Abstract
Duchenne muscular dystrophy (DMD) is caused by loss-of-function mutations in the dystrophin gene on chromosome Xp21. Disruption of the dystrophin–glycoprotein complex (DGC) on the cell membrane causes cytosolic Ca2+ influx, resulting in protease activation, mitochondrial dysfunction, and progressive myofiber degeneration, leading to muscle wasting and fragility. In addition to the function of dystrophin in the structural integrity of myofibers, a novel function of asymmetric cell division in muscular stem cells (satellite cells) has been reported. Therefore, it has been suggested that myofiber instability may not be the only cause of dystrophic degeneration, but rather that the phenotype might be caused by multiple factors, including stem cell and myofiber functions. Furthermore, it has been focused functional regulation of satellite cells by intracellular communication of extracellular vesicles (EVs) in DMD pathology. Recently, a novel molecular mechanism of DMD pathogenesis—circulating RNA molecules—has been revealed through the study of target pathways modulated by the Neutral sphingomyelinase2/Neutral sphingomyelinase3 (nSMase2/Smpd3) protein. In addition, adeno-associated virus (AAV) has been clinically applied for DMD therapy owing to the safety and long-term expression of transduction genes. Furthermore, the EV-capsulated AAV vector (EV-AAV) has been shown to be a useful tool for the intervention of DMD, because of the high efficacy of the transgene and avoidance of neutralizing antibodies. Thus, we review application of AAV and EV-AAV vectors for DMD as novel therapeutic strategy.
1. Introduction
Duchenne muscular dystrophy (DMD) is a severe and progressive muscular disorder, which mainly manifests as degeneration and regeneration in skeletal and cardiac muscles and, finally, leads to myofiber necrosis, fibrosis, and muscle weakness, through membrane fragility and disrupted cell signaling [1,2,3,4,5]. The incidence of DMD is estimated to be approximately 1 in 3500 to 10,000 newborn and adolescent males, with a prevalence of less than 10 cases per 100,000 males, which does not differ between regions [5,6,7,8,9,10,11]. Typically, the clinical symptoms of this disease are first recognized by muscular weakness, with difficulties in climbing stairs, a waddling gait, and frequent falls under five years of age, subsequently progressing into the loss of ability of independent ambulation, with wheelchair dependency around 10 years of age, eventually leading to premature death, due to respiratory and cardiac failure at approximately 20–40 years of age [5,12,13,14,15].
In DMD patients, sensitive damage and degeneration of muscle fibers, through sarcolemma destabilization and progressive loss of muscular repair ability via stem cells, concomitantly increase inflammation and fibrosis in muscular tissues, due to loss-of-function with frameshifting or nonsense mutations in the dystrophin gene on chromosome Xp21, which codes for the protein dystrophin, a myofiber membrane protein [1,16,17,18,19,20,21,22,23]. In a study of thousands of different mutations in DMD patients, the categories of each mutation were deletions, duplications, and point mutations, including small deletions and insertions, comprise of approximately 60–70%, 5–15%, and 20%, respectively [5,24,25]. In addition, two hotspot regions of these deletion and duplication clusters in the DMD patients are located at exons 45–55 (approximately 47%) and exons 3–9 (approximately 7%), respectively [5,26,27]. In addition, de novo germline mutations were observed in one-third of DMD patients; in these cases, the mothers were not somatic carriers of DMD mutations but had children with DMD [5,28,29]. The frequency of germline mutations in oocytes and sperms varies per individual but appears to be up to 14% [5,30].
The full functional dystrophin protein forms a complex known as the dystrophin-associated protein complexes (DAPCs), with ten kinds of proteins, including the dystroglycan subcomplex (α-dystroglycan and β-dystroglycan), the sarcoglycan subcomplex (α-sarcoglycan, β-sarcoglycan, γ-sarcoglycan, and δ-sarcoglycan), sarcospan, syntrophin, dystrobrevin, and neuronal nitric oxide synthase (nNOS) [5,31,32,33,34,35,36,37,38]. These differences are dependent not only on the type of tissues or cell but also on the different regions of the same myocyte on the cell membrane. It is critical for maintaining structural stability and integrity of the muscle sarcolemma by connecting the internal cytoskeletal F-actin of a muscle fiber to the surrounding extracellular matrix via its N- and C-terminal domains and transmission of forces within the muscle [5,31,32,33,34,35,36,37,38]. In DMD, DAPC disassembly is caused by dystrophin deficiency, resulting in the loss of the interaction between F-actin and the extracellular matrix, and, finally, wide-ranging dysfunction in muscle cells, resulting in high susceptibility to contraction damage [5]. The mislocation of nNOS from the sarcolemma to the sarcoplasm leads to a reduction in the total cellular level of nNOS, which decreases the production and secretion of nitric oxide to the surrounding vasculature and results in ischemic damage to muscle [5,39]. This mechanism, in which loss of sarcolemmal nNOS increases intracellular calcium concentrations via Ryanodibe receptor 1 (RyR1) leakage or nicotinamide adenine dinucleotide phosphate oxidases (NOX)-derived cytosolic reactive oxygen species (ROS) production, is supported by sarcolemmal tears (delta lesions) and rupture, which can be detected by passive leakage of muscle enzymes and microRNAs from muscles into the bloodstream [32,39,40,41].
The myofiber-specific genetic ablation of dystroglycan in mice does not result in dystrophy-like muscle degeneration; however, mice with muscle stem cells (also called satellite cells, SCs) exhibit specific loss of dystroglycan, with markedly delayed muscle regeneration [42]. In addition, SCs lacking dystrophin markedly increase the number of abnormal nonpolarized mitotic divisions and reduced asymmetric cell divisions, exacerbating dystrophic pathology [43,44]. These reports suggest that SC dysfunction is mainly involved in muscular dystrophy. In addition, it has been suggested that myofiber instability, leading to disruption of the dystrophin protein lining inside the cell membrane, is not the only cause of dystrophic degeneration but rather that the phenotype might be caused by multiple factors, including stem cell and myofiber functions. Thus, we review novel therapeutic strategy for DMD.
2. Therapy Strategy of DMD
To date, there are mainly two of treatment strategies for DMD have been explored: (1) the treatment for actual cause of DMD–dystrophin deficiency (dystriphin-targeted therapies), such as adeno-associated virus (AAV)-mediated micro/minidystrophin gene delivery, synthetic antisense oligonucleotides for exon skipping, nonsense readthrough, CRISPR-Cas9 (clustered regularly interspaced short palindromic repeat-CRISPR-associated protein 9)-mediated genome editing, protein replacement therapies, and primarily utropin; (2) the therapy of downstream pathological changes, such as transplantation of muscular stem cells, corticosteroids (prednisolone or deflazacort) with highly effective in therapy of concomitant destructive processes (namely inflammation) and an improvement in calcium homeostasis, leading to a decrease in oxidative stress in muscle tissue, and in mitochondrial function and biogenesis [45,46,47,48,49,50,51,52,53,54,55] (Figure 1). Currently, gene editing is attracting attention as a therapeutic strategy for DMD because of the restoration of the dystrophin reading frame in more than 40% of all patients with DMD [56,57,58,59,60,61,62,63]. However, it has been reported that clinical trials with this strategy have to be discontinued because of off-target effects that cause chromosomal abnormalities. In addition, the desired mutation may not be introduced in all cells, resulting in a somatic mosaic mutation, where it is difficult to undo edited parts of the genome. Additionally, as no trace of genome editing remains, it is difficult to investigate the cause of an adverse event. Therefore, the development of an additive gene therapy, as a safe protein supplementation, should be prioritized.
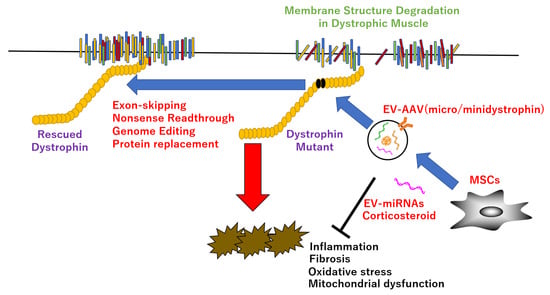
Figure 1.
Therapeutic strategy in DMD. EV-encapsulated AAV or miRNAs improve dystrophic pathology, such as inflammation, fibrosis, oxidative stress, and mitochondrial dysfunction, etc., via direct rescue of dystrophin mutant by exon-skipping, nonsense readthrough, genome editing, protein replacement, or indirect recovery of dystrophin functions by improvement of the downstream targets.
AAV vectors are derived from non-pathogenic viruses and capable of transferring genes into non-dividing cells. Moreover, because gene expression in target cells is sustainable for a long period of time, it is also excellent, in terms of safety and efficacy. Therefore, it is attracting attention as a clinically applied vector for gene therapy in DMD. Dysfunctions in skeletal and cardiac muscles of DMD model mdx mice are significantly attenuated by partial restoration of dystrophin protein using the micro-dystrophin protein, which delivers a part of the cDNA copy of dystrophin to the affected tissues, based on internally deleted dystrophins [64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79]. However, there are some problems with the administration of AAV vectors into the skeletal muscle of canine X-linked muscular dystrophy model in Japan (CXMDj), which shows severe dystrophic phenotypes without immunosuppression, resulting in insufficient transgene expression with potent innate immune responses [80,81]. Furthermore, the AAV vector has a limited carrying capacity of ~4.7 kb or less, because it is a vector derived from a small virus, whereas the muscle isoform of dystrophin, Dp427, is encoded by ~11.4 kb cDNA [82]. The time to reach the gene expression peak was long because of the single-stranded DNA genome. The gene transfer efficiency was reduced by the neutralizing antibody against the viral capsid of the AAV vector. The cost for large-scale production of AAV vectors is expensive, and large-scale protocols have not yet been established. In addition, deaths, carcinogenicity, hepatotoxicity, neurotoxicity, and thrombotic microangiopathy have been reported in clinical trials with high doses of AAV vectors, including micro-dystrophin [82,83,84,85].
In gene therapy using an exon skipping agent, the low efficiency of introduction into cardiomyocytes has become an issue [86,87]. In addition, it is impossible to treat all mutations in the dystrophin gene at present because the drug corresponds only to mutations in a limited exon region [88,89,90]. Besides, corticosteroid treatment of patients with DMD commonly showed adverse effects, such as a higher risk of developing respiratory and circulatory dysfunctions earlier, rather than later [91]. Thus, there is currently no complete and radical cure, and the establishment of new therapies is eagerly awaited.
3. Satellite and Immune Cells
A single intravenous dose of cardiac stromal cells, termed cardiosphere-derived cells (CDCs), which are cardiac progenitor/stromal cells with anti-inflammatory, antioxidant, antifibrotic, and cardiomyogenic properties [92,93], directly into the muscles of mdx mice improves the dystrophic phenotype via augmentation of cardiac and skeletal muscles [94]. Furthermore, extracellular vesicles (EVs), which are membrane microvesicles, approximately 30–100 nm in size, that are generated from multivesicular bodies of the terminal endosomal pathway, recapitulate the therapeutic benefits of the CDCs because the blockage of EV biogenesis fails to improve cardiac and skeletal muscles in vivo [93]. Both CDC and EV treatment in mdx partially reverse heart damage by attenuating fibrosis, as well as by decreasing nuclear factor-kappa B, NF-κB phosphorylation, and macrophage infiltration. Moreover, these treatments enhance cardiomyogenesis, normalize mitochondrial protein deficits, and improve the pathology of skeletal muscles by increasing force and promoting skeletal myogenesis with the activation of SCs, which proliferate and differentiate into myoblasts to form myotubes with reduced interstitial fibrosis [94].
