Abstract
For years, the gold standard for diagnosing Gaucher disease (GD) has been detecting reduced β-glucocerebrosidase (GCase) activity in peripheral blood cells combined with GBA1 mutation analysis. The use of dried blood spot (DBS) specimens offers many advantages, including easy collection, the need for a small amount of blood, and simpler transportation. However, DBS has limitations for measuring GCase activity. In this paper, we recount our cross-sectional study and publish seven years of experience using DBS samples and levels of the deacylated form of glucocerebroside, glucosylsphingosine (lyso-Gb1), for GD diagnosis. Of 444 screened subjects, 99 (22.3%) were diagnosed with GD at a median (range) age of 21 (1–78) years. Lyso-Gb levels for genetically confirmed GD patients vs. subjects negative to GD diagnosis were 252 (9–1340) ng/mL and 5.4 (1.5–16) ng/mL, respectively. Patients diagnosed with GD1 and mild GBA1 variants had lower median (range) lyso-Gb1, 194 (9–1050), compared to GD1 and severe GBA1 variants, 447 (38–1340) ng/mL, and neuronopathic GD, 325 (116–1270) ng/mL (p = 0.001). Subjects with heterozygous GBA1 variants (carrier) had higher lyso-Gb1 levels, 5.8 (2.5–15.3) ng/mL, compared to wild-type GBA1, 4.9 (1.5–16), ng/mL (p = 0.001). Lyso-Gb1 levels, median (range), were 5 (2.7–10.7) in heterozygous GBA1 carriers with Parkinson’s disease (PD), similar to lyso-Gb1 levels in subjects without PD. We call for a paradigm change for the diagnosis of GD based on lyso-Gb1 measurements and confirmatory GBA1 mutation analyses in DBS. Lyso-Gb1 levels could not be used to differentiate between heterozygous GBA1 carriers and wild type.
1. Introduction
Gaucher disease (GD), one of the most common lysosomal storage disorders, is caused by a mutant lysosomal enzyme β-glucocerebrosidase (GCase), leading to the storage of glucocerebroside (GC) and other glycolipids in various tissues; it also leads to a multi-system disease known for its great phenotypic heterogeneity [1,2]. The disease is characterized by hepatosplenomegaly, anemia, thrombocytopenia, and skeletal disease. GD has an estimated prevalence of 1:50,000–100,000 in the general population, but it has a predilection for Ashkenazi Jews, wherein 1:17 are carriers and about 1:800 are affected [3].
The current gold standard for diagnosing GD is the detection of reduced GCase activity in peripheral blood cells (traditionally compared to same-day normal controls), combined with mutation analysis at the DNA level of the glucocerebrosidase gene (GBA1), performed today by whole-gene sequence [1]. The great phenotypic heterogeneity of GD is explained, in part, by the many (>860) variants within the gene identified to date, as well as different genetic, epigenetic, and environmental factors [4]. Bone-marrow aspiration and biopsies for identifying Gaucher cells are no longer acceptable as a means to diagnose GD and should be performed only when another hematologic comorbidity is being evaluated [5].
Plasma biomarkers have been used over the years to diagnose and follow patients with GD [6,7,8]. Historical biomarkers (i.e., angiotensin-converting enzyme, ferritin, alkaline phosphatase, and high-density lipoprotein) are not specific to GD and, thus, could not be used for diagnosis [9,10,11,12,13,14]. The utility of more specific biomarkers, namely chitotriosidase and chemokine (C-C motif) ligand 18 (CCL18), is also limited, as they are not specific for GD [15,16], and one in 20 individuals is entirely deficient in chitotriosidase activity, owing to polymorphism in the CHIT1 gene [17]. In a search for more specific and sensitive biomarkers, two groups independently identified the deacylated form of glucocerebroside, glucosylsphingosine (Lyso-Gb1), as a candidate biomarker. This has been confirmed to be almost ideal, with very high specificity and sensitivity for the diagnosis of GD [18,19].
Dried blood spot (DBS) specimens are collected by applying a few drops of blood onto printed circles on absorbent filter paper. The use of DBS for diagnosis offers many advantages, including easy collection; the need for only a small amount of blood; and simpler transportation, as samples can be shipped via regular mail, at room temperature. However, DBS has limited usefulness for the measurement of GCase activity [20]. Thus, other methods for easy and reliable screening for the diagnosis of GD are needed.
