
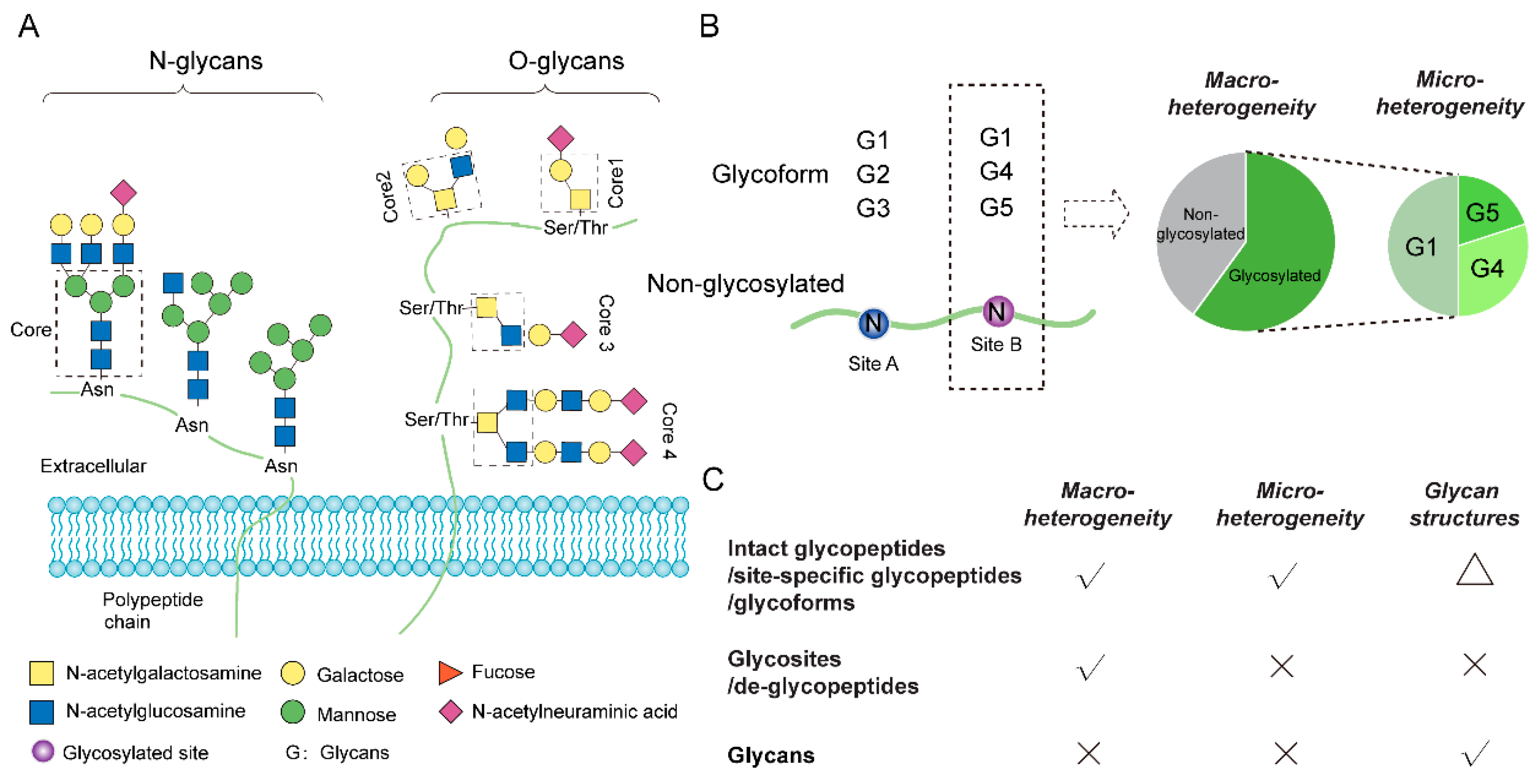
Strategies for Proteome-Wide Quantification of Glycosylation Macro- and Micro-Heterogeneity

 ,
,
Abstract
:1. Introduction
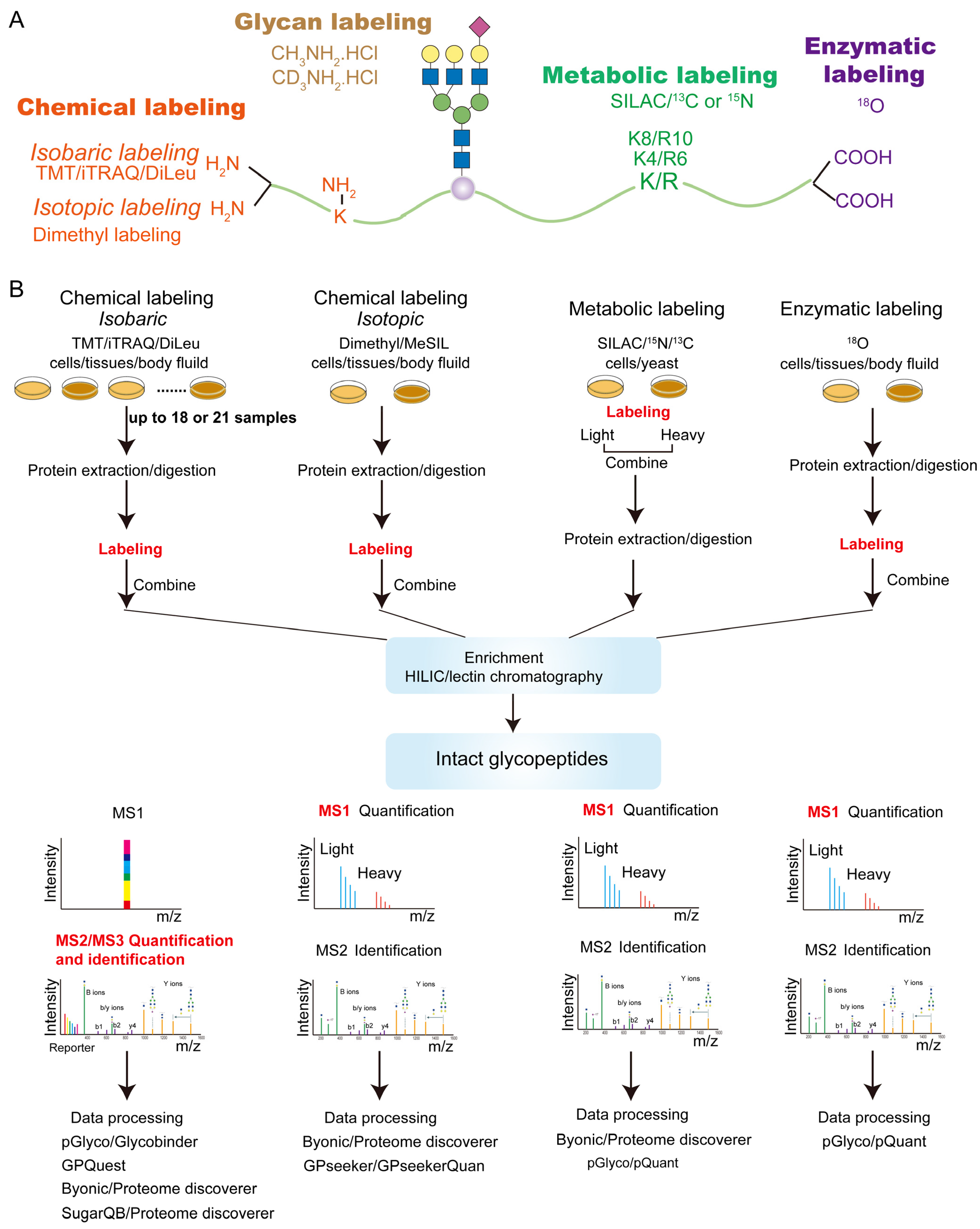
2. Labeling-Based Quantification
2.1. Isobaric Chemical Labeling
2.2. Isotopic Chemical Labeling
2.3. Metabolic Labeling
2.4. Enzymatic Labeling Using 18O Stable Isotope
2.5. Glycan Labeling
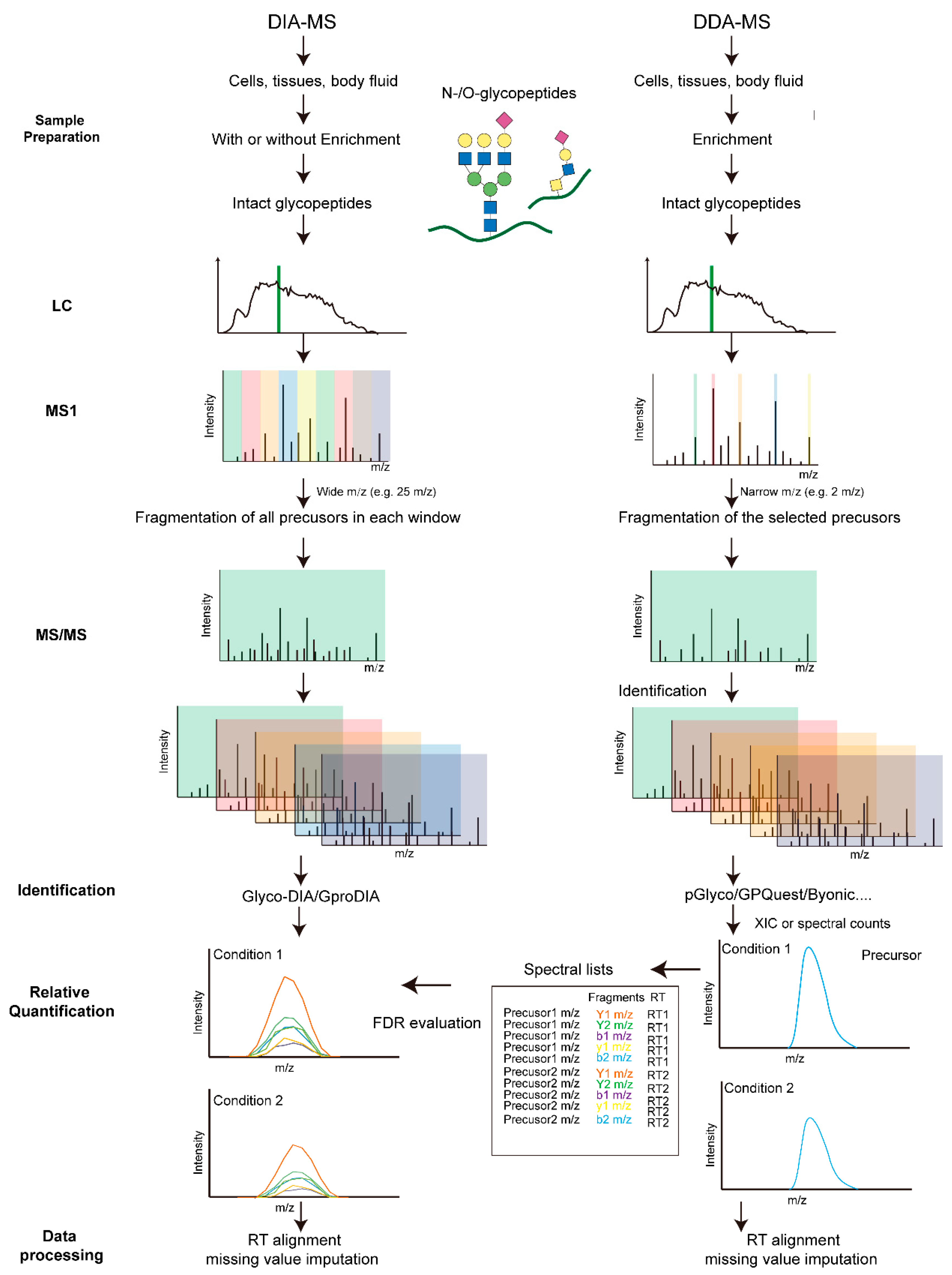
3. Label-Free Quantification
3.1. DDA-Based Label Free Quantification
3.2. DIA-Based Label-Free Quantification
4. Target Analysis Using SRM/MRM
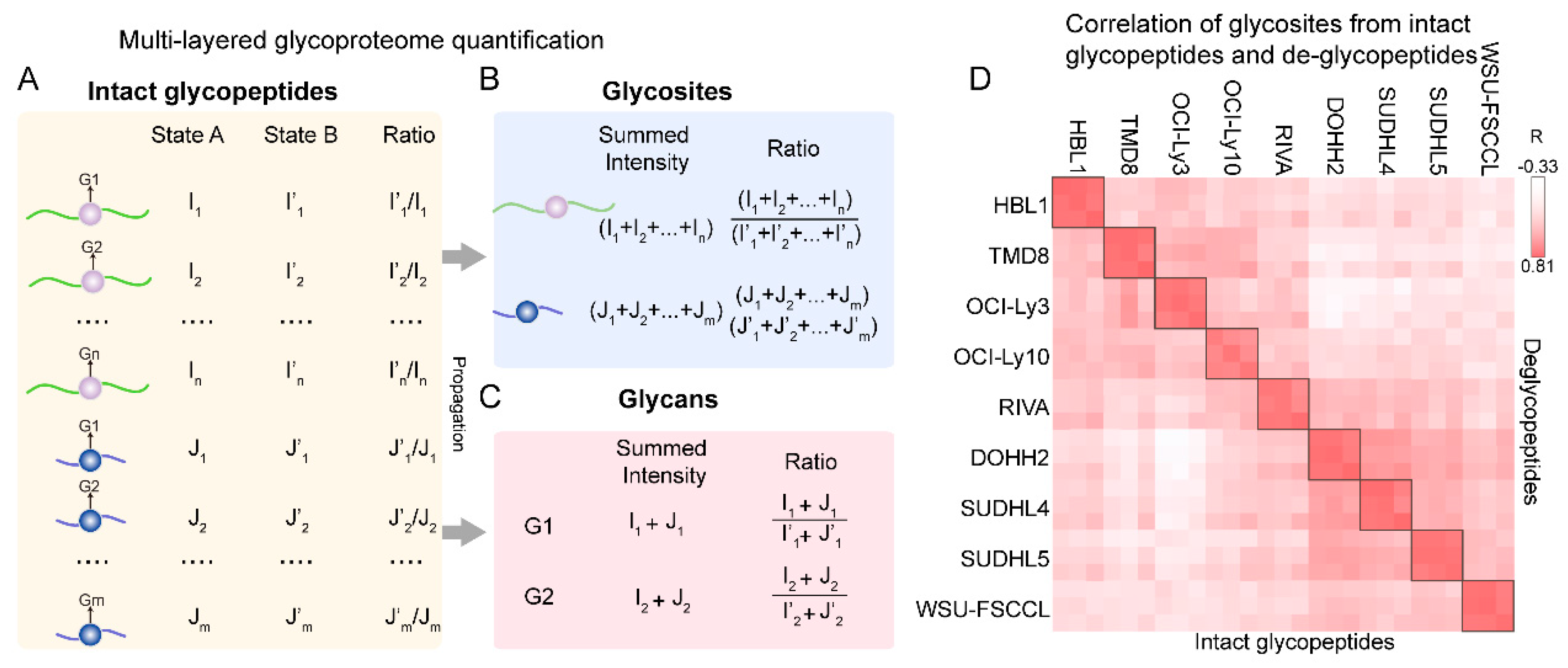
5. Multi-Layered Quantification of Glycoproteome
6. Applications of Quantitative Glycoproteomics
6.1. Cancers and Other Diseases
6.2. SARS-CoV-2
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Flynn, R.A.; Pedram, K.; Malaker, S.A.; Batista, P.J.; Smith, B.A.H.; Johnson, A.G.; George, B.M.; Majzoub, K.; Villalta, P.W.; Carette, J.E.; et al. Small RNAs are modified with N-glycans and displayed on the surface of living cells. Cell 2021, 184, 3109–3124.e22. [Google Scholar] [CrossRef] [PubMed]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Pan, J.; Shah, P.; Ao, M.; Thomas, S.N.; Liu, Y.; Chen, L.; Schnaubelt, M.; Clark, D.J.; Rodriguez, H.; et al. Clinical Proteomic Tumor Analysis, C. Integrated proteomic and glycoproteomic characterization of human high-grade serous ovarian carcinoma. Cell Rep. 2020, 33, 108276. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Hu, Y.; Sun, S.; Chen, L.; Schnaubelt, M.; Clark, D.; Ao, M.; Zhang, Z.; Chan, D.; Qian, J.; et al. Glycoproteomics-based signatures for tumor subtyping and clinical outcome prediction of high-grade serous ovarian cancer. Nat. Commun. 2020, 11, 6139. [Google Scholar] [CrossRef]
- Mereiter, S.; Polom, K.; Williams, C.; Polonia, A.; Guergova-Kuras, M.; Karlsson, N.G.; Roviello, F.; Magalhaes, A.; Reis, C.A. The Thomsen-Friedenreich antigen: A highly sensitive and specific predictor of microsatellite instability in gastric cancer. J. Clin. Med. 2018, 7, 256. [Google Scholar] [CrossRef] [Green Version]
- Pujic, I.; Perreault, H. Recent advancements in glycoproteomic studies: Glycopeptide enrichment and derivatization, characterization of glycosylation in SARS CoV2, and interacting glycoproteins. Mass. Spectrom. Rev. 2021, 2020, 1–20. [Google Scholar]
- Cao, W.; Liu, M.; Kong, S.; Wu, M.; Zhang, Y.; Yang, P. Recent advances in software tools for more generic and precise intact glycopeptide analysis. Mol. Cell. Proteom. 2021, 20, 100060. [Google Scholar] [CrossRef]
- Thaysen-Andersen, M.; Packer, N.H.; Schulz, B.L. Maturing glycoproteomics technologies provide unique structural insights into the N-glycoproteome and its regulation in health and disease. Mol. Cell. Proteom. 2016, 15, 1773–1790. [Google Scholar] [CrossRef] [Green Version]
- Xiao, K.; Tian, Z. GPSeeker enables quantitative structural N-glycoproteomics for site- and structure-specific characterization of differentially expressed N-glycosylation in hepatocellular carcinoma. J. Proteome Res. 2019, 18, 2885–2895. [Google Scholar] [CrossRef]
- Shen, J.; Jia, L.; Dang, L.; Su, Y.; Zhang, J.; Xu, Y.; Zhu, B.; Chen, Z.; Wu, J.; Lan, R.; et al. StrucGP: De novo structural sequencing of site-specific N-glycan on glycoproteins using a modularization strategy. Nat. Methods 2021, 18, 921–929. [Google Scholar] [CrossRef]
- Bantscheff, M.; Lemeer, S.; Savitski, M.M.; Kuster, B. Quantitative mass spectrometry in proteomics: Critical review update from 2007 to the present. Anal. Bioanal. Chem. 2012, 404, 939–965. [Google Scholar] [CrossRef] [PubMed]
- Marcus, K.; Eisenacher, M.; Sitek, B. Quantitative Methods in Proteomics, 2nd ed.; Humana: New York, NY, USA, 2021; pp. 1–483. [Google Scholar]
- Chen, Z.; Yu, Q.; Hao, L.; Liu, F.; Johnson, J.; Tian, Z.; Kao, W.J.; Xu, W.; Li, L. Site-specific characterization and quantitation of N-glycopeptides in PKM2 knockout breast cancer cells using DiLeu isobaric tags enabled by electron-transfer/higher-energy collision dissociation (EThcD). Analyst 2018, 143, 2508–2519. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Boyne, M.T., 2nd; Buhse, L.F.; Hill, J. Direct approach for qualitative and quantitative characterization of glycoproteins using tandem mass tags and an LTQ Orbitrap XL electron transfer dissociation hybrid mass spectrometer. Anal. Chem. 2013, 85, 1531–1539. [Google Scholar] [CrossRef]
- Zhu, H.; Qiu, C.; Ruth, A.C.; Keire, D.A.; Ye, H. A LC-MS All-in-One workflow for site-specific location, identification and quantification of N-/O- glycosylation in human chorionic gonadotropin drug products. AAPS J. 2017, 19, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, R.; Ortega, F.; Rosa-Fernandes, L.; Guimarães, V.; Quina, D.; Nahas, W.; Schwämmle, V.; Srougi, M.; Leite, K.R.M.; Thaysen-Andersen, M.; et al. Distinct urinary glycoprotein signatures in prostate cancer patients. Oncotarget 2018, 9, 33077–33097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, R.; Recuero, S.; Srougi, M.; Leite, K.R.M.; Thaysen-Andersen, M.; Palmisano, G. The complexity and dynamics of the tissue glycoproteome associated with prostate cancer progression. Mol. Cell. Proteom. 2021, 20, 100026. [Google Scholar] [CrossRef] [PubMed]
- Blazev, R.; Ashwood, C.; Abrahams, J.L.; Chung, L.H.; Francis, D.; Yang, P.; Watt, K.I.; Qian, H.; Quaife-Ryan, G.A.; Hudson, J.E.; et al. Integrated glycoproteomics identifies a role of N-glycosylation and galectin-1 on myogenesis and muscle development. Mol. Cell. Proteom. 2020, 20, 100030. [Google Scholar] [CrossRef]
