
Easily Established and Multifunctional Synthetic Nanobody Libraries as Research Tools

Abstract
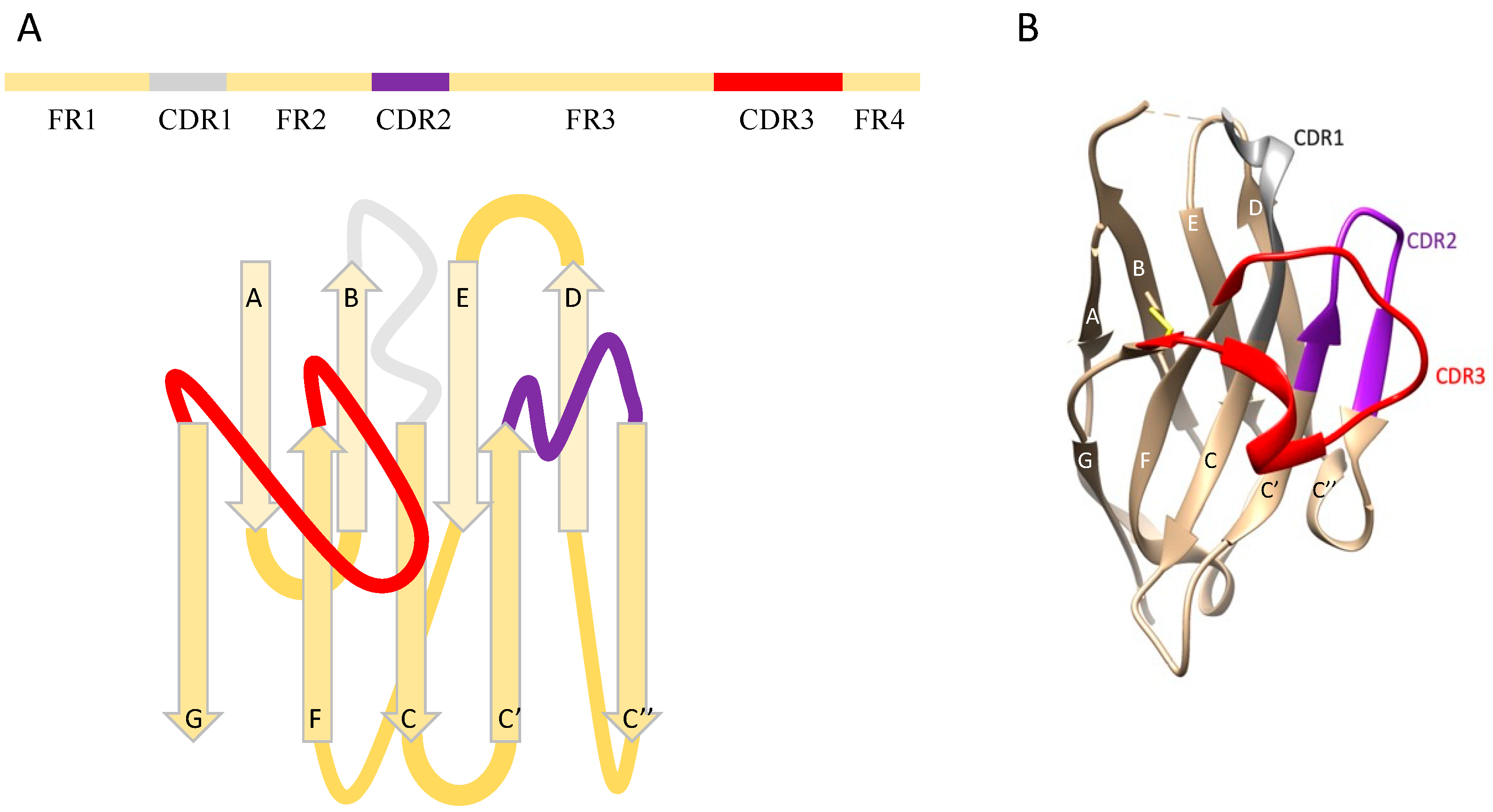
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
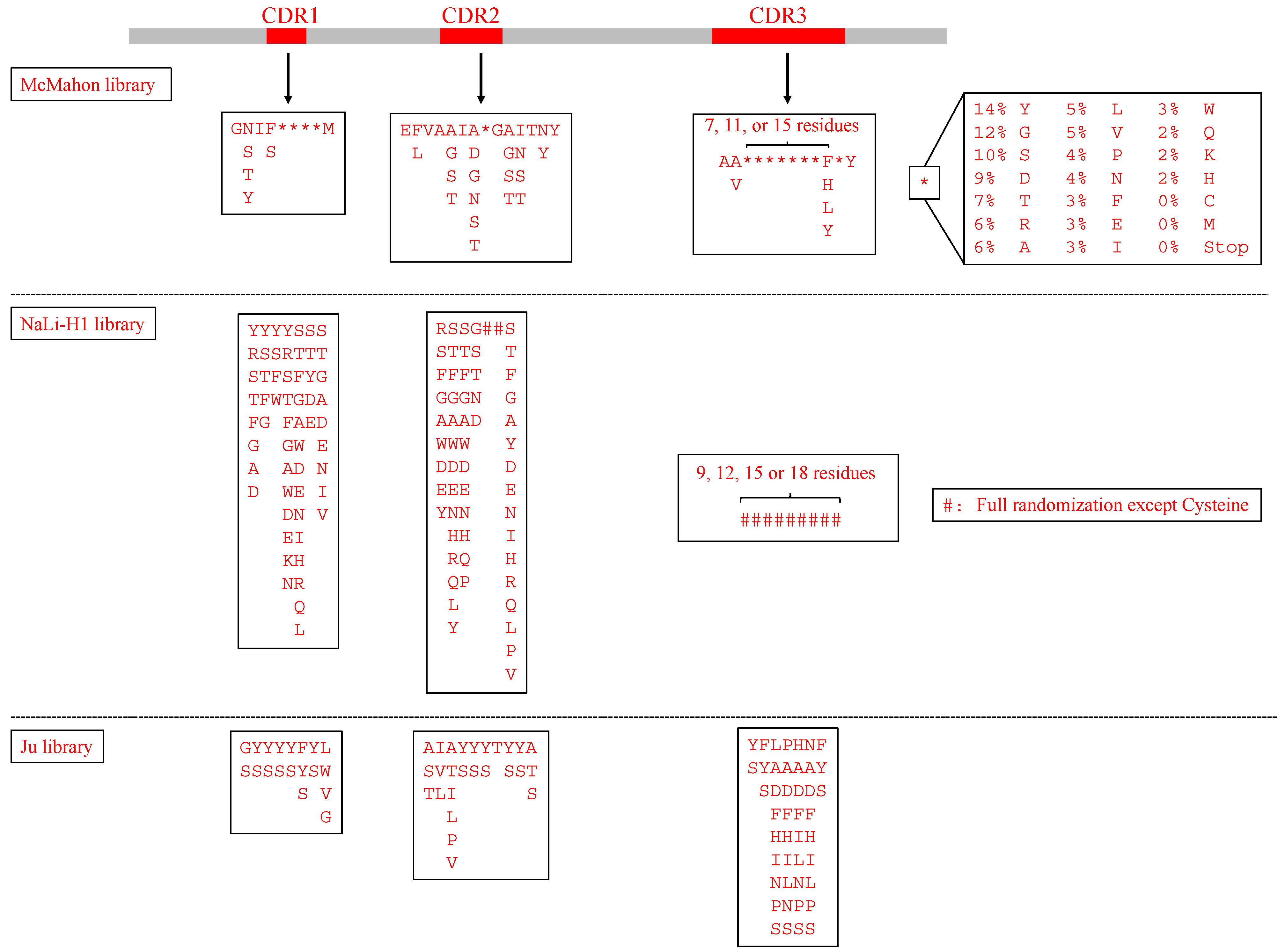
| Library Name | Framework | Randomized Region | Randomized Region Design | Biopanning Method | Antigen | Application |
|---|---|---|---|---|---|---|
| Ju library [10] | h_NbBcII10FGLA | 3 CDRs | Analyzed sequences of nanobodies found in nature and chose amino acids to enhance hydrogen bonding and hydrophobic interactions | Phage display | Interleukin-1β (IL-1β), Amyloid-β, Vascular endothelial growth factor (VEGF) | Identified nanobodies recognizing IL-1β, amyloid-β, and VEGF |
| Yan library [11] | cAbBCII10 | CDR3 only | NNK (where N = A/T/C/G, and K = G/T) | Phage display | Human prealbumin (PA), Neutrophil gelatinase-associated lipocalin (NGAL) | Developed a PA detection system |
| Wang library [12] | cAbBCII10 | 3 CDRs | NNK | Phage display | Glypican-3 (GPC3) | Identified four anti-GPC3 nanobodies as potential molecules for HCC diagnostic and therapeutic drugs |
| Wei library [13] | cAbBCII10 | 3 CDRs, and the length of CDR3 (9–20 amino acids) | CDR1+CDR2: partially randomization; CDR3: NNK | Phage display | M2 ion channel protein of influenza A virus | Showed potent neutralizing activities of nanobodies for influenza A viruses |
| NaLi-H1 [14] | hs2dAb | 3 CDRs, and the length of CDR3 (9, 12, 15 or 18 amino acids) | Analyzed the natural diversity from a llama naïve library. CDR1+CDR2: partially randomization; CDR3: fully randomization except for cysteine | Phage display | βActin, Tubulin, EGFP, mCherry | Selected nanobodies fused to a proteasome-targeting domain showed specific degradation of their targets and can be a potential tool for rapid protein knockdown in both cells and animals. |
| McMahon library [15] | a consensus framework derived from llama genes IGHV1S1-S5 | 3 CDRs, and the length of CDR3 (10, 14, or 18 amino acids) | Recapitulated the diversity of nanobodies uploaded in the wwPDB database | Yeast display | Human serum albumin, Metabolic hormone adiponectin, β2 adrenergic receptor, Human A2A adenosine receptor | Established an in vitro platform to choose conformationally selective nanobodies |