In this paper, we publish seven years of experience by using DBS samples for GD diagnosis. We believe that our data will help establish the use of lyso-Gb1 extracted from DBS samples as a simple and highly reliable method for GD screening.
2. Results
2.1. Study Cohort
In this cross-sectional study, 444 subjects were tested (117 (27%) children < 18 years). DBS samples were sent for GBA1 molecular sequencing and lyso-Gb1 quantification. A third of the cases presented with clinical features, including splenomegaly, thrombocytopenia, short stature, etc. (Table 1). The rest were tested due to a family history of GD, as part of a research study in prodromal Parkinson’s disease (PD) and for the evaluation of GBA1 mutation in patients recently diagnosed with PD.

Table 1.
Characteristics of subjects with and without a diagnosis of Gaucher disease.
The diagnosis rate of GD was 22.3% (99/444). About 50% of subjects evaluated for clinical features, and 25% of those evaluated for family history were diagnosed with GD (Table 1). The most common genotype was N370S (c.1226A > G) homozygous (Table 2a). Two patients evaluated for a recent diagnosis of PD were diagnosed with GD: one at the age of 55 years with N370S/R496H (c.1604G) variants and the second at the age of 52 years homozygous for the N370S variant. In addition, three asymptomatic subjects, all homozygous for the N370S variant, were screened as part of a prodromal PD study and were diagnosed with GD.
Table 2.
Distribution of genotypes in subjects with or without the diagnosis of Gaucher disease.
The median age at diagnosis was 21 years; almost half were diagnosed when they were younger than 20 years, a quarter were 20–30, and a quarter were older than 30 years (Figure 1). The age at diagnosis was not different between males and females. The median (range) age at diagnosis was 21 (1–78) years for those with clinical symptoms and 16 (2–42) years for those evaluated for family history.
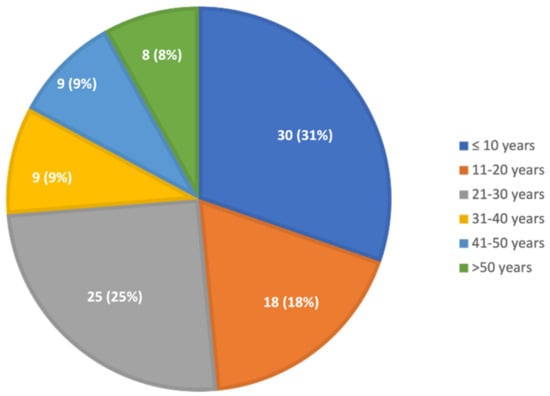
Figure 1.
Age at diagnosis of GD. Thirty children were diagnosed at the age of 0–10 years, 18 at the age of 11–20 years, 25 at the age of 21–30 years, nine at the age of 31–40 years, nine at the age of 41–50 years, four at the age of 51–60 years, and four at the age of over 61 years.
2.2. Lyso-Gb1 Levels for Diagnosis of Gaucher Disease
According to laboratory protocol, lyso-Gb1 levels less than 6.8 ng/mL are considered normal. All patients diagnosed with GD had lyso-Gb1 levels that were higher than this threshold (Table 1). Those diagnosed with GD1 and mild GBA1 variants had lower Lyso-Gb1 levels than those diagnosed with severe GBA1 variants (Table 3, Figure 2). Lyso-Gb1 levels of the diagnosed with nGD were non-significantly different from mild GD1 and severe GD1 (Figure 2).
Table 3.
Genetic variants in patients with Gaucher disease.
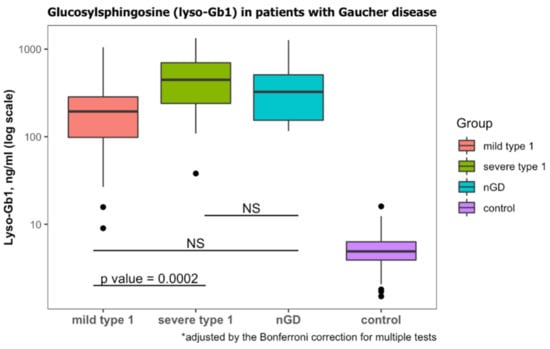
Figure 2.