- Fang, J.; Sheng, X.; Bao, H.; Zhang, Y.; Lu, H. Comparative analysis of intact glycopeptides from mannose receptor among different breast cancer subtypes using mass spectrometry. Talanta 2021, 223, 121676. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Mishra, S.; Chen, L.; Zhou, J.Y.; Chan, D.W.; Chatterjee, S.; Zhang, H. Integrated glycoprotein immobilization method for glycopeptide and glycan analysis of cardiac hypertrophy. Anal. Chem. 2015, 87, 9671–9678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, K.C.; Chen, L.; Hu, Y.; Schnaubelt, M.; Zhang, H. Developing workflow for simultaneous analyses of phosphopeptides and glycopeptides. ACS Chem. Biol. 2019, 14, 58–66. [Google Scholar] [CrossRef]
- Zhao, T.; Jia, L.; Li, J.; Ma, C.; Wu, J.; Shen, J.; Dang, L.; Zhu, B.; Li, P.; Zhi, Y.; et al. Heterogeneities of site-specific N-glycosylation in HCC tumors with low and high AFP concentrations. Front. Oncol. 2020, 10, 496. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Yang, W.; Hu, Y.; Hoti, N.; Liu, Y.; Shah, P.; Sun, S.; Clark, D.; Thomas, S.; Zhang, H. Site-specific fucosylation analysis identifying glycoproteins associated with aggressive prostate cancer cell lines using tandem affinity enrichments of intact glycopeptides followed by mass spectrometry. Anal. Chem. 2017, 89, 7623–7630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, P.; Wang, X.; Yang, W.; Toghi Eshghi, S.; Sun, S.; Hoti, N.; Chen, L.; Yang, S.; Pasay, J.; Rubin, A.; et al. Integrated proteomic and glycoproteomic analyses of prostate cancer cells reveal glycoprotein alteration in protein abundance and glycosylation. Mol. Cell. Proteom. 2015, 14, 2753–2763. [Google Scholar] [CrossRef] [Green Version]
- Stadlmann, J.; Taubenschmid, J.; Wenzel, D.; Gattinger, A.; Durnberger, G.; Dusberger, F.; Elling, U.; Mach, L.; Mechtler, K.; Penninger, J.M. Comparative glycoproteomics of stem cells identifies new players in ricin toxicity. Nature 2017, 549, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Stadlmann, J.; Hoi, D.M.; Taubenschmid, J.; Mechtler, K.; Penninger, J.M. Analysis of PNGase F-resistant N-glycopeptides using SugarQb for proteome discoverer 2.1 reveals cryptic substrate Specificities. Proteomics 2018, 18, e1700436. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; Ji, Y.; Silbern, I.; Doebele, C.; Ninov, M.; Lenz, C.; Oellerich, T.; Pan, K.T.; Urlaub, H. A streamlined pipeline for multiplexed quantitative site-specific N-glycoproteomics. Nat. Commun. 2020, 11, 5268. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; Ji, Y.; Silbern, I.; Viner, R.; Oellerich, T.; Pan, K.T.; Urlaub, H. Evaluation and optimization of High-Field Asymmetric Waveform Ion-Mobility Spectrometry for multiplexed quantitative site-specific N-glycoproteomics. Anal. Chem. 2021, 93, 8846–8855. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiao, K.; Tian, Z. Quantitative N-glycoproteomics using stable isotopic diethyl labeling. Talanta 2020, 219, 121359. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, F.; Xiao, K.; Chen, Y.; Tian, Z. Site- and structure-specific characterization of N-glycoprotein markers of MCF-7 cancer stem cells using isotopic-labelling quantitative N-glycoproteomics. Chem. Commun. 2019, 55, 7934–7937. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, F.; Chen, Y.; Tian, Z. A quantitative N-glycoproteomics study of cell-surface N-glycoprotein markers of MCF-7/ADR cancer stem cells. Anal. Bioanal. Chem. 2020, 412, 2423–2432. [Google Scholar] [CrossRef]
- Xu, F.; Wang, Y.; Xiao, K.; Hu, Y.; Tian, Z.; Chen, Y. Quantitative site- and structure-specific N-glycoproteomics characterization of differential N-glycosylation in MCF-7/ADR cancer stem cells. Clin. Proteom. 2020, 17, 3. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Xiao, K.; Wang, Y.; Tian, Z. Site- and structure-specific quantitative N-glycoproteomics study of differential N-glycosylation in MCF-7 cancer cells. J. Proteom. 2020, 212, 103594. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Xiao, K.; Tian, Z. Benchmark of site- and structure-specific quantitative tissue N-glycoproteomics for discovery of potential N-glycoprotein markers: A case study of pancreatic cancer. Glycoconj. J. 2021, 38, 213–231. [Google Scholar] [CrossRef]
- Yang, H.; Xu, F.; Chen, Y.; Tian, Z. Putative N-glycoprotein markers of MCF-7/ADR cancer stem cells from N-glycoproteomics characterization of the whole cell lysate. Talanta 2021, 232, 122437. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Sun, Z.; Zhang, L.; Cai, Y.; Peng, Y.; Cao, T.; Zhang, Y.; Lu, H. Chemical labeling for fine mapping of IgG N-glycosylation by ETD-MS. Chem. Sci. 2019, 10, 9302–9307. [Google Scholar] [CrossRef] [PubMed]
- Schjoldager, K.T.; Joshi, H.J.; Kong, Y.; Goth, C.K.; King, S.L.; Wandall, H.H.; Bennett, E.P.; Vakhrushev, S.Y.; Clausen, H. Deconstruction of O-glycosylation-GalNAc-T isoforms direct distinct subsets of the O-glycoproteome. EMBO Rep. 2015, 16, 1713–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narimatsu, Y.; Joshi, H.J.; Schjoldager, K.T.; Hintze, J.; Halim, A.; Steentoft, C.; Nason, R.; Mandel, U.; Bennett, E.P.; Clausen, H.; et al. Exploring regulation of protein O-glycosylation in isogenic human HEK293 cells by differential O-glycoproteomics. Mol. Cell. Proteom. 2019, 18, 1396–1409. [Google Scholar] [CrossRef]
- Zhang, Z.; Sun, Z.; Zhu, J.; Liu, J.; Huang, G.; Ye, M.; Zou, H. High-throughput determination of the site-specific N-sialoglycan occupancy rates by differential oxidation of glycoproteins followed with quantitative glycoproteomics analysis. Anal. Chem. 2014, 86, 9830–9837. [Google Scholar] [CrossRef]