| Sevy library [16] | Alpaca IGHV3S53 and its humanized version | 3 CDRs, and the length of CDR3 (6–18 amino acids) | Mimicked the natural occurring VHH repertoire | Yeast display | Soluble mouse Programmed cell death protein 1(PD-1) ectodomain, Amyloid-β peptide, A G-protein coupled receptor (GPCR)—MrgX1 | Used anti-mPD-1 nanobodies to block mPD-1 and mPD-L1 interaction |
| Zimmermann library [17] | 3 nanobodies in RCSB PDB database: 3K1K, 3P0G, 1ZVH | 3 CDRs, and the length of CDR3 (6, 12, or 16 amino acids) | Obtained a balance between charged, polar, aromatic, and apolar amino acids, and based on the location of different structures such as in loops, in the middle of β-sheets | Ribosome display and subsequent phage display | Maltose-binding protein (MBP), Bacterial ABC transporter IrtAB and TM287/288, Human Solute Carrier (SLC) transporter ENT1 and GlyT1 | Recognized nanobodies targeting the transient ATP-bound state of bacterial ABC transporter TM287/288; Generated conformational-selective nanobodies against flexible transporters ENT1 and GlyT1 |
2. Synthetic Nanobody Library Design
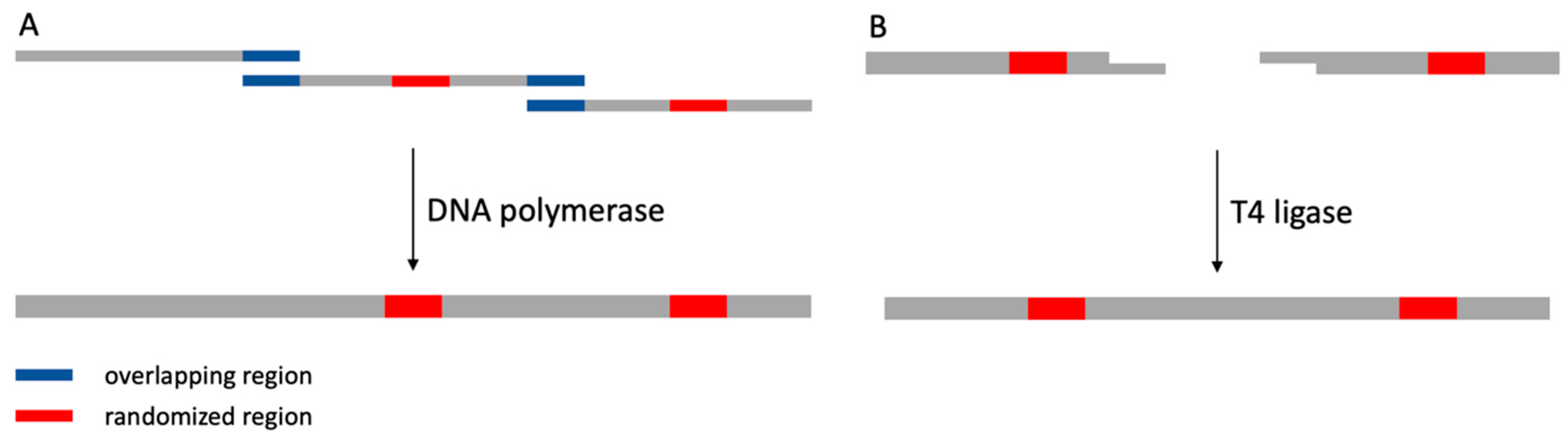
3. Construction of Synthetic Nanobody Library
4. Biopanning Method
5. Nanobody Application in Research
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hammers, C.; Songa, E.B.; Bendahman, N.; Hammers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef]
- Muyldermans, S.; Cambillau, C.; Wyns, L. Recognition of antigens by single-domain antibody fragments: The superfluous luxury of paired domains. Trends Biochem. Sci. 2001, 26, 230–235. [Google Scholar] [CrossRef]
- Mitchell, L.S.; Colwell, L.J. Analysis of nanobody paratopes reveals greater diversity than classical antibodies. Protein Eng. Des. Sel. 2018, 31, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Lauwereys, M.; Ghahroudi, M.A.; Desmyter, A.; Kinne, J.; Hölzer, W.; De Genst, E.; Wyns, L.; Muyldermans, S. Potent enzyme inhibitors derived from dromedary heavy-chain antibodies. EMBO J. 1998, 17, 3512–3520. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [Green Version]
- Duggan, S. Caplacizumab: First Global Approval. Drugs 2018, 78, 1639–1642. [Google Scholar] [CrossRef] [Green Version]
- Janssens, R.; Dekker, S.; Hendriks, R.W.; Panayotou, G.; Van Remoortere, A.; San, J.K.A.; Grosveld, F.; Drabek, D. Generation of heavy-chain-only antibodies in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 15130–15135. [Google Scholar] [CrossRef] [Green Version]