Furthermore, adult skeletal muscles have a self-repair capacity, modulated by SCs and immune cells [95]. Macrophages modify myoblast proliferation and their commitment to differentiated myocytes, as well as the formation of mature myotubes. This is done by enhancing muscle growth through the interaction of differentially activated myogenic precursor cells (MPCs) with anti-inflammatory macrophages that may exert deleterious effects, due to the resolution of damaged tissues by removal of necrotic cells [89,90,91,92]. In general, macrophage infiltration in acutely injured muscle must terminate clearing of its damaged tissues, so as to prevent the destruction of the adjacent undamaged tissues. However, in chronic inflammatory conditions of DMD, macrophages persistently accumulate and contribute to disease progression by disturbing myogenesis, with SCs coordinated by macrophage polarization [94,96,97,98,99,100,101,102,103,104,105,106,107,108,109]. In particular, loss of the Kruppel-like factor 2 (KLF2) gene, in myeloid-derived cells, enhances the inflammatory immune response to muscle injury through the recruitment of greater numbers of inflammatory Ly6C-positive monocytes, which provide monocyte-derived mature Ly6C-, CD11b-, and F4/80-positive macrophages in circulation and enable regeneration via the activation of SCs and myogenesis [103]. During muscle regeneration, the normal transition from the pro- to anti-inflammatory phase, which is mainly controlled by macrophages, is required for spatiotemporal regulation of SC differentiation, angiogenesis, and matrix remodeling [104]. The initiation of the inflammatory response induces extravasation of leukocytes, of which Ly6C-positive, Ccr2-positive, and Cx3cr1-low circulating monocytes in the bloodstream cross the endothelium because of increased vascular permeability. Subsequently, the monocytes enter the damaged muscular tissues, and neutrophils mount the proinflammatory response to attract Ly6C-positive monocytes, mainly through the Ccl2-Ccr2 axis, thus clearing the surrounding debris via efferocytosis and removing the apoptotic cells via phagocytic cells [110,111,112,113,114]. In contrast, Ly6C-positive, Ccr2-positive, and Cx3cr1-high monocytes patrol to survey vessel wall integrity [110,113]. After the debris of muscular tissue is cleansed by phagocytosis, proliferated macrophages exhibit an anti-inflammatory/restorative response, which is characterized by myogenesis, growth of new myofibers, angiogenesis, and matrix remodeling, through interactions of the macrophages with surrounding cells, as in accordance with the shift of Ly6C-positive into Ly6C-negative within the damaged tissue [98,106,115,116,117,118,119,120]. The shift in macrophage status from pro- to anti-inflammatory was regulated by mediators of the resolution of inflammation, such as annexin A1 (AnxA1), developmental endothelial locus-1 (DEL-1), glucocorticoid-induced leucine zipper (GILZ), and secretory leukocyte peptidase inhibitor (SLPI), by which recognition and engulfment of dead cells triggers the transcription of anti-inflammatory effectors, such as transforming growth factor-β (TGF-β), interleukin (IL)-10, and specialized pro-resolving mediators, concomitant with the downregulation of inflammatory cytokines [110,121,122].
Furthermore, the resolution of inflammation during skeletal muscle regeneration in vivo is controlled by several molecular pathways that regulate macrophage conversion, including AMP-activated protein kinase, alpha 1 (AMPKα1), BTB and CNC homology 1, basic leucine zipper transcription factor 1 (Bach1), insulin growth factor-1 (IGF-1), mitogen-activated protein kinase phosphatase 1/dual specificity phosphatase 1 (Mkp-1/Dusp1), peroxisome proliferator activated receptor gamma (Ppar-γ), and scavenger receptor class B, member 1 (SRBI) [99,108,109,116,123,124,125]. Among them, loss of the Bach1 gene in myeloiod cells enhances the muscular regeneration process during acute muscular damage through the conversion of inflammatory Ly-6C-high and F4/80-high marcorphages to repair Ly-6C-low and F4/80-high macrophages. This conversion is mediated by the induced expression of heme oxygenase 1 (HO-1) and regulation of many key inflammatory and repair-related genes, such as IGF-1, solute carrier family 40 member 1 (Slc40a1), IL-6, IL-10, growth differentiation factor 3 (Gdf3), Ppar-γ, Dusp1, and CCAAT/enhancer binding protein (C/EBP), beta (Cebpb) [117]. In addition, phagocytosis of muscle debris participates in macrophage skewing: M1 macrophages are decreased upon the phagocytosis of necrotic and apoptotic muscular precursor cells, with decreased tumor necrosis factor-α (TNF-α) secretion, whereas M2 macrophages increased, in accordance with TGF-β secretion [99]. In contrast, the phagocytosis of macrophages in loss-of-function, myeloid-specific AMPKα1 was impaired, leading to a defect in their phenotypic transition into M2 macrophages, in which the impaired fusion ability and abnormal proliferation of myogenic precursor cells by M2a/c macrophages finally resulted in a delay of skeletal muscle regeneration [109]. However, these deficiencies were partially rescued by the transplantation of the bone marrow of the wild-type variant [109]. In addition, monocyte/macrophage-derived IGF-I coordinates macrophage polarization and myogenesis during muscle regeneration [94,108]. Moreover, Pparγ in macrophages regulates skeletal muscle regeneration via myoblast proliferation and differentiation by transactivation and secretion of Gdf3 [116]. However, the role of Pparγ in M2 polarization remains controversial. On the one hand, Pparγ activation in adipose tissues promotes M2 polarization [126]. On the other hand, the activation of Pparγ suppresses M2 polarization via the inactivation of cytotoxic T lymphocytes [127]. In addition, the conditional deletion of the Cebpb gene in muscle fibers shows normal regeneration, but the loss of the Cebpb gene in macrophages leads to defective M2 poralization, increased inflammation, and reduced ability of muscular regeneration [99].
Under hypoxic culture conditions, CDCs secrete EVs encapsulated with a high abundance of a microRNA, termed miR-148, which promotes the regeneration effects of EVs through differentiation of myogenic precursor [94]. The recruited Ly6C-positive monocytes/macrophages with pro-inflammatory profiles activate SCs by disrupting the quiescence of their niche during the expansion phase, leading to the formation of new functional myofibers, owing to the commitment of many progenitor cells to undergo myogenic differentiation and self-renewal of a subset to return to quiescence [101,121,128,129].
4. Non-Invasive Biomarkers
Non-invasive biomarkers that evaluate the progress of DMD pathology, following therapeutic interventions, are limited. Among potential biomarkers, such as proteins, nucleic acids, metabolites, polymorphisms, mutations, RNA splicing, and epigenetics, microRNAs (miRNAs), have been intensively studied. The RNA comprises of a class of small non-coding RNA molecules, 22 nucleotides in length, that can regulate gene expression at the post-transcriptional level by destabilizing mRNA and translation silencing. They have been selected because of their: (1) high content in body fluids, including serum, plasma, tear, lymph, breast milk, urine, semen, saliva, and sweat; (2) high-stability in the blood and outside the body, due to capsulation into EVs and formation of complex with RNA-binding proteins; (3) unique expression profile, correlated with pathology progression for monitoring the initial stage of the pathological condition, such as immediately before or after the onset; (4) high-sensitivity and specificity to suppress false positives and negatives; and (5) high-throughput and cost-reduction for evaluation systems of disease levels [130,131]. As for miRNAs in DMD as biomarkers, the levels of three myomiRs, namely miR-1, miR-133a, and miR-206, which are abundant miRNAs in muscles, were increased in the sera of patients and animal models of muscular dystrophy with restored levels, due to expression of functional dystrophin protein, which was shown to be inversely correlated with disease severity in DMD patients [132,133,134,135,136]. In addition, miR-1 and miR-133 are expressed in cardiac and skeletal muscles and involved in the proliferation and differentiation processes [137,138,139,140,141]. Moreover, mir-206 is a skeletal muscle-specific miRNA, expressed in SCs, involved in muscle development and regeneration [142]. In addition, in the muscle of mdx mice, miR-1 and miR-133a levels were shown to be downregulated, whereas miR-206 level was upregulated, recovering to wild-type levels by restoration of the dystrophin protein [133,134,135,136].
5. Neutral Sphingomyelinase 2/Sphingomyelin Phosphodiesterase 3 (nSMase2/Smpd3) and DMD
DMD exhibits inflammatory responses as a common feature, during which, after muscle injury, inflammatory macrophages are recruited to secrete inflammatory cytokines and miRNAs, due to repair and regeneration [143]. In particular, these myomiRs, encapsulated within EVs, which are produced by the biogenesis of ceramide from sphingomyelin, are released from cells into circulation and controlled by the nSMase2/Smpd3-regulated secretory machinery of EVs [144,145,146,147,148,149]. Thus, to elucidate the relationship between the release of myomiRs via EV and DMD pathogenesis, GW4869 (an inhibitor of nSMase2/Smpd3) was administered to mdx mice. It has been shown that the inhibition of nSMase2/Smpd3 enzymatic activity ameliorates skeletal muscles of muscular dystrophy in mdx mice, in turn, inhibits ceramide synthesis [150]. However, there are problems with the GW4869 inhibitor, in that it has effects on other nSMase2/Smpd3 family members and relatively short-term inhibitory effects. Therefore, to investigate the effects of nSMase2/Smpd3 on dystrophic pathology, we generated mdx mice lacking the nSMase2/Smpd3 gene (mdx:Smpd3 double knockout [DKO] mice) [151]. Deletion of the nSMase2/Smpd3 gene in mdx mice reduces inflammation in dystrophic muscles, as indicated by a reduction in the infiltration of excess inflammatory cells and decrease in inflammatory cytokine expression levels, such as TNF-α, CD68, CD45, chemokine (C-C motif) receptor 5 (Ccr5), IL-1 receptor antagonist (IL-1ra), and IL-6 [151]. In addition, disruption of the nSMase2/Smpd3 gene attenuated muscle membrane permeability in dystrophic mdx mice initially but exacerbated it later on. Serum creatine kinase (CK) levels were significantly lower in 6- to 12-week-old mdx:Smpd3 DKO mice than in mdx mice. However, at 28 weeks, moderately or significantly higher serum CK levels were observed in mdx:Smpd3 DKO mice than in mdx mice. Furthermore, at 12 weeks of age, there were significantly fewer Evans blue dye (EBD: degree marker of myofiber damage)-positive muscle fibers in the tibialis anterior (TA) muscle of mdx:Smpd3 DKO mice than in that of mdx mice. However, at 20 weeks of age, the number of EBD-positive muscle fibers in the TA of some mdx:Smpd3 DKO mice was higher than that in mdx mice.
It has been reported that nSMase2/Smpd3 inhibition reduces inflammatory responses via nuclear factor (erythroid derived 2)-like 2 activation. This activation then induces antioxidant response element-controlled genes and decreases the expression of inflammatory genes, including vascular cell adhesion molecule 1, intercellular adhesion molecule 1, monocyte chemoattractant protein-1, IL-1β, IL-6, and TNF-α, by inhibiting NF-κB activation in macrophages and endothelial cells, ultimately leading to suppression of the recruitment of monocytes to the endothelium and macrophage M1 differentiation [152]. Furthermore, nSMase2/Smpd3 acts upstream of mitogen-activated protein kinase and NF-κB signaling pathways of TNF-α mediated inflammatory responses in monocytic cells/macrophages [153].
Furthermore, the systemic administration of EVs, particularly myotube-derived EVs, induced phenotypic rescue and mitigated pathological progression in dystrophic mice. This was due to improved membrane integrity, accompanied by the inhibition of intracellular influx and calcium-dependent calpain activation, which prevents the degradation of the destabilized dystrophin-associated protein complex [154]. These results suggest that, early in life, nSMase2/Smpd3 ablation may have beneficial effects, with respect to excess inflammatory responses and myofiber membrane degeneration in mdx mice but that it may have adverse effects later on. In addition, genetic ablation of nSMase2/Smpd3 improves muscle performance in mice with dystrophic phenotypes. The grip strength test demonstrated that, at 12 weeks of age, the muscle strength of mdx mice was significantly lower than that of wild-type mice, yet the muscle strength of mdx:Smpd3 DKO mice significantly recovered. In addition, in an inclined treadmill running test, mdx:Smpd3 DKO mice ran significantly longer than mdx mice at 16 and 60 weeks of age.