- Parker, B.L.; Thaysen-Andersen, M.; Fazakerley, D.J.; Holliday, M.; Packer, N.H.; James, D.E. Terminal galactosylation and sialylation switching on membrane glycoproteins upon TNF-alpha-induced insulin resistance in adipocytes. Mol. Cell. Proteom. 2016, 15, 141–153. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhou, C.; Zhang, W.; Yao, J.; Lu, H.; Dong, Q.; Zhou, H.; Qin, L. An integrative strategy for quantitative analysis of the N-glycoproteome in complex biological samples. Proteome Sci. 2014, 12, 4. [Google Scholar] [CrossRef] [Green Version]
- Quazi Shakey, B.B.; Wu, J. An approach to quantifying N-Linked glycoproteins by enzyme-catalyzed 18O3-labeling of solid-phase enriched glycopeptides. Anal. Chem. 2010, 82, 7722–7728. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cao, X.; Liu, C.; Li, W.; Zeng, W.; Li, B.; Chi, H.; Liu, M.; Qin, X.; Tang, L.; et al. N-glycopeptide signatures of IgA2 in serum from patients with hepatitis B virus-related liver diseases. Mol. Cell. Proteom. 2019, 18, 2262–2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.Q.; Zeng, W.F.; Fang, P.; Cao, W.Q.; Liu, C.; Yan, G.Q.; Zhang, Y.; Peng, C.; Wu, J.Q.; Zhang, X.J.; et al. pGlyco 2.0 enables precision N-glycoproteomics with comprehensive quality control and one-step mass spectrometry for intact glycopeptide identification. Nat. Commun. 2017, 8, 438. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Ji, G.; Wang, G.; Wei, L.; Zhang, Y.; Lu, H. One step carboxyl group isotopic labeling for quantitative analysis of intact N-glycopeptides by mass spectrometry. Chem. Commun. 2021, 57, 4154–4157. [Google Scholar] [CrossRef]
- Jiang, J.; Tian, F.; Cai, Y.; Qian, X.; Costello, C.E.; Ying, W. Site-specific qualitative and quantitative analysis of the N- and O-glycoforms in recombinant human erythropoietin. Anal. Bioanal. Chem. 2014, 406, 6265–6274. [Google Scholar] [CrossRef]
- Yang, L.; Gong, T.; Shen, H.; Pei, J.; Zhang, L.; Zhang, Q.; Huang, Y.; Hu, Z.; Pan, Z.; Yang, P.; et al. Precision N-glycoproteomic profiling of murine peritoneal macrophages after different stimulations. Front. Immunol. 2021, 12, 722293. [Google Scholar] [CrossRef]
- Woo, C.M.; Lund, P.J.; Huang, A.C.; Davis, M.M.; Bertozzi, C.R.; Pitteri, S.J. Mapping and quantification of over 2000 O-linked glycopeptides in activated human T cells with isotope-targeted glycoproteomics (Isotag). Mol. Cell. Proteom. 2018, 17, 764–775. [Google Scholar] [CrossRef] [Green Version]
- Plomp, R.; Ruhaak, L.R.; Uh, H.W.; Reiding, K.R.; Selman, M.; Houwing-Duistermaat, J.J.; Slagboom, P.E.; Beekman, M.; Wuhrer, M. Subclass-specific IgG glycosylation is associated with markers of inflammation and metabolic health. Sci. Rep. 2017, 7, 12325. [Google Scholar] [CrossRef] [Green Version]
- Bollineni, R.C.; Koehler, C.J.; Gislefoss, R.E.; Anonsen, J.H.; Thiede, B. Large-scale intact glycopeptide identification by Mascot database search. Sci. Rep. 2018, 8, 2117. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.J.; Gaunitz, S.; Wang, Z.; Strindelius, L.; Jacobson, S.C.; Clemmer, D.E.; Trinidad, J.C.; Novotny, M.V. Glycoproteomic analysis of human urinary exosomes. Anal. Chem. 2020, 92, 14357–14365. [Google Scholar] [CrossRef]
- Shu, Q.; Li, M.; Shu, L.; An, Z.; Wang, J.; Lv, H.; Yang, M.; Cai, T.; Hu, T.; Fu, Y.; et al. Large-scale identification of N-linked intact glycopeptides in human serum using HILIC enrichment and spectral library search. Mol. Cell. Proteom. 2020, 19, 672–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Jia, L.; Hao, Z.; Xu, Y.; Shen, J.; Ma, C.; Wu, J.; Zhao, T.; Zhi, Y.; Li, P.; et al. Site-specific N-glycoproteomic analysis reveals upregulated sialylation and core fucosylation during transient regeneration Loss in neonatal mouse hearts. J. Proteome Res. 2020, 19, 3191–3200. [Google Scholar] [CrossRef] [PubMed]
- Park, G.W.; Kim, J.Y.; Hwang, H.; Lee, J.Y.; Ahn, Y.H.; Lee, H.K.; Ji, E.S.; Kim, K.H.; Jeong, H.K.; Yun, K.N.; et al. Integrated GlycoProteome Analyzer (I-GPA) for automated identification and quantitation of site-specific N-glycosylation. Sci. Rep. 2016, 6, 21175. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Zheng, S.; Li, Y.; Huang, J.; Zhang, W.; Xie, Y.; Qin, W.; Qian, X. An integrated mass spectroscopy data processing strategy for fast identification, in-depth, and reproducible quantification of protein O-glycosylation in a large cohort of human urine samples. Anal. Chem. 2020, 92, 690–698. [Google Scholar] [CrossRef]
- Fang, P.; Xie, J.; Sang, S.; Zhang, L.; Liu, M.; Yang, L.; Xu, Y.; Yan, G.; Yao, J.; Gao, X.; et al. Multilayered N-Glycoproteome profiling reveals highly heterogeneous and dysregulated protein N-Glycosylation related to Alzheimer’s Disease. Anal. Chem. 2020, 92, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Ao, M.; Hu, Y.; Li, Q.K.; Zhang, H. Mapping the O-glycoproteome using site-specific extraction of O-linked glycopeptides (EXoO). Mol. Syst. Biol. 2018, 14, e8486. [Google Scholar] [CrossRef]
- Yang, G.; Hoti, N.; Chen, S.Y.; Zhou, Y.; Wang, Q.; Betenbaugh, M.; Zhang, H. One-Step enrichment of intact glycopeptides from glycoengineered chinese hamster ovary cells. Front. Chem. 2020, 8, 240. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Huttenhain, R.; Surinova, S.; Gillet, L.C.; Mouritsen, J.; Brunner, R.; Navarro, P.; Aebersold, R. Quantitative measurements of N-linked glycoproteins in human plasma by SWATH-MS. Proteomics 2013, 13, 1247–1256. [Google Scholar] [CrossRef]