- Teng, Y.; Young, J.L.; Edwards, B.; Hayes, P.; Thompson, L.; Johnston, C.; Edwards, C.; Sanders, Y.; Writer, M.; Pinto, D.; et al. Diverse human VH antibody fragments with bio-therapeutic properties from the Crescendo Mouse. New Biotechnol. 2020, 55, 65–76. [Google Scholar] [CrossRef]
- Muyldermans, S. A guide to: Generation and design of nanobodies. FEBS J. 2020, 288, 2084–2102. [Google Scholar] [CrossRef]
- Ju, M.S.; Min, S.W.; Lee, S.M.; Kwon, H.S.; Park, J.C.; Lee, J.C.; Jung, S.T. A synthetic library for rapid isolation of humanized single-domain antibodies. Biotechnol. Bioprocess Eng. 2017, 22, 239–247. [Google Scholar] [CrossRef]
- Yan, J.; Li, G.; Hu, Y.; Ou, W.; Wan, Y. Construction of a synthetic phage-displayed Nanobody library with CDR3 regions randomized by trinucleotide cassettes for diagnostic applications. J. Transl. Med. 2014, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Xu, C.; Wang, H.; Jiang, C. Identification of nanobodies against hepatocellular carcinoma marker glypican-3. Mol. Immunol. 2021, 131, 13–22. [Google Scholar] [CrossRef]
- Wei, G.; Meng, W.; Guo, H.; Pan, W.; Liu, J.; Peng, T.; Chen, L.; Chen, C.Y. Potent neutralization of influenza a virus by a single-domain antibody blocking M2 ion channel protein. PLoS ONE 2011, 6, e28309. [Google Scholar] [CrossRef]
- Moutel, S.; Bery, N.; Bernard, V.; Keller, L.; Lemesre, E.; De Marco, A.; Ligat, L.; Rain, J.C.; Favre, G.; Olichon, A.; et al. NaLi-H1: A universal synthetic library of humanized nanobodies providing highly functional antibodies and intrabodies. eLife 2016, 5, e16228. [Google Scholar] [CrossRef]
- McMahon, C.; Baier, A.S.; Pascolutti, R.; Wegrecki, M.; Zheng, S.; Ong, J.X.; Erlandson, S.C.; Hilger, D.; Rasmussen, S.G.F.; Ring, A.M.; et al. Yeast surface display platform for rapid discovery of conformationally selective nanobodies. Nat. Struct. Mol. Biol. 2018, 25, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Sevy, A.M.; Chen, M.T.; Castor, M.; Sylvia, T.; Krishnamurthy, H.; Ishchenko, A.; Hsieh, C.M. Structure- and sequence-based design of synthetic single-domain antibody libraries. Protein Eng. Des. Sel. 2020, 33, gzaa028. [Google Scholar] [CrossRef]