In addition to muscle degeneration in DMD, the loss of dystrophin in the brain has often been associated with nonprogressive cognitive deficits, behavioral disabilities, and enhanced fearfulness [155,156,157]. Thus, to investigate anxiety, emotionality, and the adaptive stress response to a novel environment in mdx:Smpd3 DKO mice, the hole-board test was performed [151]. The results exhibited a loss of the Smpd3 gene modulates anxiety behavior and stress responses, as well as the recovery of brain-derived neurotrophic factor (BDNF) expression, through exosomal miRNA in the hippocampus [151]. These findings suggest that DMD is not only a muscular disease but also a circulatory RNA disease, through intracellular communication. Furthermore, the signaling pathways modulated by the nSMase2/Smpd3 protein might be novel therapeutic targets for DMD via the regulation of the expression levels of exosomal miRNAs using this nSMase2/Smpd3 protein.
6. Adeno-Associated Virus (AAV)-Encapsulating EVs
Several clinical trials for DMD, using AAV vectors with various promoters, have been performed and are currently undergoing practical application ([15,48,66,82,158,159,160,161,162,163,164,165,166,167,168,169,170,171], Table 1). However, the risk of immune response from large doses of the AAV vector by systemic single administration to DMD patients was unresolved [172,173]. Thus, several strategies have been reported to suppress the immune response related to AAV vector administration. The first is the use of immunosuppressants, which are transduced into long-term skeletal muscles in non-human primates [174]. Second, examination of congenital and acquired immune tolerance induction methods showed that AAV vector-mediated microdystrophin, which is part of the full-length dystrophin gene, improved canine DMD pathology by the induction of immune tolerance. Moreover, systemic injection of the AAV9 vector with MSCs, which is a novel solution for treating DMD, into CXMDj of severe dystrophic phenotypes improves gene transfer and dystrophic phenotypes in skeletal muscle and heart functions by immune modulation [81,175]. Finally, vectors can be developed to avoid immune system attacks [81].

Table 1.
AAV vector serotypes used in Clinical trials for DMD.
In addition, MSCs have been shown to improve pulmonary, cardiac, and distal skeletal muscle functions by their ability to fuse with dystrophic muscle, anti-inflammatory activities, and trophic factors that augment the activity of endogenous repair cells [176,177]. Furthermore, systemic administration of continuous IL-10 expressing MSCs by AAV vector into the CXMDj model indicated improvement of running capacity and recovery of tetanic force [178]. These findings showed the possibility of reducing the AAV administration dose in DMD patients to approximately 1/100 dose, with multiple administrations. However, some problems associated with gene therapy using AAV vectors remain. First, the systemic administration of AAV vectors carries a risk of carcinogenicity, hepatotoxicity, neurotoxicity, and thrombotic microangiopathy [172,173]. In addition, modification of the AAV capsid for construction of novel AAV vectors has unpredictable immunotoxicity and can damage non-target organs or exhibit unknown cross-reactivity of neutralizing antibody reaction for the AAV capsid in humans, with possible decreased productivity of the AAV vectors [179,180,181,182]. In addition to gene therapy, which is a treatment method for supplying or adding the genome editing strategy, which repairs mutations in abnormal genes or causes a loss of function of specific genes, has been explored [183,184,185]. Although the low efficiency of modifying target genes for the development of new in vivo technologies has been overcome, undesired effects have also increased, including the following: (1) off-target effects, (2) side effects with abnormal chromosome cleavege, and (3) carcinogenicity with suppression effects of p53 by homologous recombination [61,186,187,188]. In fact, clinical trials using genome editing have been discontinued because of suspicion of the development of chromosomal abnormalities. Therefore, as one of the novel gene therapy strategies, a drug delivery system for the AAV-encapsulating EV, which contains the AAV vector within the EV, was reported to be useful in the intervention of various target tissues in some diseases [189,190,191,192,193,194,195,196,197,198,199,200]. Furthermore, the effectiveness of gene therapy with AAV vectors is greatly affected by neutralizing antibodies [81,201,202,203]. Therefore, the induction of immune tolerance to AAV vectors has become an important issue for efficient and safe gene transduction. Nevertheless, it is possible to induce a reduction in immunogenicity to the AAV vector by encapsulating the AAV vector within EVs, which is naturally present in the body (as a natural transporter); it is expected that AAV vector transduction is more efficient than AAV vector administration alone [189,194,204]. In addition, AAV vectors administrated in vivo are consumed by innate immunity plasmacytoid dendritic cells, as mediated by Toll-like receptor 9 [205]. Therefore, attempts have been made to suppress these immune responses [81,174,177]. Furthermore, AAV-encapsulating EVs can also be introduced into in vivo barrier tissues such as the brain, eyes, and inner ear [189,190,191,192,193,194,195,196,197,198,199,200]. Therefore, repeated administration of AAV vectors would be possible because of dose reduction for in vivo administration with immunosuppression and/or EVs.
Although certain AAV serotypes, such as AAV9, efficiently, target muscle fibers, transduction of SCs has less reported. However, it was recently suggested that gene editing to the dys reading frame in the mdx mice was induced by transduction of AAV vector into the SCs [206,207]. Also, it was known that AAV vectors show their low immunogenicity, leading to persistence and long-term transgene expression of the vector, due to the reduced ability of AAV vectors to activate antigen-presenting cells [208]. However, neo-antigens, including viral capsid and the transgene product introduced by AAV in mdx muscle vectors elicit cellular and humoral immune responses, which can be reduced using immune modulatory treatments targeting T cells, B cells, and pro-inflammatory processes [208,209]. It was indicated that treatment with the immune modulatory drugs, including cytotoxic T-lymphocyte-associated protein 4 (CTLA4)-linked IgG2a, Rituximab, Prednisolone, and VBP6 enhanced the beneficial effects of AAV-microdystrophin therapy for force generation [208].
Also, interaction between the SCs and EVs was critical event for myogenesis and angiogenesis during skeletal muscle regeneration [120,210]. In addition with this reports, EVs from muscular progenitor cells (MPCs) treated with a histone deacetylase inhibitor (HDACi), which has been shown to increase muscle regeneration in mdx mice, were enriched myoangiogenesis-related miRNAs, such as miR-181a, miR-17, miR-210, miR-107, miR-19b, Let-7e-5p, miR-26a, and miR-103 compared with EVs from untreated MPCs [211]. Further, EVs released by MSCs such as fibro-adipogenic progenitors (FAPs) transfer miRNAs, which cooperatively target therapeutic pathways related with regeneration, fibrosis, and inflammation, to SCs [211]. Since increase of miR-206 level within the EVs released by FAPs in muscles indicated the essential roles of the miR-206 for the EV-induced expansion of MPCs and regeneration of dystrophic muscles [211]. These reports suggest that miRNA transduction via the EVs into MPCs will be novel therapeutic intervention in muscular dystrophy.
7. Conclusions
Currently, some therapeutic strategies for DMD have been clinically applied, but no curative treatment for DMD has been yet established. Although AAV vectors are considered to be a promising therapeutic strategy for treating DMD, because of their safety and usefulness, their use still has concerns, such as adverse effects caused by a single high-dose administration. However, it was clarified that the induction of immune tolerance by MSCs improved the therapeutic effects by alleviating the action of immune cells on AAV vectors. Furthermore, AAV-encapsulated EVs could be a novel therapeutic strategy for DMD because they can escape from neutralizing antibodies.
Author Contributions
Y.M., Y.H., K.H. and T.O. drafted the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by DAIICHI SANKYO COMPANY, LIMITED, Joint Research Fund.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
The preparation of this manuscript was supported by Hayashita-Kinoh H. and Nitahara-Kasahara Y.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Monaco, A.P.; Feener, C.C.; Kunkel, L.M. Conservation of the Duchenne muscular dystrophy gene in mice and humans. Science 1987, 238, 347–350. [Google Scholar] [CrossRef]
- Hoffman, E.P. The discovery of dystrophin, the protein product of the Duchenne muscular dystrophy gene. FEBS J. 2020, 287, 3879–3887. [Google Scholar] [CrossRef]
- Shirokova, N.; Niggli, E. Cardiac phenotype of Duchenne Muscular Dystrophy: Insights from cellular studies. J. Mol. Cell Cardiol. 2013, 58, 217–224. [Google Scholar] [CrossRef]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Colvin, M.K.; Cripe, L.; Herron, A.R.; Kennedy, A.; Kinnett, K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Considerations Working Group. Lancet Neurol. 2018, 17, 251–267. [Google Scholar] [CrossRef]
- Mah, J.K.; Korngut, L.; Dykeman, J.; Day, L.; Pringsheim, T.; Jette, N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul. Disord. 2014, 24, 482–491. [Google Scholar] [CrossRef]
- Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.M.; Duval, B.; Aoyagi, A.; et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD Global Database: Analysis of more than 7000 Duchenne muscular dystrophy mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef]