- Xu, Y.; Bailey, U.M.; Schulz, B.L. Automated measurement of site-specific N-glycosylation occupancy with SWATH-MS. Proteomics 2015, 15, 2177–2186. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, Z.; Guo, L.; Zhu, Z.; Zhang, Y. Proteome-wide analysis of N-Glycosylation stoichiometry using SWATH technology. J. Proteome Res. 2017, 16, 3830–3840. [Google Scholar] [CrossRef]
- Zacchi, L.F.; Schulz, B.L. SWATH-MS glycoproteomics reveals consequences of defects in the glycosylation machinery. Mol. Cell. Proteom. 2016, 15, 2435–2447. [Google Scholar] [CrossRef] [Green Version]
- Sanda, M.; Goldman, R. Data independent analysis of IgG glycoforms in samples of unfractionated human plasma. Anal. Chem. 2016, 88, 10118–10125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanda, M.; Zhang, L.; Edwards, N.J.; Goldman, R. Site-specific analysis of changes in the glycosylation of proteins in liver cirrhosis using data-independent workflow with soft fragmentation. Anal. Bioanal. Chem. 2017, 409, 619–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.H.; Krisp, C.; Packer, N.H.; Molloy, M.P. Development of a data independent acquisition mass spectrometry workflow to enable glycopeptide analysis without predefined glycan compositional knowledge. J. Proteom. 2018, 172, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Mao, Y.; Clausen, H.; Vakhrushev, S.Y. Glyco-DIA: A method for quantitative O-glycoproteomics with in silico-boosted glycopeptide libraries. Nat. Methods 2019, 16, 902–910. [Google Scholar] [CrossRef]
- Yang, Y.; Yan, G.; Kong, S.; Wu, M.; Yang, P.; Cao, W.; Qiao, L. GproDIA enables data-independent acquisition glycoproteomics with comprehensive statistical control. Nat. Commun. 2021, 12, 6073. [Google Scholar] [CrossRef] [PubMed]
- Song, E.; Zhu, R.; Hammoud, Z.T.; Mechref, Y. LC-MS/MS quantitation of esophagus disease blood serum glycoproteins by enrichment with hydrazide chemistry and lectin affinity chromatography. J. Proteome Res. 2014, 13, 4808–4820. [Google Scholar] [CrossRef] [Green Version]
- Benicky, J.; Sanda, M.; Pompach, P.; Wu, J.; Goldman, R. Quantification of fucosylated hemopexin and complement factor H in plasma of patients with liver disease. Anal. Chem. 2014, 86, 10716–10723. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Park, G.W.; Jeong, J.E.; Ji, E.S.; An, H.J.; Kim, J.Y.; Yoo, J.S. Parallel reaction monitoring with multiplex immunoprecipitation of N-glycoproteins in human serum for detection of hepatocellular carcinoma. Anal. Bioanal. Chem. 2019, 411, 3009–3019. [Google Scholar] [CrossRef]
- Yin, H.; Zhu, J.; Wang, M.; Yao, Z.P.; Lubman, D.M. Quantitative analysis of alpha-1-antitrypsin glycosylation isoforms in HCC patients using LC-HCD-PRM-MS. Anal. Chem. 2020, 92, 8201–8208. [Google Scholar] [CrossRef]
- Morales-Betanzos, C.A.; Lee, H.; Gonzalez Ericsson, P.I.; Balko, J.M.; Johnson, D.B.; Zimmerman, L.J.; Liebler, D.C. Quantitative mass spectrometry analysis of PD-L1 protein expression, N-glycosylation and expression stoichiometry with PD-1 and PD-L2 in human melanoma. Mol. Cell. Proteom. 2017, 16, 1705–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, W.; Sanda, M.; Wu, J.; Koomen, J.; Goldman, R. Quantitative analysis of immunoglobulin subclasses and subclass specific glycosylation by LC-MS-MRM in liver disease. J. Proteom. 2015, 116, 24–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Cai, Z.; Bomgarden, R.D.; Pike, I.; Kuhn, K.; Rogers, J.C.; Roberts, T.M.; Gygi, S.P.; Paulo, J.A. TMTpro-18plex: The expanded and complete set of TMTpro reagents for sample multiplexing. J. Proteome Res. 2021, 20, 2964–2972. [Google Scholar] [CrossRef] [PubMed]
- Frost, D.C.; Feng, Y.; Li, L. 21-plex diLeu isobaric tags for high-throughput quantitative proteomics. Anal. Chem. 2020, 92, 8228–8234. [Google Scholar] [CrossRef]
- Ren, Y.; He, Y.; Lin, Z.; Zi, J.; Yang, H.; Zhang, S.; Lou, X.; Wang, Q.; Li, S.; Liu, S. Reagents for isobaric labeling peptides in quantitative proteomics. Anal. Chem. 2018, 90, 12366–12371. [Google Scholar] [CrossRef]
- Bern, M.; Kil, Y.J.; Becker, C. Byonic: Advanced peptide and protein identification software. Curr. Protoc. Bioinformatics 2012, 40, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, R.; Chernykh, A.; Alagesan, K.; Bern, M.; Cao, W.; Chalkley, R.J.; Cheng, K.; Choo, M.S.; Edwards, N.; Goldman, R.; et al. Community evaluation of glycoproteomics informatics solutions reveals high-performance search strategies for serum glycopeptide analysis. Nat. Methods 2021, 18, 1304–1316. [Google Scholar] [CrossRef]
- Toghi Eshghi, S.; Shah, P.; Yang, W.; Li, X.; Zhang, H. GPQuest: A spectral aibrary matching algorithm for site-specific assignment of tandem mass spectra to intact N-glycopeptides. Anal. Chem. 2015, 87, 5181–5188. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Shah, P.; Eshghi, S.T.; Yang, W.; Trikannad, N.; Yang, S.; Chen, L.; Aiyetan, P.; Hoti, N.; Zhang, Z.; et al. Comprehensive analysis of protein glycosylation by solid-phase extraction of N-linked glycans and glycosite-containing peptides. Nat. Biotechnol. 2016, 34, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Robinson, R.C.; Poulsen, N.A.; Barile, D. Multiplexed bovine milk oligosaccharide analysis with aminoxy tandem mass tags. PLoS ONE 2018, 13, e0196513. [Google Scholar] [CrossRef]
- Ow, S.Y.; Salim, M.; Noirel, J.; Evans, C.; Rehman, I.; Wright, P.C. iTRAQ underestimation in simple and complex mixtures: “The Good, the Bad and the Ugly”. J. Proteome Res. 2009, 8, 5347–5355. [Google Scholar] [CrossRef] [PubMed]