- Zimmermann, I.; Egloff, P.; Hutter, C.A.J.; Arnold, F.M.; Stohler, P.; Bocquet, N.; Hug, M.N.; Huber, S.; Siegrist, M.; Hetemann, L.; et al. Synthetic single domain antibodies for the conformational trapping of membrane proteins. eLife 2018, 7, e34317. [Google Scholar] [CrossRef]
- Saerens, D.; Pellis, M.; Loris, R.; Pardon, E.; Dumoulin, M.; Matagne, A.; Wyns, L.; Muyldermans, S.; Conrath, K. Identification of a universal VHH framework to graft non-canonical antigen-binding loops of camel single-domain antibodies. J. Mol. Biol. 2005, 352, 597–607. [Google Scholar] [CrossRef]
- Vincke, C.; Loris, R.; Saerens, D.; Martinez-Rodriguez, S.; Muyldermans, S.; Conrath, K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J. Biol. Chem. 2009, 284, 3273–3284. [Google Scholar] [CrossRef] [Green Version]
- Monegal, A.; Olichon, A.; Bery, N.; Filleron, T.; Favre, G.; Marco, A. De Single domain antibodies with VH hallmarks are positively selected during panning of llama (Lama glama) naïve libraries. Dev. Comp. Immunol. 2012, 36, 150–156. [Google Scholar] [CrossRef]
- Olichon, A.; Surrey, T. Selection of genetically encoded fluorescent single domain antibodies engineered for efficient expression in Escherichia coli. J. Biol. Chem. 2007, 282, 36314–36320. [Google Scholar] [CrossRef] [Green Version]
- Nevoltris, D.; Lombard, B.; Dupuis, E.; Mathis, G.; Chames, P.; Baty, D. Conformational nanobodies reveal tethered epidermal growth factor receptor involved in EGFR/ErbB2 predimers. ACS Nano 2015, 9, 1388–1399. [Google Scholar] [CrossRef]
- Bannas, P.; Hambach, J.; Koch-Nolte, F. Nanobodies and nanobody-based human heavy chain antibodies as antitumor therapeutics. Front. Immunol. 2017, 8, 1603. [Google Scholar] [CrossRef]
- Kunz, P.; Zinner, K.; Mücke, N.; Bartoschik, T.; Muyldermans, S.; Hoheisel, J.D. The structural basis of nanobody unfolding reversibility and thermoresistance. Sci. Rep. 2018, 8, 7934. [Google Scholar] [CrossRef]
- Dunbar, J.; Krawczyk, K.; Leem, J.; Baker, T.; Fuchs, A.; Georges, G.; Shi, J.; Deane, C.M. SAbDab: The structural antibody database. Nucleic Acids Res. 2014, 42, D1140–D1146. [Google Scholar] [CrossRef]
- Zuo, J.; Li, J.; Zhang, R.; Xu, L.; Chen, H.; Jia, X.; Su, Z.; Zhao, L.; Huang, X.; Xie, W. Institute collection and analysis of Nanobodies (iCAN): A comprehensive database and analysis platform for nanobodies. BMC Genom. 2017, 18, 797. [Google Scholar] [CrossRef] [Green Version]