- Ryder, S.; Leadley, R.M.; Armstrong, N.; Westwood, M.; de Kock, S.; Butt, T.; Jain, M.; Kleijnen, J. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: An evidence review. Orphanet J. Rare Dis. 2017, 12, 79. [Google Scholar] [CrossRef]
- Mercuri, E.; Bönnemann, C.G.; Muntoni, F. Muscular dystrophies. Lancet 2019, 394, 2025–2038. [Google Scholar] [CrossRef]
- Passamano, L.; Taglia, A.; Palladino, A.; Viggiano, E.; D’Ambrosio, P.; Scutifero, M.; Rosaria Cecio, M.; Torre, V.; DE Luca, F.; Picillo, E.; et al. Improvement of survival in Duchenne Muscular Dystrophy: Retrospective analysis of 835 patients. Acta Myol. 2012, 31, 121–125. [Google Scholar]
- Mendell, J.R.; Rodino-Klapac, L.R. Duchenne muscular dystrophy: CRISPR/Cas9 treatment. Cell Res. 2016, 26, 513–514. [Google Scholar] [CrossRef]
- McGreevy, J.W.; Hakim, C.H.; McIntosh, M.A.; Duan, D. Animal models of Duchenne muscular dystrophy: From basic mechanisms to gene therapy. Dis. Model Mech. 2015, 8, 195–213. [Google Scholar] [CrossRef]
- Chamberlain, J.R.; Chamberlain, J.S. Progress toward gene therapy for Duchenne muscular dystrophy. Mol. Ther. 2017, 25, 1125–1131. [Google Scholar] [CrossRef]
- Ervasti, J.M.; Ohlendieck, K.; Kahl, S.D.; Gaver, M.G.; Campbell, K.P. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 1990, 345, 315–319. [Google Scholar] [CrossRef]
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002, 82, 291–329. [Google Scholar] [CrossRef]
- Walcher, T.; Steinbach, P.; Spiess, J.; Kunze, M.; Gradinger, R.; Walcher, D.; Bernhardt, P. Detection of long-term progression of myocardial fibrosis in Duchenne muscular dystrophy in an affected family: A cardiovascular magnetic resonance study. Eur. J. Radiol. 2011, 80, 115–119. [Google Scholar] [CrossRef]
- Farini, A.; Gowran, A.; Bella, P.; Sitzia, C.; Scopece, A.; Castiglioni, E.; Rovina, D.; Nigro, P.; Villa, C.; Fortunato, F.; et al. Fibrosis rescue improves cardiac function in dystrophin-deficient mice and duchenne patient-specific cardiomyocytes by immunoproteasome modulation. Am. J. Pathol. 2019, 189, 339–353. [Google Scholar] [CrossRef]
- Pichavant, C.; Aartsma-Rus, A.; Clemens, P.R.; Davies, K.E.; Dickson, G.; Takeda, S.; Wilton, S.D.; Wolff, J.A.; Wooddell, C.I.; Xiao, X.; et al. Current status of pharmaceutical and genetic therapeutic approaches to treat DMD. Mol. Ther. 2011, 19, 830–840. [Google Scholar] [CrossRef]
- Gao, Q.Q.; McNally, E.M. the dystrophin complex: Structure, function, and implications for therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [CrossRef] [PubMed]
- Verhaart, I.E.C.; Aartsma-Rus, A. herapeutic developments for Duchenne muscular dystrophy. Nat. Rev. Neurol. 2019, 15, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; van Deutekom, J.C.; Fokkema, I.F.; Van Ommen, G.J.; Den Dunnen, J.T. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006, 34, 135–144. [Google Scholar] [CrossRef]
- Magri, F.; Govoni, A.; D’Angelo, M.G.; del Bo, R.; Ghezzi, S.; Sandra, G.; Turconi, A.C.; Sciacco, M.; Ciscato, P.; Bordoni, A.; et al. Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up. J. Neurol. 2011, 258, 1610–1623. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Fueki, N.; Shiba, N.; Motoki, H.; Miyazaki, D.; Nishizawa, H.; Echigoya, Y.; Yokota, T.; Aoki, Y.; Takeda, S. Deletion of exons 3-9 encompassing a mutational hot spot in the DMD gene presents an asymptomatic phenotype, indicating a target region for multiexon skipping therapy. J. Hum. Genet. 2016, 61, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Shiba, N.; Miyazaki, D.; Nishizawa, H.; Inaba, Y.; Fueki, N.; Maruyama, R.; Echigoya, Y.; Yokota, T. Comparison of the phenotypes of patients harboring in-frame deletions starting at exon 45 in the Duchenne muscular dystrophy gene indicates potential for the development of exon skipping therapy. J. Hum. Genet. 2017, 62, 459–463. [Google Scholar] [CrossRef]
- Chen, W.J.; Lin, Q.F.; Zhang, Q.J.; He, J.; Liu, X.Y.; Lin, M.T.; Murong, S.X.; Liou, C.W.; Wang, N. Molecular analysis of the dystrophin gene in 407 Chinese patients with Duchenne/Becker muscular dystrophy by the combination of multiplex ligation-dependent probe amplification and Sanger sequencing. Clin. Chim. Acta 2013, 423, 35–38. [Google Scholar] [CrossRef]
- Garcia, S.; de Haro, T.; Zafra-Ceres, M.; Poyatos, A.; Gomez-Capilla, J.A.; Gomez-Llorente, C. Identification of de novo mutations of Duchenne/Becker muscular dystrophies in southern Spain. Int. J. Med. Sci. 2014, 11, 988–993. [Google Scholar] [CrossRef][Green Version]
- Helderman-van den Enden, A.T.; de Jong, R.; den Dunnen, J.T.; Houwing-Duistermaat, J.J.; Kneppers, A.L.; Ginjaar, H.B.; Breuning, M.H.; Bakker, E. Recurrence risk due to germ line mosaicism: Duchenne and Becker muscular dystrophy. Clin. Genet. 2009, 75, 465–472. [Google Scholar] [CrossRef]
- Ervasti, J.M.; Sonnemann, K.J. Biology of the striated muscle dystrophin-glycoprotein complex. Int. Rev. Cytol. 2008, 265, 191–225. [Google Scholar] [CrossRef] [PubMed]
- Morizumi, H.; Hizawa, K.; Nunomura, S.; Ii, K. Comparative study of alterations of skeletal muscle in Duchenne muscular dystrophy and polymyositis. Acta Pathol. Jpn. 1984, 34, 1221–1242. [Google Scholar] [CrossRef] [PubMed]
- Omairi, S.; Hau, K.L.; Collins-Hooper, H.; Scott, C.; Vaiyapuri, S.; Torelli, S.; Montanaro, F.; Matsakas, A.; Patel, K. Regulation of the dystrophin-associated glycoprotein complex composition by the metabolic properties of muscle fibres. Sci. Rep. 2019, 9, 2770. [Google Scholar] [CrossRef] [PubMed]
- Gumerson, J.D.; Michele, D.E. The dystrophin-glycoprotein complex in the prevention of muscle damage. J. Biomed. Biotechnol. 2011, 2011, 210797. [Google Scholar] [CrossRef] [PubMed]
- Ibraghimov-Beskrovnaya, O.; Ervasti, J.M.; Leveille, C.J.; Slaughter, C.A.; Sernett, S.W.; Campbell, K.P. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 1992, 355, 696–702. [Google Scholar] [CrossRef]
- Campbell, K.P.; Kahl, S.D. Association of dystrophin and an integral membrane glycoprotein. Nature 1989, 338, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Ozawa, E. Glycoprotein complex anchoring dystrophin to sarcolemma. J. Biochem. 1990, 108, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Ervasti, J.M.; Campbell, K.P. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 1993, 122, 809–823. [Google Scholar] [CrossRef]
- Sander, M.; Chavoshan, B.; Harris, S.A.; Iannaccone, S.T.; Stull, J.T.; Thomas, G.D.; Victor, R.G. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2000, 97, 13818–13823. [Google Scholar] [CrossRef]
- Perry, M.M.; Muntoni, F. Noncoding RNAs and Duchenne muscular dystrophy. Epigenomics 2016, 8, 1527–1537. [Google Scholar] [CrossRef]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of dystrophin disrupts skeletal muscle signaling: Roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef] [PubMed]
- Pestronk, A.; Parhad, I.M.; Drachman, D.B.; Price, D.L. Membrane myopathy: Morphological similarities to Duchenne muscular dystrophy. Muscle Nerve 1982, 5, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Cohn, R.D.; Henry, M.D.; Michele, D.E.; Barresi, R.; Saito, F.; Moore, S.A.; Flanagan, J.D.; Skwarchuk, M.W.; Robbins, M.E.; Mendell, J.R.; et al. Disruption of DAG1 in differentiated skeletal muscle reveals a role for dystroglycan in muscle regeneration. Cell 2002, 110, 639–648. [Google Scholar] [CrossRef]
- Dumont, N.A.; Wang, Y.X.; von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef]
- Doorenweerd, N. Combining genetics, neuropsychology and neuroimaging to improve understanding of brain involvement in Duchenne muscular dystrophy—A narrative review. Neuromuscul. Disord. 2020, 30, 437–442. [Google Scholar] [CrossRef]
- Mázala, D.A.; Novak, J.S.; Hogarth, M.W.; Nearing, M.; Adusumalli, P.; Tully, C.B.; Habib, N.F.; Gordish-Dressman, H.; Chen, Y.W.; Jaiswal, J.K.; et al. TGF-β-driven muscle degeneration and failed regeneration underlie disease onset in a DMD mouse model. JCI Insight 2020, 5, e135703. [Google Scholar] [CrossRef]
- Shieh, P.B. Emerging strategies in the treatment of duchenne muscular dystrophy. Neurotherapeutics 2018, 15, 840–848. [Google Scholar] [CrossRef]
- Waldrop, M.A.; Flanigan, K.M. Update in Duchenne and Becker muscular dystrophy. Curr. Opin. Neurol. 2019, 32, 722–727. [Google Scholar] [CrossRef]
- Sun, C.; Shen, L.; Zhang, Z.; Xie, X. Therapeutic strategies for duchenne muscular dystrophy: An update. Genes 2020, 11, 837. [Google Scholar] [CrossRef]
- Péladeau, C.; Adam, N.J.; Jasmin, B.J. Celecoxib treatment improves muscle function in mdx mice and increases utrophin A expression. Celecoxib treatment improves muscle function in mdx mice and increases utrophin A expression. FASEB J. 2018, 32, 5090–5103. [Google Scholar] [CrossRef]
- Spurney, C.F.; Rocha, C.T.; Henricson, E.; Florence, J.; Mayhew, J.; Gorni, K.; Pasquali, L.; Pestronk, A.; Martin, G.R.; Hu, F.; et al. CINRG pilot trial of coenzyme Q10 in steroid-treated Duchenne muscular dystrophy. Muscle Nerve 2011, 44, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Servais, L.; Straathof, C.S.M.; Schara, U.; Klein, A.; Leinonen, M.; Hasham, S.; Meier, T.; De Waele, L.; Gordish-Dressman, H.; McDonald, C.M.; et al. Long-term data with idebenone on respiratory function outcomes in patients with Duchenne muscular dystrophy. Neuromuscul. Disord. 2020, 30, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Dubinin, M.V.; Starinets, V.S.; Talanov, E.Y.; Mikheeva, I.B.; Belosludtseva, N.V.; Belosludtsev, K.N. alisporivir improves mitochondrial function in skeletal muscle of mdx mice but suppresses mitochondrial dynamics and biogenesis. Int. J. Mol. Sci. 2021, 22, 9780. [Google Scholar] [CrossRef] [PubMed]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Belosludtseva, N.V.; Belosludtsev, K.N. the effect of deflazacort treatment on the functioning of skeletal muscle mitochondria in duchenne muscular dystrophy. Int. J. Mol. Sci. 2020, 21, 8763. [Google Scholar] [CrossRef] [PubMed]
- Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.P.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 2008, 14, 442–447. [Google Scholar] [CrossRef]