- Kovalchik, K.A.; Colborne, S.; Spencer, S.E.; Sorensen, P.H.; Chen, D.D.Y.; Morin, G.B.; Hughes, C.S. RawTools: Rapid and Dynamic Interrogation of Orbitrap Data Files for Mass Spectrometer System Management. J. Proteome Res. 2019, 18, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Song, C.Q.; Yuan, Z.F.; Fu, Y.; Chi, H.; Wang, L.H.; Fan, S.B.; Zhang, K.; Zeng, W.F.; He, S.M.; et al. pQuant improves quantitation by keeping out interfering signals and evaluating the accuracy of calculated ratios. Anal. Chem. 2014, 86, 5286–5294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Qiu, C.; Gryniewicz-Ruzicka, C.M.; Keire, D.A.; Ye, H. Multiplexed comparative analysis of intact glycopeptides using electron-transfer dissociation and synchronous precursor selection based triple-stage mass spectrometry. Anal. Chem. 2020, 92, 7547–7555. [Google Scholar] [CrossRef] [PubMed]
- Steentoft, C.; Vakhrushev, S.Y.; Vester-Christensen, M.B.; Schjoldager, K.T.; Kong, Y.; Bennett, E.P.; Mandel, U.; Wandall, H.; Levery, S.B.; Clausen, H. Mining the O-glycoproteome using zinc-finger nuclease-glycoengineered SimpleCell lines. Nat. Methods 2011, 8, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Liao, R.; Gao, Y.; Chen, M.; Li, L.; Hu, X. A ubiquitous but overlooked side reaction in dimethyl labeling of peptides. Anal. Chem. 2018, 90, 13533–13540. [Google Scholar] [CrossRef]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, J.; Takao, T.; Hori, H.; Besada, V.; Rodriguez, R.; Padron, G.; Shimonishi, Y. A method for determination of N-glycosylation sites in glycoproteins by collision-induced dissociation analysis in fast atom bombardment mass spectrometry: Identification of the positions of carbohydrate-linked asparagine in recombinant a-amylase by treatment with peptide-N-glycosidase F in 180-labeled water. Anal. Biochem. 1992, 205, 151–158. [Google Scholar]
- Kuster, B.; Mann, M. 18O-labeling of N-glycosylation sites to improve the identification of gel-separated glycoproteins using peptide mass mapping and database searching. Anal. Chem. 1999, 71, 1431–1440. [Google Scholar] [CrossRef]
- Kaji, H.; Saito, H.; Yamauchi, Y.; Shinkawa, T.; Taoka, M.; Hirabayashi, J.; Kasai, K.-I.; Takahashi, N.; Isobe, T. Lectin affinity capture, isotope-coded tagging and mass spectrometry to identify N-linked glycoproteins. Nat. Biotechnol. 2003, 21, 667–672. [Google Scholar] [CrossRef]
- Liu, Z.; Cao, J.; He, Y.; Qiao, L.; Xu, C.; Lu, H.; Yang, P. Tandem 18O stable isotope labeling for quantification of N-Glycoproteome. J. Proteome Res. 2009, 9, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Hang, H.C.; Yu, C.; Kato, D.L.; Bertozzi, C.R. A metabolic labeling approach toward proteomic analysis of mucin-type O-linked glycosylation. Proc. Natl. Acad. Sci. USA 2003, 100, 14846–14851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laughlin, S.T.; Baskin, J.M.; Amacher, S.L.; Bertozzi, C.R. In vivo imaging of membrane-associated glycans in developing zebrafish. Science 2008, 320, 664–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Smeekens, J.M.; Wu, R. Systematic and site-specific analysis of N-sialoglycosylated proteins on the cell surface by integrating click chemistry and MS-based proteomics. Chem. Sci. 2015, 6, 4681–4689. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Tang, G.X.; Wu, R. Site-specific quantification of surface N-glycoproteins in statin-treated liver cells. Anal. Chem. 2016, 88, 3324–3332. [Google Scholar] [CrossRef]
- Xiao, H.; Wu, R. Quantitative investigation of human cell surface N-glycoprotein dynamics. Chem. Sci. 2017, 8, 268–277. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Wu, R. Simultaneous quantitation of glycoprotein degradation and synthesis rates by integrating isotope labeling, chemical enrichment, and multiplexed proteomics. Anal. Chem. 2017, 89, 10361–10367. [Google Scholar] [CrossRef]
- Zhu, J.; Chen, Z.; Zhang, J.; An, M.; Wu, J.; Yu, Q.; Skilton, S.J.; Bern, M.; Ilker Sen, K.; Li, L.; et al. Differential Quantitative Determination of Site-Specific Intact N-Glycopeptides in Serum Haptoglobin between Hepatocellular Carcinoma and Cirrhosis Using LC-EThcD-MS/MS. J. Proteome Res. 2019, 18, 359–371. [Google Scholar] [CrossRef]
- Lee, J.Y.; Lee, H.K.; Park, G.W.; Hwang, H.; Jeong, H.K.; Yun, K.N.; Ji, E.S.; Kim, K.H.; Kim, J.S.; Kim, J.W.; et al. Characterization of Site-Specific N-Glycopeptide Isoforms of alpha-1-Acid Glycoprotein from an Interlaboratory Study Using LC-MS/MS. J. Proteome Res. 2016, 15, 4146–4164. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Haynes, S.E.; Nesvizhskii, A.I. IonQuant enables accurate and sensitive label-free quantification with FDR-controlled match-between-runs. Mol. Cell. Proteom. 2021, 20, 100077. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Shen, S.; Li, J.; Hu, Q.; Nie, L.; Tu, C.; Wang, X.; Poulsen, D.J.; Orsburn, B.C.; Wang, J.; et al. IonStar enables high-precision, low-missing-data proteomics quantification in large biological cohorts. Proc. Natl. Acad. Sci. USA 2018, 115, E4767–E4776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, P.; Praissman, J.L.; Grant, O.C.; Cai, Y.; Xiao, T.; Rosenbalm, K.E.; Aoki, K.; Kellman, B.P.; Bridger, R.; Barouch, D.H.; et al. Virus-receptor interactions of glycosylated SARS-CoV-2 spike and human ACE2 receptor. Cell Host Microbe 2020, 28, 586–601.e6. [Google Scholar] [CrossRef]
- Zhang, F.; Ge, W.; Ruan, G.; Cai, X.; Guo, T. Data-Independent Acquisition Mass Spectrometry-Based Proteomics and Software Tools: A Glimpse in 2020. Proteomics 2020, 20, e1900276. [Google Scholar] [CrossRef] [PubMed]