- Nimrod, G.; Fischman, S.; Austin, M.; Herman, A.; Keyes, F.; Leiderman, O.; Hargreaves, D.; Strajbl, M.; Breed, J.; Klompus, S.; et al. Computational Design of Epitope-Specific Functional Antibodies. Cell Rep. 2018, 25, 2121–2131. [Google Scholar] [CrossRef] [Green Version]
- Cawez, F.; Duray, E.; Hu, Y.; Vandenameele, J.; Romão, E.; Vincke, C.; Dumoulin, M.; Galleni, M.; Muyldermans, S.; Vandevenne, M. Combinatorial Design of a Nanobody that Specifically Targets Structured RNAs. J. Mol. Biol. 2018, 430, 1652–1670. [Google Scholar] [CrossRef]
- Kadkhodaei, S.; Memari, H.R.; Abbasiliasi, S.; Rezaei, M.A.; Movahedi, A.; Shun, T.J.; Ariff, A. Bin Multiple overlap extension PCR (MOE-PCR): An effective technical shortcut to high throughput synthetic biology. RSC Adv. 2016, 6, 66682–66694. [Google Scholar] [CrossRef]
- Greenwood, J.; Willis, A.E.; Perham, R.N. Multiple display of foreign peptides on a filamentous bacteriophage. Peptides from Plasmodium falciparum circumsporozoite protein as antigens. J. Mol. Biol. 1991, 220, 821–827. [Google Scholar] [CrossRef]
- Hoogenboom, H.R.; Griffiths, A.D.; Johnson, K.S.; Chiswell, D.J.; Hudson, P.; Winter, G. Multi-subunit proteins on the surface of filamentous phage: Methodologies for displaying antibody (Fab) heavy and light chains. Nucleic Acids Res. 1991, 19, 4133–4137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazan, J.; Całkosiñski, I.; Gamian, A. Phage displaya powerful technique for immunotherapy: 1. Introduction and potential of therapeutic applications. Hum. Vaccines Immunother. 2012, 8, 1817–1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmen, S.; Jermutus, L. Concepts in antibody phage display. Brief. Funct. Genom. Proteom. 2002, 1, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Alfaleh, M.A.; Alsaab, H.O.; Mahmoud, A.B.; Alkayyal, A.A.; Jones, M.L.; Mahler, S.M.; Hashem, A.M. Phage Display Derived Monoclonal Antibodies: From Bench to Bedside. Front. Immunol. 2020, 11, 1986. [Google Scholar] [CrossRef]
- Fellouse, F.A.; Esaki, K.; Birtalan, S.; Raptis, D.; Cancasci, V.J.; Koide, A.; Jhurani, P.; Vasser, M.; Wiesmann, C.; Kossiakoff, A.A.; et al. High-throughput Generation of Synthetic Antibodies from Highly Functional Minimalist Phage-displayed Libraries. J. Mol. Biol. 2007, 373, 924–940. [Google Scholar] [CrossRef]