- Stocco, A.; Smolina, N.; Sabatelli, P.; Šileikytė, J.; Artusi, E.; Mouly, V.; Cohen, M.; Forte, M.; Schiavone, M.; Bernardi, P. Treatment with a triazole inhibitor of the mitochondrial permeability transition pore fully corrects the pathology of sapje zebrafish lacking dystrophin. Pharmacol. Res. 2021, 165, 105421. [Google Scholar] [CrossRef]
- Angelini, G.; Mura, G.; Messina, G. Therapeutic approaches to preserve the musculature in Duchenne Muscular Dystrophy: The importance of the secondary therapies. Exp. Cell Res. 2021, 410, 112968. [Google Scholar] [CrossRef]
- Soblechero-Martín, P.; Albiasu-Arteta, E.; Anton-Martinez, A.; de la Puente-Ovejero, L.; Garcia-Jimenez, I.; González-Iglesias, G.; Larrañaga-Aiestaran, I.; López-Martínez, A.; Poyatos-García, J.; Ruiz-Del-Yerro, E.; et al. Duchenne muscular dystrophy cell culture models created by CRISPR/Cas9 gene editing and their application in drug screening. Sci. Rep. 2021, 11, 18188. [Google Scholar] [CrossRef]
- Olson, E.N. Toward the correction of muscular dystrophy by gene editing. Proc. Natl. Acad. Sci. USA 2021, 118, e2004840117. [Google Scholar] [CrossRef]
- Chen, M.; Shi, H.; Gou, S.; Wang, X.; Li, L.; Jin, Q.; Wu, H.; Zhang, H.; Li, Y.; Wang, L.; et al. In vivo genome editing in mouse restores dystrophin expression in Duchenne muscular dystrophy patient muscle fibers. Genome Med. 2021, 13, 57. [Google Scholar] [CrossRef]
- Gee, P.; Lung, M.S.Y.; Okuzaki, Y.; Sasakawa, N.; Iguchi, T.; Makita, Y.; Hozumi, H.; Miura, Y.; Yang, L.F.; Iwasaki, M.; et al. Extracellular nanovesicles for packaging of CRISPR-Cas9 protein and sgRNA to induce therapeutic exon skipping. Nat. Commun. 2020, 11, 1334. [Google Scholar] [CrossRef]
- Morisaka, H.; Yoshimi, K.; Okuzaki, Y.; Gee, P.; Kunihiro, Y.; Sonpho, E.; Xu, H.; Sasakawa, N.; Naito, Y.; Nakada, S.; et al. CRISPR-Cas3 induces broad and unidirectional genome editing in human cells. Nat. Commun. 2019, 10, 5302. [Google Scholar] [CrossRef]
- Min, Y.L.; Li, H.; Rodriguez-Caycedo, C.; Mireault, A.A.; Huang, J.; Shelton, J.M.; McAnally, J.R.; Amoasii, L.; Mammen, P.P.A.; Bassel-Duby, R.; et al. CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci. Adv. 2019, 5, eaav4324. [Google Scholar] [CrossRef]
- Potter, R.A.; Griffin, D.A.; Heller, K.N.; Peterson, E.L.; Clark, E.K.; Mendell, J.R.; Rodino-Klapac, L.R. Dose-escalation study of systemically delivered rAAVrh74.MHCK7.micro-dystrophin in the mdx mouse model of duchenne muscular dystrophy. Hum. Gene Ther. 2021, 32, 375–389. [Google Scholar] [CrossRef]
- Hakim, C.H.; Clément, N.; Wasala, L.P.; Yang, H.T.; Yue, Y.; Zhang, K.; Kodippili, K.; Adamson-Small, L.; Pan, X.; Schneider, J.S.; et al. Micro-dystrophin AAV vectors made by transient transfection and herpesvirus system are equally potent in treating mdx mouse muscle disease. Mol. Ther. Methods Clin. Dev. 2020, 18, 664–678. [Google Scholar] [CrossRef]
- Ramos, J.N.; Hollinger, K.; Bengtsson, N.E.; Allen, J.M.; Hauschka, S.D.; Chamberlain, J.S. Development of novel micro-dystrophins with enhanced functionality. Mol. Ther. 2019, 27, 623–635. [Google Scholar] [CrossRef]
- Wasala, N.B.; Shin, J.H.; Lai, Y.; Yue, Y.; Montanaro, F.; Duan, D. Cardiac-specific expression of deltaH2-R15 mini-dystrophin normalized all electrocardiogram abnormalities and the end-diastolic volume in a 23-month-old mouse model of duchenne dilated cardiomyopathy. Hum. Gene Ther. 2018, 29, 737–748. [Google Scholar] [CrossRef]
- Hakim, C.H.; Wasala, N.B.; Pan, X.; Kodippili, K.; Yue, Y.; Zhang, K.; Yao, G.; Haffner, B.; Duan, S.X.; Ramos, J.; et al. A five-repeat micro-dystrophin gene ameliorated dystrophic phenotype in the severe DBA/2J-mdx model of duchenne muscular dystrophy. Mol. Ther. Methods Clin. Dev. 2017, 6, 216–230. [Google Scholar] [CrossRef]
- Bostick, B.; Shin, J.H.; Yue, Y.; Wasala, N.B.; Lai, Y.; Duan, D. AAV micro-dystrophin gene therapy alleviates stress-induced cardiac death but not myocardial fibrosis in >21-m-old mdx mice, an end-stage model of Duchenne muscular dystrophy cardiomyopathy. J. Mol. Cell Cardiol. 2012, 53, 217–222. [Google Scholar] [CrossRef]
- Bostick, B.; Shin, J.H.; Yue, Y.; Duan, D. AAV-microdystrophin therapy improves cardiac performance in aged female mdx mice. Mol. Ther. 2011, 19, 1826–1832. [Google Scholar] [CrossRef]
- Shin, J.H.; Nitahara-Kasahara, Y.; Hayashita-Kinoh, H.; Ohshima-Hosoyama, S.; Kinoshita, K.; Chiyo, T.; Okada, H.; Okada, T.; Takeda, S. Improvement of cardiac fibrosis in dystrophic mice by rAAV9-mediated microdystrophin transduction. Gene Ther. 2011, 18, 910–919. [Google Scholar] [CrossRef]
- Bostick, B.; Yue, Y.; Lai, Y.; Long, C.; Li, D.; Duan, D. Adeno-associated virus serotype-9 microdystrophin gene therapy ameliorates electrocardiographic abnormalities in mdx mice. Hum. Gene Ther. 2008, 19, 851–856. [Google Scholar] [CrossRef]
- Wang, Z.; Kuhr, C.S.; Allen, J.M.; Blankinship, M.; Gregorevic, P.; Chamberlain, J.S.; Tapscott, S.J.; Storb, R. Sustained AAV-mediated dystrophin expression in a canine model of Duchenne muscular dystrophy with a brief course of immunosuppression. Mol. Ther. 2007, 15, 1160–1166. [Google Scholar] [CrossRef]
- Yoshimura, M.; Sakamoto, M.; Ikemoto, M.; Mochizuki, Y.; Yuasa, K.; Miyagoe-Suzuki, Y.; Takeda, S. AAV vector-mediated microdystrophin expression in a relatively small percentage of mdx myofibers improved the mdx phenotype. Mol. Ther. 2004, 10, 821–828. [Google Scholar] [CrossRef]
- Yue, Y.; Li, Z.; Harper, S.Q.; Davisson, R.L.; Chamberlain, J.S.; Duan, D. Microdystrophin gene therapy of cardiomyopathy restores dystrophin-glycoprotein complex and improves sarcolemma integrity in the mdx mouse heart. Circulation 2003, 108, 1626–1632. [Google Scholar] [CrossRef]
- Watchko, J.; O’Day, T.; Wang, B.; Zhou, L.; Tang, Y.; Li, J.; Xiao, X. Adeno-associated virus vector-mediated minidystrophin gene therapy improves dystrophic muscle contractile function in mdx mice. Hum. Gene Ther. 2002, 13, 1451–1460. [Google Scholar] [CrossRef]
- Wang, B.; Li, J.; Xiao, X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc. Natl. Acad. Sci. USA 2000, 97, 13714–13719. [Google Scholar] [CrossRef]
- Danilov, K.A.; Vassilieva, S.G.; Polikarpova, A.V.; Starikova, A.V.; Shmidt, A.A.; Galkin, I.I.; Tsitrina, A.A.; Egorova, T.V.; Orlov, S.N.; Kotelevtsev, Y.V. In vitro assay for the efficacy assessment of AAV vectors expressing microdystrophin. Exp. Cell Res. 2020, 392, 112033. [Google Scholar] [CrossRef]
- Jørgensen, L.H.; Larochelle, N.; Orlopp, K.; Dunant, P.; Dudley, R.W.; Stucka, R.; Thirion, C.; Walter, M.C.; Laval, S.H.; Lochmüller, H. Efficient and fast functional screening of microdystrophin constructs in vivo and in vitro for therapy of duchenne muscular dystrophy. Hum. Gene Ther. 2009, 20, 641–650. [Google Scholar] [CrossRef]
- Yuasa, K.; Yoshimura, M.; Urasawa, N.; Ohshima, S.; Howell, J.M.; Nakamura, A.; Hijikata, T.; Miyagoe-Suzuki, Y.; Takeda, S. Injection of a recombinant AAV serotype 2 into canine skeletal muscles evokes strong immune responses against transgene product. Gene Ther. 2007, 14, 1249–1260. [Google Scholar] [CrossRef]
- Ohshima, S.; Shin, J.H.; Yuasa, K.; Nishiyama, A.; Kira, J.; Okada, T.; Takeda, S. Transduction efficiency and immune response associated with the administration of AAV8 vector into dog skeletal muscle. Mol. Ther. 2009, 17, 73–80. [Google Scholar] [CrossRef]
- Duan, D. Systemic AAV Micro-dystrophin gene therapy for duchenne muscular dystrophy. Mol. Ther. 2018, 26, 2337–2356. [Google Scholar] [CrossRef]
- Agarwal, S. High-dose AAV gene therapy deaths. Nat. Biotechnol. 2020, 38, 910. [Google Scholar] [CrossRef]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging issues in AAV-mediated in vivo gene therapy. Mol. Ther. Methods Clin. Dev. 2017, 8, 87–104. [Google Scholar] [CrossRef]
- Chandler, R.J.; LaFave, M.C.; Varshney, G.K.; Trivedi, N.S.; Carrillo-Carrasco, N.; Senac, J.S.; Wu, W.; Hoffmann, V.; Elkahloun, A.G.; Burgess, S.M.; et al. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J. Clin. Investig. 2015, 125, 870–880. [Google Scholar] [CrossRef]
- Shimizu-Motohashi, Y.; Murakami, T.; Kimura, E.; Komaki, H.; Watanabe, N. Exon skipping for Duchenne muscular dystrophy: A systematic review and meta-analysis. Orphanet J. Rare Dis. 2018, 13, 93. [Google Scholar] [CrossRef]
- Niks, E.H.; Aartsma-Rus, A. Exon skipping: A first in class strategy for Duchenne muscular dystrophy. Expert Opin. Biol. Ther. 2017, 17, 225–236. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Straub, V.; Hemmings, R.; Haas, M.; Schlosser-Weber, G.; Stoyanova-Beninska, V.; Mercuri, E.; Muntoni, F.; Sepodes, B.; Vroom, E.; et al. Development of exon skipping therapies for duchenne muscular dystrophy: A critical review and a perspective on the outstanding issues. Nucleic Acid Ther. 2017, 27, 251–259. [Google Scholar] [CrossRef]
- Echigoya, Y.; Lim, K.R.Q.; Nakamura, A.; Yokota, T. Multiple exon skipping in the duchenne muscular dystrophy hot spots: Prospects and challenges. J. Pers. Med. 2018, 8, 41. [Google Scholar] [CrossRef]
- Kim, S.; Zhu, Y.; Romitti, P.A.; Fox, D.J.; Sheehan, D.W.; Valdez, R.; Matthews, D.; Barber, B.J.; MD STARnet. Associations between timing of corticosteroid treatment initiation and clinical outcomes in Duchenne muscular dystrophy. Neuromuscul. Disord. 2017, 27, 730–737. [Google Scholar] [CrossRef]
- Marbán, E. A mechanistic roadmap for the clinical application of cardiac cell therapies. Nat. Biomed. Eng. 2018, 2, 353–361. [Google Scholar] [CrossRef]
- Aminzadeh, M.A.; Rogers, R.G.; Gouin, K.; Fournier, M.; Tobin, R.E.; Guan, X.; Childers, M.K.; Andres, A.M.; Taylor, D.J.; Ibrahim, A.; et al. Reversal of Cardiac and Skeletal Manifestations of Duchenne Muscular Dystrophy by Catdiosphere-Derived Cells and Their EVs in mdx Dystrophic Mice and in Human DUCHENNE Cardiomyocytes. bioRxiv Website. 2017. Available online: https://www.biorxiv.org/content/biorxiv/early/2017/04/20/128900.full.pdf (accessed on 22 November 2021).