- Yeo, K.Y.B.; Chrysanthopoulos, P.K.; Nouwens, A.S.; Marcellin, E.; Schulz, B.L. High-performance targeted mass spectrometry with precision data-independent acquisition reveals site-specific glycosylation macroheterogeneity. Anal. Biochem. 2016, 510, 106–113. [Google Scholar] [CrossRef] [Green Version]
- Pan, K.T.; Chen, C.C.; Urlaub, H.; Khoo, K.H. Adapting data-independent acquisition for mass spectrometry-based protein site-specific N-glycosylation analysis. Anal. Chem. 2017, 89, 4532–4539. [Google Scholar] [CrossRef]
- Li, K.W.; Gonzalez-Lozano, M.A.; Koopmans, F.; Smit, A.B. Recent Developments in Data Independent Acquisition (DIA) Mass Spectrometry: Application of Quantitative Analysis of the Brain Proteome. Front. Mol. Neurosci. 2020, 13, 564446. [Google Scholar] [CrossRef] [PubMed]
- Hong, Q.; Lebrilla, C.B.; Miyamoto, S.; Ruhaak, L.R. Absolute quantitation of immunoglobulin G and its glycoforms using multiple reaction monitoring. Anal. Chem. 2013, 85, 8585–8593. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y. Quantitative N-glycoproteome, phosphoproteome and ubiquitinome analyses for studying B-cell receptor signaling in B-cell lymphoma. Ph.D. Thesis, Naturwissenschaften Johann Wolfgang Goethe-Universität Frankfurt am Main, Frankfurt am Main, Germany, 2021. [Google Scholar]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.M.; Zhou, M.T.; Li, S.W.; Zhen, X.C.; Yang, S. Glycoproteins as diagnostic and prognostic biomarkers for neurodegenerative diseases: A glycoproteomic approach. J. Neurosci. Res. 2021, 99, 1308–1324. [Google Scholar] [CrossRef]
- Yang, K.; Yang, Z.; Chen, X.; Li, W. The significance of sialylation on the pathogenesis of Alzheimer’s disease. Brain Res. Bull. 2021, 173, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.; Hackett, W.E.; Zhong, L.; Wan, X.F.; Zaia, J. Measuring site-specific glycosylation similarity between influenza a virus variants with statistical certainty. Mol. Cell. Proteom. 2020, 19, 1533–1545. [Google Scholar] [CrossRef] [PubMed]
- Steffen, U.; Koeleman, C.A.; Sokolova, M.V.; Bang, H.; Kleyer, A.; Rech, J.; Unterweger, H.; Schicht, M.; Garreis, F.; Hahn, J.; et al. IgA subclasses have different effector functions associated with distinct glycosylation profiles. Nat. Commun. 2020, 11, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojcik, I.; Senard, T.; de Graaf, E.L.; Janssen, G.M.C.; de Ru, A.H.; Mohammed, Y.; van Veelen, P.A.; Vidarsson, G.; Wuhrer, M.; Falck, D. Site-specific glycosylation mapping of Fc gamma receptor IIIb from neutrophils of individual healthy donors. Anal. Chem. 2020, 92, 13172–13181. [Google Scholar] [CrossRef]
- Keser, T.; Tijardovic, M.; Gornik, I.; Lukic, E.; Lauc, G.; Gornik, O.; Novokmet, M. High-throughput and site-specific N-glycosylation analysis of human alpha-1-acid glycoprotein offers a great potential for new biomarker discovery. Mol. Cell. Proteom. 2021, 20, 100044. [Google Scholar] [CrossRef]
- Shade, K.C.; Conroy, M.E.; Washburn, N.; Kitaoka, M.; Huynh, D.J.; Laprise, E.; Patil, S.U.; Shreffler, W.G.; Anthony, R.M. Sialylation of immunoglobulin E is a determinant of allergic pathogenicity. Nature 2020, 582, 265–270. [Google Scholar] [CrossRef]
- Momcilovic, A.; de Haan, N.; Hipgrave Ederveen, A.L.; Bondt, A.; Koeleman, C.A.M.; Falck, D.; de Neef, L.A.; Mesker, W.E.; Tollenaar, R.; de Ru, A.; et al. Simultaneous immunoglobulin A and G glycopeptide profiling for high-throughput applications. Anal. Chem. 2020, 92, 4518–4526. [Google Scholar] [CrossRef] [Green Version]
- De Leoz, M.L.A.; Duewer, D.L.; Fung, A.; Liu, L.; Yau, H.K.; Potter, O.; Staples, G.O.; Furuki, K.; Frenkel, R.; Hu, Y.; et al. NIST interlaboratory study on glycosylation analysis of monoclonal antibodies: Comparison of results from diverse analytical methods. Mol. Cell. Proteom. 2019, 19, 11–30. [Google Scholar] [CrossRef] [Green Version]
- Buettner, A.; Maier, M.; Bonnington, L.; Bulau, P.; Reusch, D. Multi-attribute monitoring of complex erythropoetin beta glycosylation by GluC liquid chromatography-mass spectrometry peptide mapping. Anal. Chem. 2020, 92, 7574–7580. [Google Scholar] [CrossRef]
- Chandler, K.B.; Mehta, N.; Leon, D.R.; Suscovich, T.J.; Alter, G.; Costello, C.E. Multi-isotype glycoproteomic characterization of serum antibody heavy chains reveals isotype- and subclass-specific N-Glycosylation profiles. Mol. Cell. Proteom. 2019, 18, 686–703. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Xiao, K.; Tian, Z. Site- and structure-specific characterization of the human urinary N-glycoproteome with site-determining and structure-diagnostic product ions. Rapid. Commun. Mass. Spectrom. 2021, 35, e8952. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yu, Q.; Yu, Q.; Johnson, J.; Shipman, R.; Zhong, X.; Huang, J.; Asthana, S.; Carlsson, C.; Okonkwo, O.; et al. In-depth site-specific analysis of N-glycoproteome in human cerebrospinal fluid and glycosylation landscape changes in Alzheimer’s Disease. Mol. Cell. Proteom. 2021, 20, 100081. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Lin, Y.H.; Dingess, K.A.; Mank, M.; Stahl, B.; Heck, A.J.R. Quantitative longitudinal inventory of the N-Glycoproteome of human milk from a single donor reveals the highly variable repertoire and dynamic site-specific changes. J. Proteome Res. 2020, 19, 1941–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.; Guan, S.; Wang, X.; Zhao, J.; Gao, M.; Zhang, X. Deconstruction of heterogeneity of size-dependent exosome subpopulations from human urine by profiling N-Glycoproteomics and phosphoproteomics simultaneously. Anal. Chem. 2020, 92, 9239–9246. [Google Scholar] [CrossRef] [PubMed]