- Alfaleh, M.; Jones, M.; Howard, C.; Mahler, S. Strategies for Selecting Membrane Protein-Specific Antibodies using Phage Display with Cell-Based Panning. Antibodies 2017, 6, 10. [Google Scholar] [CrossRef]
- Jones, M.L.; Alfaleh, M.A.; Kumble, S.; Zhang, S.; Osborne, G.W.; Yeh, M.; Arora, N.; Hou, J.J.C.; Howard, C.B.; Chin, D.Y.; et al. Targeting membrane proteins for antibody discovery using phage display. Sci. Rep. 2016, 6, 26240. [Google Scholar] [CrossRef]
- Leow, C.H.; Cheng, Q.; Fischer, K.; McCarthy, J. The Development of Single Domain Antibodies for Diagnostic and Therapeutic Applications. In Antibody Engineering; IntechOpen: London, UK, 2018; pp. 175–204. [Google Scholar]
- Löfblom, J. Bacterial display in combinatorial protein engineering. Biotechnol. J. 2011, 6, 1115–1129. [Google Scholar] [CrossRef]
- Kajiwara, K.; Aoki, W.; Ueda, M. Evaluation of the yeast surface display system for screening of functional nanobodies. AMB Express 2020, 10, 51. [Google Scholar] [CrossRef] [Green Version]
- Gai, S.A.; Wittrup, K.D. Yeast surface display for protein engineering and characterization. Curr. Opin. Struct. Biol. 2007, 17, 467–473. [Google Scholar] [CrossRef]
- Salema, V.; Fernández, L.Á. Escherichia coli surface display for the selection of nanobodies. Microb. Biotechnol. 2017, 10, 1468–1484. [Google Scholar] [CrossRef] [PubMed]
- Salema, V.; Marín, E.; Martínez-Arteaga, R.; Ruano-Gallego, D.; Fraile, S.; Margolles, Y.; Teira, X.; Gutierrez, C.; Bodelón, G.; Fernández, L.Á. Selection of Single Domain Antibodies from Immune Libraries Displayed on the Surface of E. coli Cells with Two β-Domains of Opposite Topologies. PLoS ONE 2013, 8, e75126. [Google Scholar] [CrossRef]
- Dower, W.J.; Miller, J.F.; Ragsdale, C.W. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 1988, 16, 6127–6145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanes, J.; Plückthun, A. In vitro selection and evolution of functional proteins by using ribosome display. Proc. Natl. Acad. Sci. USA 1997, 94, 4937–4942. [Google Scholar] [CrossRef] [Green Version]
- Zahnd, C.; Amstutz, P.; Plückthun, A. Ribosome display: Selecting and evolving proteins in vitro that specifically bind to a target. Nat. Methods 2007, 4, 269–279. [Google Scholar] [CrossRef]