- Rogers, R.G.; Fournier, M.; Sanchez, L.; Ibrahim, A.G.; Aminzadeh, M.A.; Lewis, M.I.; Marbán, E. Disease-modifying bioactivity of intravenous cardiosphere-derived cells and EVs in mdx mice. JCI Insight 2019, 4, e130202. [Google Scholar] [CrossRef]
- Juhas, M.; Abutaleb, N.; Wang, J.T.; Ye, J.; Shaikh, Z.; Sriworarat, C.; Qian, Y.; Bursac, N. Incorporation of macrophages into engineered skeletal muscle enables enhanced muscle regeneration. Nat. Biomed. Eng. 2018, 2, 942–954. [Google Scholar] [CrossRef]
- Chazaud, B.; Sonnet, C.; Lafuste, P.; Bassez, G.; Rimaniol, A.C.; Poron, F.; Authier, F.J.; Dreyfus, P.A.; Gherardi, R.K. Satellite cells attract monocytes and use macrophages as a support to escape apoptosis and enhance muscle growth. J. Cell Biol. 2003, 163, 1133–1143. [Google Scholar] [CrossRef]
- Bencze, M.; Negroni, E.; Vallese, D.; Yacoub-Youssef, H.; Chaouch, S.; Wolff, A.; Aamiri, A.; Di Santo, J.P.; Chazaud, B.; Butler-Browne, G.; et al. Proinflammatory macrophages enhance the regenerative capacity of human myoblasts by modifying their kinetics of proliferation and differentiation. Mol. Ther. 2012, 20, 2168–2179. [Google Scholar] [CrossRef]
- Saclier, M.; Yacoub-Youssef, H.; Mackey, A.L.; Arnold, L.; Ardjoune, H.; Magnan, M.; Sailhan, F.; Chelly, J.; Pavlath, G.K.; Mounier, R.; et al. Differentially activated macrophages orchestrate myogenic precursor cell fate during human skeletal muscle regeneration. Stem Cells 2013, 31, 384–396. [Google Scholar] [CrossRef]
- Ruffell, D.; Mourkioti, F.; Gambardella, A.; Kirstetter, P.; Lopez, R.G.; Rosenthal, N.; Nerlov, C. A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc. Natl. Acad. Sci. USA 2009, 106, 17475–17480. [Google Scholar] [CrossRef]
- Porter, J.D.; Guo, W.; Merriam, A.P.; Khanna, S.; Cheng, G.; Zhou, X.; Andrade, F.H.; Richmonds, C.; Kaminski, H.J. Persistent over-expression of specific CC class chemokines correlates with macrophage and T-cell recruitment in mdx skeletal muscle. Neuromuscul. Disord. 2003, 13, 223–235. [Google Scholar] [CrossRef]
- McArthur, S.; Juban, G.; Gobbetti, T.; Desgeorges, T.; Theret, M.; Gondin, J.; Toller-Kawahisa, J.E.; Reutelingsperger, C.P.; Chazaud, B.; Perretti, M.; et al. Annexin A1 drives macrophage skewing to accelerate muscle regeneration through AMPK activation. J. Clin. Investig. 2020, 130, 1156–1167. [Google Scholar] [CrossRef]
- Zhang, C.; Cheng, N.; Qiao, B.; Zhang, F.; Wu, J.; Liu, C.; Li, Y.; Du, J. Age-related decline of interferon-gamma responses in macrophage impairs satellite cell proliferation and regeneration. J. Cachexia Sarcopenia Muscle 2020, 11, 1291–1305. [Google Scholar] [CrossRef]
- Manoharan, P.; Song, T.; Radzyukevich, T.L.; Sadayappan, S.; Lingrel, J.B.; Heiny, J.A. KLF2 in myeloid lineage cells regulates the innate immune response during skeletal muscle injury and regeneration. iScience 2019, 17, 334–346. [Google Scholar] [CrossRef]
- Madaro, L.; Torcinaro, A.; De Bardi, M.; Contino, F.F.; Pelizzola, M.; Diaferia, G.R.; Imeneo, G.; Bouchè, M.; Puri, P.L.; de Santa, F. Macrophages fine tune satellite cell fate in dystrophic skeletal muscle of mdx mice. PLoS Genet. 2019, 15, e1008408. [Google Scholar] [CrossRef]
- Tidball, J.G.; Welc, S.S. Macrophage-Derived IGF-1 Is a potent coordinator of myogenesis and inflammation in regenerating muscle. Mol. Ther. 2015, 23, 1134–1135. [Google Scholar] [CrossRef]
- Arnold, L.; Henry, A.; Poron, F.; Baba-Amer, Y.; van Rooijen, N.; Plonquet, A.; Gherardi, R.K.; Chazaud, B. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 2007, 204, 1057–1069. [Google Scholar] [CrossRef]
- Du, H.; Shih, C.H.; Wosczyna, M.N.; Mueller, A.A.; Cho, J.; Aggarwal, A.; Rando, T.A.; Feldman, B.J. Macrophage-released ADAMTS1 promotes muscle stem cell activation. Nat. Commun. 2017, 8, 669. [Google Scholar] [CrossRef]
- Tonkin, J.; Temmerman, L.; Sampson, R.D.; Gallego-Colon, E.; Barberi, L.; Bilbao, D.; Schneider, M.D.; Musarò, A.; Rosenthal, N. Monocyte/macrophage-derived IGF-1 orchestrates murine skeletal muscle regeneration and modulates autocrine polarization. Mol. Ther. 2015, 23, 1189–1200. [Google Scholar] [CrossRef]
- Mounier, R.; Théret, M.; Arnold, L.; Cuvellier, S.; Bultot, L.; Göransson, O.; Sanz, N.; Ferry, A.; Sakamoto, K.; Foretz, M.; et al. AMPKalpha1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab. 2013, 18, 251–264. [Google Scholar] [CrossRef]
- Chazaud, B. Inflammation and skeletal muscle regeneration: Leave it to the macrophages! Trends Immunol. 2020, 41, 481–492. [Google Scholar] [CrossRef]
- Martinez, C.O.; McHale, M.J.; Wells, J.T.; Ochoa, O.; Michalek, J.E.; McManus, L.M.; Shireman, P.K. Regulation of skeletal muscle regeneration by CCR2-activating chemokines is directly related to macrophage recruitment. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R832–R842. [Google Scholar] [CrossRef]
- Ochoa, O.; Sun, D.; Reyes-Reyna, S.M.; Waite, L.L.; Michalek, J.E.; McManus, L.M.; Shireman, P.K. Delayed angiogenesis and VEGF production in CCR2-/- mice during impaired skeletal muscle regeneration. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R651–R661. [Google Scholar] [CrossRef]
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404. [Google Scholar] [CrossRef]
- Sugimoto, M.A.; Vago, J.P.; Perretti, M.; Teixeira, M.M. Mediators of the resolution of the inflammatory response. Trends Immunol. 2019, 40, 212–227. [Google Scholar] [CrossRef]
- Lemos, D.R.; Babaeijandaghi, F.; Low, M.; Chang, C.K.; Lee, S.T.; Fiore, D.; Zhang, R.H.; Natarajan, A.; Nedospasov, S.A.; Rossi, F.M. Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF-mediated apoptosis of fibro/adipogenic progenitors. Nat. Med. 2015, 21, 786–794. [Google Scholar] [CrossRef]
- Varga, T.; Mounier, R.; Patsalos, A.; Gogolák, P.; Peloquin, M.; Horvath, A.; Pap, A.; Daniel, B.; Nagy, G.; Pintye, E.; et al. Macrophage PPARgamma, a lipid activated transcription factor controls the growth factor GDF3 and skeletal muscle regeneration. Immunity 2016, 45, 1038–1051. [Google Scholar] [CrossRef]
- Zhao, W.; Lu, H.; Wang, X.; Ransohoff, R.M.; Zhou, L. CX3CR1 deficiency delays acute skeletal muscle injury repair by impairing macrophage functions. FASEB J. 2016, 30, 380–393. [Google Scholar] [CrossRef]
- Latroche, C.; Weiss-Gayet, M.; Muller, L.; Gitiaux, C.; Leblanc, P.; Liot, S.; Ben-Larbi, S.; Abou-Khalil, R.; Verger, N.; Bardot, P.; et al. Coupling between myogenesis and angiogenesis during skeletal muscle regeneration is stimulated by restorative macrophages. Stem Cell Rep. 2017, 9, 2018–2033. [Google Scholar] [CrossRef]
- Giannakis, N.; Sansbury, B.E.; Patsalos, A.; Hays, T.T.; Riley, C.O.; Han, X.; Spite, M.; Nagy, L. Dynamic changes to lipid mediators support transitions among macrophage subtypes during muscle regeneration. Nat. Immunol. 2019, 20, 626–636. [Google Scholar] [CrossRef]
- Juban, G.; Saclier, M.; Yacoub-Youssef, H.; Kernou, A.; Arnold, L.; Boisson, C.; Ben Larbi, S.; Magnan, M.; Cuvellier, S.; Théret, M.; et al. AMPK activation regulates LTBP4-dependent TGF-beta1 secretion by pro-inflammatory macrophages and controls fibrosis in duchenne muscular dystrophy. Cell Rep. 2018, 25, 2163–2176.e6. [Google Scholar] [CrossRef]
- Watanabe, S.; Alexander, M.; Misharin, A.V.; Budinger, G.R.S. The role of macrophages in the resolution of inflammation. J. Clin. Investig. 2019, 129, 2619–2628. [Google Scholar] [CrossRef]
- Gordon, S.; Plüddemann, A. Macrophage clearance of apoptotic cells: A critical assessment. Front. Immunol. 2018, 9, 127. [Google Scholar] [CrossRef]
- Perdiguero, E.; Sousa-Victor, P.; Ruiz-Bonilla, V.; Jardí, M.; Caelles, C.; Serrano, A.L.; Muñoz-Cánoves, P. p38/MKP-1-regulated AKT coordinates macrophage transitions and resolution of inflammation during tissue repair. J. Cell Biol. 2011, 195, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Patsalos, A.; Tzerpos, P.; Halasz, L.; Nagy, G.; Pap, A.; Giannakis, N.; Lyroni, K.; Koliaraki, V.; Pintye, E.; Dezso, B.; et al. The BACH1-HMOX1 regulatory axis is indispensable for proper macrophage subtype specification and skeletal muscle regeneration. J. Immunol. 2019, 203, 1532–1547. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Qu, C.; Li, T.; Cui, W.; Wang, X.; Du, J. Phagocytosis mediated by scavenger receptor class BI promotes macrophage transition during skeletal muscle regeneration. J. Biol. Chem. 2019, 294, 15672–15685. [Google Scholar] [CrossRef]
- Bouhlel, M.A.; Derudas, B.; Rigamonti, E.; Dièvart, R.; Brozek, J.; Haulon, S.; Zawadzki, C.; Jude, B.; Torpier, G.; Marx, N.; et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007, 6, 137–143. [Google Scholar] [CrossRef]
- Van Ginderachter, J.A.; Meerschaut, S.; Liu, Y.; Brys, L.; De Groeve, K.; Hassanzadeh Ghassabeh, G.; Raes, G.; de Baetselier, P. Peroxisome proliferator-activated receptor gamma (PPARgamma) ligands reverse CTL suppression by alternatively activated (M2) macrophages in cancer. Blood 2006, 108, 525–535. [Google Scholar] [CrossRef]
- Baghdadi, M.B.; Tajbakhsh, S. Regulation and phylogeny of skeletal muscle regeneration. Dev. Biol. 2018, 433, 200–209. [Google Scholar] [CrossRef]
- Wosczyna, M.N.; Rando, T.A. A muscle stem cell support group: Coordinated cellular responses in muscle regeneration. Dev. Cell 2018, 46, 135–143. [Google Scholar] [CrossRef]
- Zayed, Y.; Qi, X.; Peng, C. Identification of novel MicroRNAs and characterization of MicroRNA expression profiles in zebrafish ovarian follicular cells. Front. Endocrinol. 2019, 10, 518. [Google Scholar] [CrossRef]
- Marozzo, R.; Pegoraro, V.; Angelini, C. MiRNAs, myostatin, and muscle MRI imaging as biomarkers of clinical features in becker muscular dystrophy. Diagnostics 2020, 10, 713. [Google Scholar] [CrossRef]
- Mizuno, H.; Nakamura, A.; Aoki, Y.; Ito, N.; Kishi, S.; Yamamoto, K.; Sekiguchi, M.; Takeda, S.; Hashido, K. Identification of muscle-specific microRNAs in serum of muscular dystrophy animal models: Promising novel blood-based markers for muscular dystrophy. PLoS ONE 2011, 6, e18388. [Google Scholar] [CrossRef]