- Andaluz Aguilar, H.; Iliuk, A.B.; Chen, I.H.; Tao, W.A. Sequential phosphoproteomics and N-glycoproteomics of plasma-derived extracellular vesicles. Nat. Protoc. 2020, 15, 161–180. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Rathinavel, A.K.; Radhakrishnan, P. Altered glycosylation in cancer: A promising target for biomarkers and therapeutics. Biochim. Biophys. Acta. Rev. Cancer 2021, 1875, 188464. [Google Scholar] [CrossRef] [PubMed]
- Shu, H.; Zhang, L.; Chen, Y.; Guo, Y.; Li, L.; Chen, F.; Cao, Z.; Yan, G.; Lu, C.; Liu, C.; et al. Quantification of intact O-Glycopeptides on haptoglobin in sera of patients with hepatocellular carcinoma and liver cirrhosis. Front. Chem. 2021, 9, 705341. [Google Scholar] [CrossRef]
- Jia, L.; Li, J.; Li, P.; Liu, D.; Li, J.; Shen, J.; Zhu, B.; Ma, C.; Zhao, T.; Lan, R.; et al. Site-specific glycoproteomic analysis revealing increased core-fucosylation on FOLR1 enhances folate uptake capacity of HCC cells to promote EMT. Theranostics 2021, 11, 6905–6921. [Google Scholar] [CrossRef]
- Lilja, H.; Ulmert, D.; Vickers, A.J. Prostate-specific antigen and prostate cancer: Prediction, detection and monitoring. Nat. Rev. Cancer 2008, 8, 268–278. [Google Scholar] [CrossRef]
- Gabriele, C.; Prestagiacomo, L.E.; Cuda, G.; Gaspari, M. Mass Spectrometry-Based Glycoproteomics and Prostate Cancer. Int. J. Mol. Sci. 2021, 22, 5222. [Google Scholar] [CrossRef]
- van der Burgt, Y.E.M.; Siliakus, K.M.; Cobbaert, C.M.; Ruhaak, L.R. HILIC-MRM-MS for linkage-specific separation of sialylated glycopeptides to quantify prostate-specific antigen proteoforms. J. Proteome Res. 2020, 19, 2708–2716. [Google Scholar] [CrossRef] [PubMed]
- Wanyama, F.M.; Blanchard, V. Glycomic-based biomarkers for ovarian cancer: Advances and challenges. Diagnostics 2021, 11, 643. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Cabral, H. Abnormal glycosylation of cancer stem cells and targeting strategies. Front. Oncol. 2021, 11, 649338. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-C.; Lu, Y.-T.; Yeh, T.-S.; Chan, Y.-H.; Dash, S.; Yu, J.-S. Identification of fucosylated SERPINA1 as a novel plasma marker for pancreatic cancer using lectin affinity capture coupled with iTRAQ-based quantitative glycoproteomics. Int. J. Mol. Sci. 2021, 22, 6079. [Google Scholar] [CrossRef] [PubMed]
- Duarte, H.O.; Rodrigues, J.G.; Gomes, C.; Hensbergen, P.J.; Ederveen, A.L.H.; de Ru, A.H.; Mereiter, S.; Polonia, A.; Fernandes, E.; Ferreira, J.A.; et al. ST6Gal1 targets the ectodomain of ErbB2 in a site-specific manner and regulates gastric cancer cell sensitivity to trastuzumab. Oncogene 2021, 40, 3719–3733. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ma, C.; Chin, L.S.; Li, L. Integrative glycoproteomics reveals protein N-glycosylation aberrations and glycoproteomic network alterations in Alzheimer’s disease. Sci. Adv. 2020, 6, eabc5802. [Google Scholar] [CrossRef]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369, 330–333. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, H.; Wang, H. Glycans of SARS-CoV-2 spike protein in virus infection and antibody production. Front. Mol. Biosci. 2021, 8, 629873. [Google Scholar] [CrossRef]
- Tian, W.; Li, D.; Zhang, N.; Bai, G.; Yuan, K.; Xiao, H.; Gao, F.; Chen, Y.; Wong, C.C.L.; Gao, G.F. O-glycosylation pattern of the SARS-CoV-2 spike protein reveals an “O-Follow-N” rule. Cell Res. 2021, 31, 1123–1125. [Google Scholar] [CrossRef]
- Dong, X.; Chen, C.; Yan, J.; Zhang, X.; Li, X.; Liang, X. Comprehensive O-Glycosylation Analysis of the SARS-CoV-2 Spike Protein with Biomimetic Trp-Arg Materials. Anal. Chem. 2021, 93, 10444–10452. [Google Scholar] [CrossRef]
- Bagdonaite, I.; Thompson, A.J.; Wang, X.; Sogaard, M.; Fougeroux, C.; Frank, M.; Diedrich, J.K.; Yates, J.R., 3rd; Salanti, A.; Vakhrushev, S.Y.; et al. Site-Specific O-Glycosylation Analysis of SARS-CoV-2 Spike Protein Produced in Insect and Human Cells. Viruses 2021, 13, 551. [Google Scholar] [CrossRef] [PubMed]
- Shajahan, A.; Supekar, N.T.; Gleinich, A.S.; Azadi, P. Deducing the N- and O-glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. Glycobiology 2020, 30, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Brun, J.; Vasiljevic, S.; Gangadharan, B.; Hensen, M.; Chandran, A.; Hill, M.L.; Kiappes, J.L.; Dwek, R.A.; Alonzi, D.S.; Struwe, W.B.; et al. Assessing Antigen Structural Integrity through Glycosylation Analysis of the SARS-CoV-2 Viral Spike. ACS Cent. Sci. 2021, 7, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, W.; Mao, Y.; Chen, Y.; Wang, S.; Zhong, Y.; Su, T.; Gong, M.; Du, D.; Lu, X.; et al. Site-specific N-glycosylation Characterization of Recombinant SARS-CoV-2 Spike Proteins. Mol. Cell. Proteom. 2020, 20, 100058. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, Z.; Hu, W.; Hao, P.; Yang, S. Impact of Expressing Cells on Glycosylation and Glycan of the SARS-CoV-2 Spike Glycoprotein. ACS Omega 2021, 6, 15988–15999. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Ren, K.; Zhang, X.; Chen, J.; Jiang, Z.; Jiang, J.; Ji, F.; Ouyang, X.; Li, L. Mass Spectrometry Analysis of Newly Emerging Coronavirus HCoV-19 Spike Protein and Human ACE2 Reveals Camouflaging Glycans and Unique Post-Translational Modifications. Engineering 2020, 7, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Shajahan, A.; Archer-Hartmann, S.; Supekar, N.T.; Gleinich, A.S.; Heiss, C.; Azadi, P. Comprehensive characterization of N- and O- glycosylation of SARS-CoV-2 human receptor angiotensin converting enzyme 2. Glycobiology 2021, 31, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Mehdipour, A.R.; Hummer, G. Dual nature of human ACE2 glycosylation in binding to SARS-CoV-2 spike. Proc. Natl. Acad. Sci. USA 2021, 118, e2100425118. [Google Scholar] [CrossRef]
