeters, K.; Santens, P.; De Deyn, P.P.; et al. CHMP2B C-truncating mutations in frontotemporal lobar degeneration are associated with an aberrant endosomal phenotype in vitro. Hum. Mol. Genet. 2008, 17, 313–322. [Google Scholar] [CrossRef]
- van Blitterswijk, M.; Vlam, L.; van Es, M.A.; van der Pol, W.L.; Hennekam, E.A.; Dooijes, D.; Schelhaas, H.J.; van der Kooi, A.J.; de Visser, M.; Veldink, J.H.; et al. Genetic overlap between apparently sporadic motor neuron diseases. PLoS ONE 2012, 7, e48983. [Google Scholar] [CrossRef]
- Skibinski, G.; Parkinson, N.J.; Brown, J.M.; Chakrabarti, L.; Lloyd, S.L.; Hummerich, H.; Nielsen, J.E.; Hodges, J.R.; Spillantini, M.G.; Thusgaard, T.; et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 2005, 37, 806–808. [Google Scholar] [CrossRef]
- Narain, P.; Pandey, A.; Gupta, S.; Gomes, J.; Bhatia, R.; Vivekanandan, P. Targeted next-generation sequencing reveals novel and rare variants in Indian patients with amyotrophic lateral sclerosis. Neurobiol. Aging 2018, 71, 265-e9. [Google Scholar] [CrossRef]
- Momeni, P.; Rogaeva, E.; Van Deerlin, V.; Yuan, W.; Grafman, J.; Tierney, M.; Huey, E.; Bell, J.; Morris, C.M.; Kalaria, R.N.; et al. Genetic variability in CHMP2B and frontotemporal dementia. Neurodegener. Dis. 2006, 3, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.E.; Ferraiuolo, L.; Goodall, E.F.; Heath, P.R.; Higginbottom, A.; Mortiboys, H.; Hollinger, H.C.; Hartley, J.A.; Brockington, A.; Burness, C.E.; et al. Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS ONE 2010, 5, e9872. [Google Scholar] [CrossRef] [PubMed]
- Malik, B.; Devine, H.; Patani, R.; La Spada, A.R.; Hanna, M.G.; Greensmith, L. Gene expression analysis reveals early dysregulation of disease pathways and links Chmp7 to pathogenesis of spinal and bulbar muscular atrophy. Sci. Rep. 2019, 9, 3539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lusk, C.P.; Ader, N.R. CHMPions of repair: Emerging perspectives on sensing and repairing the nuclear envelope barrier. Curr. Opin. Cell Biol. 2020, 64, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.; Schultz, S.W.; Bellanger, A.; Jones, C.M.; Petersen, L.I.; Raiborg, C.; Skarpen, E.; Pedurupillay, C.R.J.; Kjos, I.; Kip, E.; et al. Unrestrained ESCRT-III drives micronuclear catastrophe and chromosome fragmentation. Nat. Cell Biol. 2020, 22, 856–867. [Google Scholar] [CrossRef]
- von Appen, A.; LaJoie, D.; Johnson, I.E.; Trnka, M.J.; Pick, S.M.; Burlingame, A.L.; Ullman, K.S.; Frost, A. LEM2 phase separation promotes ESCRT-mediated nuclear envelope reformation. Nature 2020, 582, 115–118. [Google Scholar] [CrossRef]
- Coyne, A.N.; Rothstein, J.D. The ESCRT-III protein VPS4, but not CHMP4B or CHMP2B, is pathologically increased in familial and sporadic ALS neuronal nuclei. Acta Neuropathol. Commun. 2021, 9, 127. [Google Scholar] [CrossRef]
- Gu, M.; LaJoie, D.; Chen, O.S.; von Appen, A.; Ladinsky, M.S.; Redd, M.J.; Nikolova, L.; Bjorkman, P.J.; Sundquist, W.I.; Ullman, K.S.; et al. LEM2 recruits CHMP7 for ESCRT-mediated nuclear envelope closure in fission yeast and human cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2166–E2175. [Google Scholar] [CrossRef] [Green Version]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [Green Version]
- Hayes, L.R.; Duan, L.; Bowen, K.; Kalab, P.; Rothstein, J.D. C9orf72 arginine-rich dipeptide repeat proteins disrupt karyopherin-mediated nuclear import. eLife 2020, 9, e51685. [Google Scholar] [CrossRef]