- Saerens, D.; Frederix, F.; Reekmans, G.; Conrath, K.; Jans, K.; Brys, L.; Huang, L.; Bosmans, E.; Maes, G.; Borghs, G.; et al. Engineering camel single-domain antibodies and immobilization chemistry for human prostate-specific antigen sensing. Anal. Chem. 2005, 77, 7547–7555. [Google Scholar] [CrossRef]
- Schellenberg, M.J.; Petrovich, R.M.; Malone, C.C.; Williams, R.S. Selectable high-yield recombinant protein production in human cells using a GFP/YFP nanobody affinity support. Protein Sci. 2018, 27, 1083–1092. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Wang, X.; Chen, Q.; Yun, Y.; Tang, Z.; Liu, X. Nanobody-alkaline phosphatase fusion protein-based enzyme-linked immunosorbent assay for one-step detection of ochratoxin a in rice. Sensors 2018, 18, 4044. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Y.; Wang, K.; Lu, Q.; Ji, P.; Liu, B.; Zhu, J.; Liu, Q.; Sun, Y.; Zhang, J.; Zhou, E.M.; et al. Nanobody-horseradish peroxidase fusion protein as an ultrasensitive probe to detect antibodies against Newcastle disease virus in the immunoassay. J. Nanobiotechnol. 2019, 17, 35. [Google Scholar] [CrossRef] [Green Version]
- Mu, Y.; Jia, C.; Zheng, X.; Zhu, H.; Zhang, X.; Xu, H.; Liu, B.; Zhao, Q.; Zhou, E.M. A nanobody-horseradish peroxidase fusion protein-based competitive ELISA for rapid detection of antibodies against porcine circovirus type 2. J. Nanobiotechnol. 2021, 19, 34. [Google Scholar] [CrossRef]
- Warne, T.; Edwards, P.C.; Dorfe, A.S.; Leslie, A.G.W.; Tate, C.G. Molecular basis for high-affinity agonist binding in GPCRs. Science 2019, 778, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.G.F.; Choi, H.J.; Fung, J.J.; Pardon, E.; Casarosa, P.; Chae, P.S.; Devree, B.T.; Rosenbaum, D.M.; Thian, F.S.; Kobilka, T.S.; et al. Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature 2011, 469, 175–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dmitriev, O.Y.; Lutsenko, S.; Muyldermans, S. Nanobodies as probes for protein dynamics in vitro and in cells. J. Biol. Chem. 2016, 291, 3767–3775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Nokhrin, S.; Hassanzadeh-Ghassabeh, G.; Yu, C.H.; Yang, H.; Barry, A.N.; Tonelli, M.; Markley, J.L.; Muyldermans, S.; Dmitriev, O.Y.; et al. Interactions between metal-binding domains modulate intracellular targeting of Cu(I)-ATPase ATP7B, as revealed by nanobody binding. J. Biol. Chem. 2014, 289, 32682–32693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Nafría, J.; Lee, Y.; Bai, X.; Carpenter, B.; Tate, C.G. Cryo-EM structure of the adenosine A2A receptor coupled to an engineered heterotrimeric G protein. eLife 2018, 7, e35946. [Google Scholar] [CrossRef] [PubMed]