- Supekar, N.T.; Shajahan, A.; Gleinich, A.S.; Rouhani, D.; Heiss, C.; Azadi, P. SARS-CoV-2 Nucleocapsid protein is decorated with multiple N- and O-glycans. bioRxiv 2020. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Reagent | Principle | Sample | Multiplexity | MS Level | Advantages | Disadvantages | Ref. |
|---|---|---|---|---|---|---|---|---|
| Isobaric chemical labeling | TMT/ iTRAQ /DiLeu/ IBT | React with amine on peptides | Cells, tissue, fluid | 2, 4, 6, 10, 11, 16, 18, 21 | MS2 or MS3 | Enhanced signal intensity in MS and MS/MS; high multiplexing capability; simple data analysis; reduced measurement time; applicable to any sample; reduced run-to-run variations; low missing values | Expensive for commercial reagents; Does not allow in vivo labeling | [3,4,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28] |
| Isotopic chemical labeling | Dimethyl/Diethyl | React with the carboxyl groups of peptides | Cells, tissue, fluid | 3 | MS1 | Low costs; simple in handling; applicable to any sample types | Incomplete labeling complicates data analysis; side reactions; limited multiplexing capability (up to 2-plex); not suitable for in vivo labeling | [9,29,30,31,32,33,34,35,36,37,38,39] |
| Metabolic labeling | SILAC | Metabolic labeling with amino acids containing stable heavy isotopes when culturing cells | Cells | 2 or 3 | MS1 | Allow in vivo labeling, minimize system errors; applicable to cells but can be expanded to tissues or model organisms using internal standards (e.g., superSILAC) | High costs; not applicable to many biological materials; limited multiplexity; complicated MS1 spectra of glycopeptides; over-sequencing of same glycopeptides | [40] |
| Enzymatic labeling using 18O stable isotope | 18O water | Introduce 18O atoms into the carboxyl termini of intact glycopeptides during tryptic digestion | Cells, tissue, fluid | 2 | MS1 | Low costs; simple in handling; applicable to any sample (cells, animal or human tissue) | Incomplete labeling complicates data analysis. Limited multiplexing capability (up to 2-plex); not suitable for in vivo labeling | [41,42,43] |
| Glycan labeling | 15N/13C | Metabolic labeling when culturing with 15N or 13C media | yeast | 2 | MS1 | Can be used for the evaluation of FDR of glycopeptide search engine. | Complicated data analysis | [44] |
| Glycan labeling | Methylamine stable isotope labeling (MeSIL) | Label the carboxyl groups on both the sialic acid and the peptides | Cells, tissue, fluid | 2 | MS1 | Label intact N-glycopeptides by one-step reaction easily with high labeling efficiency; distinction of neutral and sialylated glycopeptides | No description | [45] |
| DDA-based LFQ | XIC/ intensity | XIC or intensity of glycopeptides across runs | Cells, tissue, fluid | No limited sample numbers | MS1 | No labeling required; applicable to any sample; simplified sample handling; | Huge variations in replicate measurements; longer data acquisition time; requires more computationally sophisticated data analysis; severe missing values | [46,47,48,49,50,51,52,53,54,55,56] |
| DDA-based LFQ | Spectra counts | The number of identified glycopeptide spectra matches | Cells, tissue, fluid | No limited sample numbers | MS1 | No labeling required; applicable to any sample types; simplified sample handling; | Requires large sample size (spectral counts) to confidently predict small changes in expression; lower accuracy than labeling and XIC-based LFQ methods; severe missing values | [57,58] |
| DIA-based LFQ | DIA-label free | XIC of glycopeptides | Cells, tissue, fluid | No limited sample numbers | MS1 | No labeling required; applicable to any sample types; simplified sample handling; higher sensitivity, reproducibility and less missing values than DDA; | Needs constructing the sample specific glycopeptides spectra libraries | [59,60,61,62,63,64,65,66,67] |
| Target analysis | SRM /MRM /PRM | Monitor the target precursor and product ions | Cells, tissue, fluid | No limited sample numbers | MS1 | Very high sensitivity, reproducibility | The number of precursor ions to be monitored is limited by the scan speed of MS | [68,69,70,71,72,73] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, P.; Ji, Y.; Oellerich, T.; Urlaub, H.; Pan, K.-T. Strategies for Proteome-Wide Quantification of Glycosylation Macro- and Micro-Heterogeneity. Int. J. Mol. Sci. 2022, 23, 1609. https://doi.org/10.3390/ijms23031609
Fang P, Ji Y, Oellerich T, Urlaub H, Pan K-T. Strategies for Proteome-Wide Quantification of Glycosylation Macro- and Micro-Heterogeneity. International Journal of Molecular Sciences. 2022; 23(3):1609. https://doi.org/10.3390/ijms23031609
Chicago/Turabian StyleFang, Pan, Yanlong Ji, Thomas Oellerich, Henning Urlaub, and Kuan-Ting Pan. 2022. "Strategies for Proteome-Wide Quantification of Glycosylation Macro- and Micro-Heterogeneity" International Journal of Molecular Sciences 23, no. 3: 1609. https://doi.org/10.3390/ijms23031609
APA StyleFang, P., Ji, Y., Oellerich, T., Urlaub, H., & Pan, K.-T. (2022). Strategies for Proteome-Wide Quantification of Glycosylation Macro- and Micro-Heterogeneity. International Journal of Molecular Sciences, 23(3), 1609. https://doi.org/10.3390/ijms23031609