- Cacchiarelli, D.; Legnini, I.; Martone, J.; Cazzella, V.; D’Amico, A.; Bertini, E.; Bozzoni, I. miRNAs as serum biomarkers for Duchenne muscular dystrophy. EMBO Mol. Med. 2011, 3, 258–265. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Zhao, L.; Zhang, D.; Yao, X.; Zhang, H.; Wang, Y.C.; Wang, X.Y.; Xia, H.; Yan, J.; et al. Circulating muscle-specific miRNAs in duchenne muscular dystrophy patients. Mol. Ther. Nucleic Acids 2014, 3, e177. [Google Scholar] [CrossRef]
- Roberts, T.C.; Blomberg, K.E.; McClorey, G.; El Andaloussi, S.; Godfrey, C.; Betts, C.; Coursindel, T.; Gait, M.J.; Smith, C.I.; Wood, M.J. Expression analysis in multiple muscle groups and serum reveals complexity in the microRNA transcriptome of the mdx mouse with implications for therapy. Mol. Ther. Nucleic Acids 2012, 14, e39. [Google Scholar] [CrossRef]
- Cacchiarelli, D.; Martone, J.; Girardi, E.; Cesana, M.; Incitti, T.; Morlando, M.; Nicoletti, C.; Santini, T.; Sthandier, O.; Barberi, L.; et al. MicroRNAs involved in molecular circuitries relevant for the Duchenne muscular dystrophy pathogenesis are controlled by the dystrophin/nNOS pathway. Cell Metab. 2010, 12, 341–351. [Google Scholar] [CrossRef]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar] [CrossRef]
- Xu, C.; Lu, Y.; Pan, Z.; Chu, W.; Luo, X.; Lin, H.; Xiao, J.; Shan, H.; Wang, Z.; Yang, B. The muscle-specific microRNAs miR-1 and miR-133 produce opposing effects on apoptosis by targeting HSP60, HSP70 and caspase-9 in cardiomyocytes. J. Cell Sci. 2007, 120, 3045–3052. [Google Scholar] [CrossRef]
- Townley-Tilson, W.H.; Callis, T.E.; Wang, D. MicroRNAs 1, 133, and 206: Critical factors of skeletal and cardiac muscle development, function, and disease. Int. J. Biochem. Cell Biol. 2010, 42, 1252–1255. [Google Scholar] [CrossRef]
- Huang, Z.P.; Neppl, R.L., Jr.; Wang, D.Z. Application of microRNA in cardiac and skeletal muscle disease gene therapy. Methods Mol. Biol. 2011, 709, 197–210. [Google Scholar] [CrossRef]
- Huang, Z.P.; Espinoza-Lewis, R.; Wang, D.Z. Determination of miRNA targets in skeletal muscle cells. Methods Mol. Biol. 2012, 798, 475–490. [Google Scholar] [CrossRef]
- Ma, G.; Wang, Y.; Li, Y.; Cui, L.; Zhao, Y.; Zhao, B.; Li, K. MiR-206, a key modulator of skeletal muscle development and disease. Int. J. Biol. Sci. 2015, 11, 345–352. [Google Scholar] [CrossRef]
- Howard, Z.M.; Lowe, J.; Blatnik, A.J., 3rd; Roberts, D.; Burghes, A.H.M.; Bansal, S.S.; Rafael-Fortney, J.A. Early Inflammation in Muscular Dystrophy Differs between Limb and Respiratory Muscles and Increases with Dystrophic Severity. Am. J. Pathol. 2021, 191, 730–747. [Google Scholar] [CrossRef]
- Salehi, M.; Sharifi, M. Exosomal miRNAs as novel cancer biomarkers: Challenges and opportunities. J. Cell Physiol. 2018, 233, 6370–6380. [Google Scholar] [CrossRef]
- Kacperska, M.J.; Walenczak, J.; Tomasik, B. Plasmatic microRNA as potential biomarkers of multiple sclerosis: Literature review. Adv. Clin. Exp. Med. 2016, 25, 775–779. [Google Scholar] [CrossRef]
- Arantes, L.M.R.B.; de Carvalho, A.C.; Melendez, M.E.; Lopes Carvalho, A. Serum, plasma and saliva biomarkers for head and neck cancer. Expert Rev. Mol. Diagn. 2018, 18, 85–112. [Google Scholar] [CrossRef]
- Barceló, M.; Mata, A.; Bassas, L.; Larriba, S. Exosomal microRNAs in seminal plasma are markers of the origin of azoospermia and can predict the presence of sperm in testicular tissue. Hum. Reprod. 2018, 33, 1087–1098. [Google Scholar] [CrossRef]
- Wolenski, F.S.; Shah, P.; Sano, T.; Shinozawa, T.; Bernard, H.; Gallacher, M.J.; Wyllie, S.D.; Varrone, G.; Cicia, L.A.; Carsillo, M.E.; et al. Identification of microRNA biomarker candidates in urine and plasma from rats with kidney or liver damage. J. Appl. Toxicol. 2017, 37, 278–286. [Google Scholar] [CrossRef]
- Rubio, M.; Bustamante, M.; Hernandez-Ferrer, C.; Fernandez-Orth, D.; Pantano, L.; Sarria, Y.; Piqué-Borras, M.; Vellve, K.; Agramunt, S.; Carreras, R.; et al. Circulating miRNAs, isomiRs and small RNA clusters in human plasma and breast milk. PLoS ONE 2018, 13, e0193527. [Google Scholar] [CrossRef]
- Matsuzaka, Y.; Tanihata, J.; Komaki, H.; Ishiyama, A.; Oya, Y.; Rüegg, U.; Takeda, S.I.; Hashido, K. Characterization and functional analysis of extracellular vesicles and muscle-abundant miRNAs (miR-1, miR-133a, and miR-206) in C2C12 myocytes and mdx mice. PLoS ONE 2016, 11, e0167811. [Google Scholar] [CrossRef]
- Matsuzaka, Y.; Tanihata, J.; Ooshima, Y.; Yamada, D.; Sekiguchi, M.; Miyatake, S.; Aoki, Y.; Terumitsu, M.; Yashiro, R.; Komaki, H.; et al. The nSMase2/Smpd3 gene modulates the severity of muscular dystrophy and the emotional stress response in mdx mice. BMC Med. 2020, 18, 343. [Google Scholar] [CrossRef]
- Lallemand, T.; Rouahi, M.; Swiader, A.; Grazide, M.H.; Geoffre, N.; Alayrac, P.; Recazens, E.; Coste, A.; Salvayre, R.; Nègre-Salvayre, A.; et al. nSMase2 (Type 2-Neutral Sphingomyelinase) Deficiency or inhibition by GW4869 reduces inflammation and atherosclerosis in Apoe(-/-) mice. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1479–1492. [Google Scholar] [CrossRef]
- Al-Rashed, F.; Ahmad, Z.; Thomas, R.; Melhem, M.; Snider, A.J.; Obeid, L.M.; Al-Mulla, F.; Hannun, Y.A.; Ahmad, R. Neutral sphingomyelinase 2 regulates inflammatory responses in monocytes/macrophages induced by TNF-alpha. Sci. Rep. 2020, 10, 16802. [Google Scholar] [CrossRef]
- Leng, L.; Dong, X.; Gao, X.; Ran, N.; Geng, M.; Zuo, B.; Wu, Y.; Li, W.; Yan, H.; Han, G.; et al. EV-mediated improvement in membrane integrity and muscle function in dystrophic mice. Mol. Ther. 2021, 29, 1459–1470. [Google Scholar] [CrossRef]
- Sekiguchi, M.; Zushida, K.; Yoshida, M.; Maekawa, M.; Kamichi, S.; Yoshida, M.; Sahara, Y.; Yuasa, S.; Takeda, S.; Wada, K. A deficit of brain dystrophin impairs specific amygdala GABAergic transmission and enhances defensive behaviour in mice. Brain 2009, 132, 124–135. [Google Scholar] [CrossRef]
- Remmelink, E.; Aartsma-Rus, A.; Smit, A.B.; Verhage, M.; Loos, M.; van Putten, M. Cognitive flexibility deficits in a mouse model for the absence of full-length dystrophin. Genes Brain Behav. 2016, 15, 558–567. [Google Scholar] [CrossRef]
- Vaillend, C.; Chaussenot, R. Relationships linking emotional, motor, cognitive and GABAergic dysfunctions in dystrophin-deficient mdx mice. Hum. Mol. Genet. 2017, 26, 1041–1055. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.A.; Rodino-Klapac, L.R.; Goodspeed, K.; Gray, S.J.; Kay, C.N.; Boye, S.L.; Boye, S.E.; George, L.A.; Salabarria, S.; et al. Current clinical applications of in vivo gene therapy with AAVs. Mol. Ther. 2021, 29, 464–488. [Google Scholar] [CrossRef]
- Buscara, L.; Gross, D.A.; Daniele, N. Of rAAV and Men: From Genetic Neuromuscular Disorder Efficacy and Toxicity Preclinical Studies to Clinical Trials and Back. J. Pers. Med. 2020, 10, 258. [Google Scholar] [CrossRef]
- Mariot, V.; Le Guiner, C.; Barthélémy, I.; Montus, M.; Blot, S.; Torelli, S.; Morgan, J.; Muntoni, F.; Voit, T.; Dumonceaux, J. Myostatin is a quantifiable biomarker for monitoring pharmaco-gene therapy in duchenne muscular dystrophy. Mol. Ther. Methods Clin. Dev. 2020, 18, 415–421. [Google Scholar] [CrossRef]
- Asher, D.R.; Thapa, K.; Dharia, S.D.; Khan, N.; Potter, R.A.; Rodino-Klapac, L.R.; Mendell, J.R. Clinical development on the frontier: Gene therapy for duchenne muscular dystrophy. Expert Opin. Biol. Ther. 2020, 20, 263–274. [Google Scholar] [CrossRef]
- Boehler, J.F.; Ricotti, V.; Gonzalez, J.P.; Soustek-Kramer, M.; Such, L.; Brown, K.J.; Schneider, J.S.; Morris, C.A. Membrane recruitment of nNOSµ in microdystrophin gene transfer to enhance durability. Neuromuscul. Disord. 2019, 29, 735–741. [Google Scholar] [CrossRef]
- Aguti, S.; Malerba, A.; Zhou, H. The progress of AAV-mediated gene therapy in neuromuscular disorders. Expert Opin. Biol. Ther. 2018, 18, 681–693. [Google Scholar] [CrossRef]
- Duan, D. Micro-dystrophin gene therapy goes systemic in Duchenne muscular dystrophy patients. Hum. Gene Ther. 2018, 29, 733–736. [Google Scholar] [CrossRef]
- Liu, X.; Liu, M.; Wu, L.; Liang, D. Gene therapy for hemophilia and Duchenne muscular dystrophy in China. Hum. Gene Ther. 2018, 29, 146–150. [Google Scholar] [CrossRef]
- Hollinger, K.; Chamberlain, J.S. Viral vector-mediated gene therapies. Curr. Opin. Neurol. 2015, 28, 522–527. [Google Scholar] [CrossRef][Green Version]
- Kawecka, K.; Theodoulides, M.; Hasoglu, Y.; Jarmin, S.; Kymalainen, H.; Le-Heron, A.; Popplewell, L.; Malerba, A.; Dickson, G.; Athanasopoulos, T. Adeno-associated virus (AAV) mediated dystrophin gene transfer studies and exon skipping strategies for Duchenne muscular dystrophy (DMD). Curr. Gene Ther. 2015, 15, 395–415. [Google Scholar] [CrossRef]
- Jarmin, S.; Kymalainen, H.; Popplewell, L.; Dickson, G. New developments in the use of gene therapy to treat Duchenne muscular dystrophy. Expert Opin. Biol. Ther. 2014, 14, 209–230. [Google Scholar] [CrossRef]
- Mendell, J.R.; Rodino-Klapac, L.; Sahenk, Z.; Malik, V.; Kaspar, B.K.; Walker, C.M.; Clark, K.R. Gene therapy for muscular dystrophy: Lessons learned and path forward. Neurosci. Lett. 2012, 527, 90–99. [Google Scholar] [CrossRef]
- Bowles, D.E.; McPhee, S.W.; Li, C.; Gray, S.J.; Samulski, J.J.; Camp, A.S.; Li, J.; Wang, B.; Monahan, P.E.; Rabinowitz, J.E.; et al. Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector. Mol. Ther. 2012, 20, 443–455. [Google Scholar] [CrossRef]
- Rodino-Klapac, L.R.; Janssen, P.M.; Montgomery, C.L.; Coley, B.D.; Chicoine, L.G.; Clark, K.R.; Mendell, J.R. A translational approach for limb vascular delivery of the micro-dystrophin gene without high volume or high pressure for treatment of Duchenne muscular dystrophy. J. Transl. Med. 2007, 5, 45. [Google Scholar] [CrossRef]
- Bolt, M.W.; Brady, J.T.; Whiteley, L.O.; Khan, K.N. Development challenges associated with rAAV-based gene therapies. J. Toxicol. Sci. 2021, 46, 57–68. [Google Scholar] [CrossRef]
- Kay, M.A. AAV vectors and tumorigenicity. Nat. Biotechnol. 2007, 25, 1111–1113. [Google Scholar] [CrossRef]