- Uchański, T.; Masiulis, S.; Fischer, B.; Kalichuk, V.; López-Sánchez, U.; Zarkadas, E.; Weckener, M.; Sente, A.; Ward, P.; Wohlkönig, A.; et al. Megabodies expand the nanobody toolkit for protein structure determination by single-particle cryo-EM. Nat. Methods 2021, 18, 60–68. [Google Scholar] [CrossRef]
- Uchański, T.; Pardon, E.; Steyaert, J. Nanobodies to study protein conformational states. Curr. Opin. Struct. Biol. 2020, 60, 117–123. [Google Scholar] [CrossRef]
- Rothbauer, U.; Zolghadr, K.; Tillib, S.; Nowak, D.; Schermelleh, L.; Gahl, A.; Backmann, N.; Conrath, K.; Muyldermans, S.; Cardoso, M.C.; et al. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat. Methods 2006, 3, 887–889. [Google Scholar] [CrossRef]
- Traenkle, B.; Rothbauer, U. Under the microscope: Single-domain antibodies for live-cell imaging and super-resolution microscopy. Front. Immunol. 2017, 8, 1030. [Google Scholar] [CrossRef] [Green Version]
- Ries, J.; Kaplan, C.; Platonova, E.; Eghlidi, H.; Ewers, H. A simple, versatile method for GFP-based super-resolution microscopy via nanobodies. Nat. Methods 2012, 9, 582–584. [Google Scholar] [CrossRef]
- Caussinus, E.; Kanca, O.; Affolter, M. Fluorescent fusion protein knockout mediated by anti-GFP nanobody. Nat. Struct. Mol. Biol. 2012, 19, 117–122. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, B.; Yang, D. Easily Established and Multifunctional Synthetic Nanobody Libraries as Research Tools. Int. J. Mol. Sci. 2022, 23, 1482. https://doi.org/10.3390/ijms23031482
Liu B, Yang D. Easily Established and Multifunctional Synthetic Nanobody Libraries as Research Tools. International Journal of Molecular Sciences. 2022; 23(3):1482. https://doi.org/10.3390/ijms23031482
Chicago/Turabian StyleLiu, Bingying, and Daiwen Yang. 2022. "Easily Established and Multifunctional Synthetic Nanobody Libraries as Research Tools" International Journal of Molecular Sciences 23, no. 3: 1482. https://doi.org/10.3390/ijms23031482
APA StyleLiu, B., & Yang, D. (2022). Easily Established and Multifunctional Synthetic Nanobody Libraries as Research Tools. International Journal of Molecular Sciences, 23(3), 1482. https://doi.org/10.3390/ijms23031482