- Yavuz, S.; Santarella-Mellwig, R.; Koch, B.; Jaedicke, A.; Mattaj, I.W.; Antonin, W. NLS-mediated NPC functions of the nucleoporin Pom121. FEBS Lett. 2010, 584, 3292–3298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funakoshi, T.; Clever, M.; Watanabe, A.; Imamoto, N. Localization of Pom121 to the inner nuclear membrane is required for an early step of interphase nuclear pore complex assembly. Mol. Biol. Cell 2011, 22, 1058–1069. [Google Scholar] [CrossRef] [PubMed]
- Borah, S.; Thaller, D.J.; Hakhverdyan, Z.; Rodriguez, E.C.; Isenhour, A.W.; Rout, M.P.; King, M.C.; Lusk, C.P. Heh2/Man1 may be an evolutionarily conserved sensor of NPC assembly state. Mol. Biol. Cell 2021, 32, 1359–1373. [Google Scholar] [CrossRef] [PubMed]
- Vidak, S.; Serebryannyy, L.A.; Misteli, T. Activation of endoplasmic reticulum stress via clustering of inner nuclear membrane proteins. BioRixv 2021. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 182–194. [Google Scholar] [CrossRef] [Green Version]
- Igaz, L.M.; Kwong, L.K.; Xu, Y.; Truax, A.C.; Uryu, K.; Neumann, M.; Clark, C.M.; Elman, L.B.; Miller, B.L.; Grossman, M.; et al. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am. J. Pathol. 2008, 173, 182–194. [Google Scholar] [CrossRef] [Green Version]
- Chou, C.C.; Zhang, Y.; Umoh, M.E.; Vaughan, S.W.; Lorenzini, I.; Liu, F.; Sayegh, M.; Donlin-Asp, P.G.; Chen, Y.H.; Duong, D.M.; et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci. 2018, 21, 228–239. [Google Scholar] [CrossRef] [Green Version]
- Mosalaganti, S.; Kosinski, J.; Albert, S.; Schaffer, M.; Strenkert, D.; Salome, P.A.; Merchant, S.S.; Plitzko, J.M.; Baumeister, W.; Engel, B.D.; et al. In situ architecture of the algal nuclear pore complex. Nat. Commun. 2018, 9, 2361. [Google Scholar] [CrossRef] [Green Version]
- Zimmerli, C.E.; Allegretti, M.; Rantos, V.; Goetz, S.K.; Obarska-Kosinska, A.; Zagoriy, I.; Halavatyi, A.; Hummer, G.; Mahamid, J.; Kosinski, J.; et al. Nuclear pores dilate and constrict in cellulo. Science 2021, 374, eabd9776. [Google Scholar] [CrossRef]
- Akey, C.W.; Singh, D.; Ouch, C.; Echeverria, I.; Nudelman, I.; Varberg, J.M.; Yu, Z.; Fang, F.; Shi, Y.; Wang, J.; et al. Comprehensive structure and functional adaptations of the yeast nuclear pore complex. BioRixv 2021, 1–46. [Google Scholar] [CrossRef]
- Mahamid, J.; Pfeffer, S.; Schaffer, M.; Villa, E.; Danev, R.; Cuellar, L.K.; Forster, F.; Hyman, A.A.; Plitzko, J.M.; Baumeister, W. Visualizaing the molecular sociology at the HeLa cell nuclear periphery. Science 2016, 351, 969–972. [Google Scholar] [CrossRef] [PubMed]
- Zila, V.; Margiotta, E.; Turonova, B.; Muller, T.G.; Zimmerli, C.E.; Mattei, S.; Allegretti, M.; Borner, K.; Rada, J.; Muller, B.; et al. Cone-shaped HIV-1 capsids are transported through intact nuclear pores. Cell 2021, 184, 1032–1046.e18. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandra, S.; Lusk, C.P. Emerging Connections between Nuclear Pore Complex Homeostasis and ALS. Int. J. Mol. Sci. 2022, 23, 1329. https://doi.org/10.3390/ijms23031329
Chandra S, Lusk CP. Emerging Connections between Nuclear Pore Complex Homeostasis and ALS. International Journal of Molecular Sciences. 2022; 23(3):1329. https://doi.org/10.3390/ijms23031329
Chicago/Turabian StyleChandra, Sunandini, and C. Patrick Lusk. 2022. "Emerging Connections between Nuclear Pore Complex Homeostasis and ALS" International Journal of Molecular Sciences 23, no. 3: 1329. https://doi.org/10.3390/ijms23031329