- Ishii, A.; Okada, H.; Hayashita-Kinoh, H.; Shin, J.H.; Tamaoka, A.; Okada, T.; Takeda, S. rAAV8 and rAAV9-mediated long-term muscle transduction with tacrolimus (FK506) in non-human primates. Mol. Ther. Methods Clin. Dev. 2020, 18, 44–49. [Google Scholar] [CrossRef]
- Hayashita-Kinoh, H.; Yugeta, N.; Okada, H.; Nitahara-Kasahara, Y.; Chiyo, T.; Okada, T.; Takeda, S. Intra-amniotic rAAV-mediated microdystrophin gene transfer improves canine X-linked muscular dystrophy and may induce immune tolerance. Mol. Ther. 2015, 23, 627–637. [Google Scholar] [CrossRef]
- Hayashita-Kinoh, H.; Guillermo, P.H.; Nitahara-Kasahara, Y.; Kuraoka, M.; Okada, H.; Chiyo, T.; Takeda, S.; Okada, T. Improved transduction of canine X-linked muscular dystrophy with rAAV9-microdystrophin via multipotent MSC pretreatment. Mol. Ther. Methods Clin. Dev. 2020, 20, 133–141. [Google Scholar] [CrossRef]
- Ichim, T.E.; Alexandrescu, D.T.; Solano, F.; Lara, F.; Campion Rde, N.; Paris, E.; Woods, E.J.; Murphy, M.P.; Dasanu, C.A.; Patel, A.N.; et al. Mesenchymal stem cells as anti-inflammatories: Implications for treatment of Duchenne muscular dystrophy. Cell Immunol. 2010, 260, 75–82. [Google Scholar] [CrossRef]
- Shen, O.Y.; Chen, Y.F.; Xu, H.T.; Lee, C.W. The efficacy of naive versus modified mesenchymal stem cells in improving muscle function in duchenne muscular dystrophy: A systematic review. Biomedicines 2021, 9, 1097. [Google Scholar] [CrossRef]
- Nitahara-Kasahara, Y.; Kuraoka, M.; Oda, Y.; Hayashita-Kinoh, H.; Takeda, S.; Okada, T. Enhanced cell survival and therapeutic benefits of IL-10-expressing multipotent mesenchymal stromal cells for muscular dystrophy. Stem Cell Res. Ther. 2021, 12, 105. [Google Scholar] [CrossRef]
- Barnes, C.; Scheideler, O.; Schaffer, D. Engineering the AAV capsid to evade immune responses. Curr. Opin. Biotechnol. 2019, 60, 99–103. [Google Scholar] [CrossRef]
- Ogden, P.J.; Kelsic, E.D.; Sinai, S.; Church, G.M. Comprehensive AAV capsid fitness landscape reveals a viral gene and enables machine-guided design. Science 2019, 366, 1139–1143. [Google Scholar] [CrossRef]
- Bryant, D.H.; Bashir, A.; Sinai, S.; Jain, N.K.; Ogden, P.J.; Riley, P.F.; Church, G.M.; Colwell, L.J.; Kelsic, E.D. Deep diversification of an AAV capsid protein by machine learning. Nat. Biotechnol. 2021, 39, 691–696. [Google Scholar] [CrossRef]
- Büning, H.; Huber, A.; Zhang, L.; Meumann, N.; Hacker, U. Engineering the AAV capsid to optimize vector-host-interactions. Curr. Opin. Pharmacol. 2015, 24, 94–104. [Google Scholar] [CrossRef]
- Nelson, C.E.; Hakim, C.H.; Ousterout, D.G.; Thakore, P.I.; Moreb, E.A.; Castellanos Rivera, R.M.; Madhavan, S.; Pan, X.; Ran, F.A.; Yan, W.X.; et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016, 351, 403–407. [Google Scholar] [CrossRef]
- Hakim, C.H.; Kumar, S.R.P.; Pérez-López, D.O.; Wasala, N.B.; Zhang, D.; Yue, Y.; Teixeira, J.; Pan, X.; Zhang, K.; Million, E.D.; et al. Cas9-specific immune responses compromise local and systemic AAV CRISPR therapy in multiple dystrophic canine models. Nat. Commun. 2021, 12, 6769. [Google Scholar] [CrossRef]
- Nelson, C.E.; Wu, Y.; Gemberling, M.P.; Oliver, M.L.; Waller, M.A.; Bohning, J.D.; Robinson-Hamm, J.N.; Bulaklak, K.; Castellanos Rivera, R.M.; Collier, J.H.; et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019, 25, 427–432. [Google Scholar] [CrossRef]
- Shojaei Baghini, S.; Gardanova, Z.; Zekiy, A.O.; Shomali, N.; Tosan, F.; Jarahian, M. Optimizing sgRNA to improve CRISPR/Cas9 knockout efficiency: Special focus on human and animal cell. Front. Bioeng. Biotechnol. 2021, 9, 775309. [Google Scholar] [CrossRef]
- Yaméogo, P.; Duchêne, B.L.; Majeau, N.; Tremblay, J.P. CRISPR-SCReT (CRISPR-stop codon read through) method to control Cas9 expression for gene editing. Gene Ther. 2021, in press. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, C.; Li, H.; Wang, P.; Gao, Y.; Mokadam, N.A.; Ma, J.; Arnold, W.D.; Han, R. Efficient precise in vivo base editing in adult dystrophic mice. Nat. Commun. 2021, 12, 3719. [Google Scholar] [CrossRef]
- Liu, B.; Li, Z.; Huang, S.; Yan, B.; He, S.; Chen, F.; Liang, Y. AAV-containing exosomes as a novel vector for improved gene delivery to lung cancer cells. Front. Cell Dev. Biol. 2021, 9, 707607. [Google Scholar] [CrossRef]
- Wang, W.; Liu, J.; Yang, M.; Qiu, R.; Li, Y.; Bian, S.; Hao, B.; Lei, B. Intravitreal injection of an exosome-associated adeno-associated viral vector enhances retinoschisin 1 gene transduction in the mouse retina. Hum. Gene Ther. 2021, 32, 707–716. [Google Scholar] [CrossRef]
- Khan, N.; Maurya, S.; Bammidi, S.; Jayandharan, G.R. AAV6 vexosomes mediate robust suicide gene delivery in a murine model of hepatocellular carcinoma. Mol. Ther. Methods Clin. Dev. 2020, 17, 497–504. [Google Scholar] [CrossRef]
- Breuer, C.B.; Hanlon, K.S.; Natasan, J.S.; Volak, A.; Meliani, A.; Mingozzi, F.; Kleinstiver, B.P.; Moon, J.J.; Maguire, C.A. In vivo engineering of lymphocytes after systemic exosome-associated AAV delivery. Sci. Rep. 2020, 10, 4544. [Google Scholar] [CrossRef]
- Schiller, L.T.; Lemus-Diaz, N.; Rinaldi Ferreira, R.; Böker, K.O.; Gruber, J. Enhanced production of exosome-associated AAV by overexpression of the tetraspanin CD9. Mol. Ther. Methods Clin. Dev. 2018, 9, 278–287. [Google Scholar] [CrossRef]
- Meliani, A.; Boisgerault, F.; Fitzpatrick, Z.; Marmier, S.; Leborgne, C.; Collaud, F.; Simon Sola, M.; Charles, S.; Ronzitti, G.; Vignaud, A.; et al. Enhanced liver gene transfer and evasion of preexisting humoral immunity with exosome-enveloped AAV vectors. Blood Adv. 2017, 1, 2019–2031. [Google Scholar] [CrossRef]
- György, B.; Maguire, C.A. Extracellular vesicles: Nature’s nanoparticles for improving gene transfer with adeno-associated virus vectors. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2018, 10, e1488. [Google Scholar] [CrossRef]
- Wassmer, S.J.; Carvalho, L.S.; György, B.; Vandenberghe, L.H.; Maguire, C.A. Exosome-associated AAV2 vector mediates robust gene delivery into the murine retina upon intravitreal injection. Sci. Rep. 2017, 7, 45329. [Google Scholar] [CrossRef]
- Martin, D.M.; Raphael, Y. It’s All in the delivery: Improving AAV transfection efficiency with exosomes. Mol. Ther. 2017, 25, 309–311. [Google Scholar] [CrossRef][Green Version]
- György, B.; Sage, C.; Indzhykulian, A.A.; Scheffer, D.I.; Brisson, A.R.; Tan, S.; Wu, X.; Volak, A.; Mu, D.; Tamvakologos, P.I.; et al. Rescue of hearing by gene delivery to inner-ear hair cells using exosome-associated AAV. Mol. Ther. 2017, 25, 379–391. [Google Scholar] [CrossRef]
- Hudry, E.; Martin, C.; Gandhi, S.; György, B.; Scheffer, D.I.; Mu, D.; Merkel, S.F.; Mingozzi, F.; Fitzpatrick, Z.; Dimant, H.; et al. Exosome-associated AAV vector as a robust and convenient neuroscience tool. Gene Ther. 2016, 23, 819. [Google Scholar] [CrossRef]
- Maguire, C.A.; Balaj, L.; Sivaraman, S.; Crommentuijn, M.H.; Ericsson, M.; Mincheva-Nilsson, L.; Baranov, V.; Gianni, D.; Tannous, B.A.; Sena-Esteves, M.; et al. Microvesicle-associated AAV vector as a novel gene delivery system. Mol. Ther. 2012, 20, 960–971. [Google Scholar] [CrossRef]
- Leborgne, C.; Latournerie, V.; Boutin, S.; Desgue, D.; Quéré, A.; Pignot, E.; Collaud, F.; Charles, S.; Simon Sola, M.; Masat, E. Prevalence and long-term monitoring of humoral immunity against adeno-associated virus in Duchenne Muscular Dystrophy patients. Cell Immunol. 2019, 342, 103780. [Google Scholar] [CrossRef]
- Rapti, K.; Grimm, D. Adeno-associated viruses (AAV) and host immunity—A race between the hare and the hedgehog. Front. Immunol. 2021, 12, 753467. [Google Scholar] [CrossRef]
- Chu, W.S.; Ng, J. Immunomodulation in administration of rAAV: Preclinical and clinical adjuvant pharmacotherapies. Front. Immunol. 2021, 12, 658038. [Google Scholar] [CrossRef]
- Cheng, M.; Dietz, L.; Gong, Y.; Eichler, F.; Nammour, J.; Ng, C.; Grimm, D.; Maguire, C.A. Neutralizing antibody evasion and transduction with purified extracellular vesicle-enveloped AAV vectors. Hum. Gene Ther. 2021, in press. [Google Scholar] [CrossRef]
- Muhuri, M.; Maeda, Y.; Ma, H.; Ram, S.; Fitzgerald, K.A.; Tai, P.W.; Gao, G. Overcoming innate immune barriers that impede AAV gene therapy vectors. J. Clin. Investig. 2021, 131, e143780. [Google Scholar] [CrossRef]
- Kwon, J.B.; Ettyreddy, A.R.; Vankara, A.; Bohning, J.D.; Devlin, G.; Hauschka, S.D.; Asokan, A.; Gersbach, C.A. In vivo gene editing of muscle stem cells with adeno-associated viral vectors in a mouse model of Duchenne muscular dystrophy. Mol. Ther. Methods Clin. Dev. 2020, 19, 320–329. [Google Scholar] [CrossRef]
- Nance, M.E.; Shi, R.; Hakim, C.H.; Wasala, N.B.; Yue, Y.; Pan, X.; Zhang, T.; Robinson, C.A.; Duan, S.X.; Yao, G.; et al. AAV9 edits muscle stem cells in normal and dystrophic adult mice. Mol. Ther. 2019, 27, 1568–1585. [Google Scholar] [CrossRef]
- Li, N.; Parkes, J.E.; Spathis, R.; Morales, M.; Mcdonald, J.; Kendra, R.M.; Ott, E.M.; Brown, K.J.; Lawlor, M.W.; Nagaraju, K. The effect of immunomodulatory treatments on anti-dystrophin immune response after AAV gene therapy in dystrophin deficient mdx mice. J. Neuromuscul. Dis. 2021, 8, S325–S340. [Google Scholar] [CrossRef]
- Yuasa, K.; Sakamoto, M.L.; Miyagoe-Suzuki, Y.; Tanouchi, A.; Yamamoto, H.; Li, J.; Chamberlain, J.S.; Xiao, X.; Takeda, S. Adeno-associated virus vector-mediated gene transfer into dystrophin-deficient skeletal muscles evokes enhanced immune response against the transgene product. Gene Ther. 2002, 9, 1576–1588. [Google Scholar] [CrossRef]
- Xuan, W.; Khan, M.; Ashraf, M. Pluripotent stem cell-induced skeletal muscle progenitor cells with givinostat promote myoangiogenesis and restore dystrophin in injured Duchenne dystrophic muscle. Stem Cell Res. Ther. 2021, 12, 131. [Google Scholar] [CrossRef]
- Sandonà, M.; Consalvi, S.; Tucciarone, L.; De Bardi, M.; Scimeca, M.; Angelini, D.F.; Buffa, V.; D’Amico, A.; Bertini, E.S.; Cazzaniga, S.; et al. HDAC inhibitors tune miRNAs in extracellular vesicles of dystrophic muscle-resident mesenchymal cells. EMBO Rep. 2020, 21, e50863. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).