Lyso-Gb1 levels at diagnosis of GD. Classification of mild type 1, severe type 1 and neuronopathic GD (nGD) was based on genotypes. Lyso-Gb1 levels of subjects with wild-type GBA1 were considered to be the control. NS, non-significant. Black dots represent the outliers.
In order to determine a lyso-Gb1 cutoff level for the diagnosis of GD, we computed a ROC curve and searched for the cutoff with the maximum sensitivity for the diagnosis of GD (Figure 3). The cutoff found in our data was 9 ng/mL (sensitivity 100%, CI 96–100%). The specificity of this cutoff was 91.3% (95% CI 87.8–94.1%).
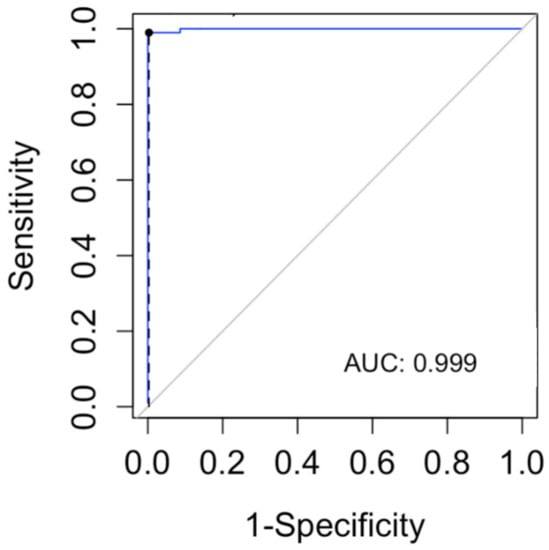
Figure 3.
The receiver operating characteristic (ROC) curve was used to evaluate the lyso-Gb1 ability for classifying Gaucher disease vs. no Gaucher disease. The area under the curve (AUC) was calculated.
Lyso-Gb1 levels above 9 ng/mL without clinically relevant bi-allelic GBA1 variants were more common in heterozygote GBA1 carriers than in the wild type (p = 0.036) (Table S2). No association was found with age, sex, or cause of testing.
2.3. Lyso-Gb1 Levels for Diagnosis Heterozygous GBA1 Carrier against Wild Type
In the cohort, 188 (42.3%) subjects were identified with a heterozygous GBA1 carrier status (Table 2b). Those subjects were older than subjects with wild type and were mainly screened due to a family history of GD, a diagnosis of PD, or as part of research in prodromal PD (Table 4). Lyso-Gb1 was significantly higher in carriers than those with the wild type (Table 4). Carriers with PD had slightly lower lyso-Gb1, but this difference did not reach statistical difference (Figure 4).
Table 4.
Characteristics of carriers vs. those with wild type GBA1.
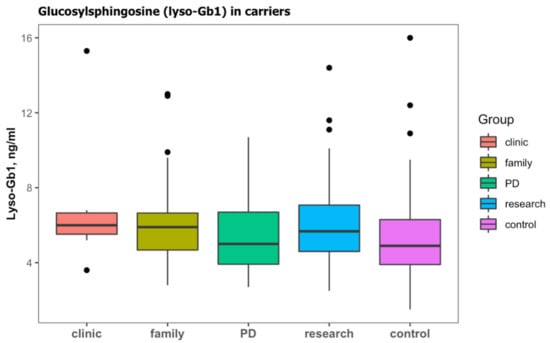
Figure 4.
Lyso-Gb1 levels in subjects with monoallelic variants in GBA1 (carriers) according to the cause for sending a sample; clinical features (clinic), family history of GD (family), Parkinson’s disease (PD), and subjects enrolled on a study of prodromal Parkinson’s disease (research). Lyso-Gb1 levels of subjects with wild-type GBA1 were considered to be the control. Black dots represent the outliers.
In order to determine a lyso-Gb1 cutoff level for the diagnosis of subjects with carrier status in GBA1 vs. wild type, we computed a ROC curve and searched for the cutoff with the maximum sensitivity for the diagnosis of subjects with monoallelic variants in GBA1 (Figure 5). The cutoff found in our data was 2.5 ng/mL (sensitivity 100%, CI 98–100%). The specificity of this cutoff was 6% (95% CI 2.7–11.1%).
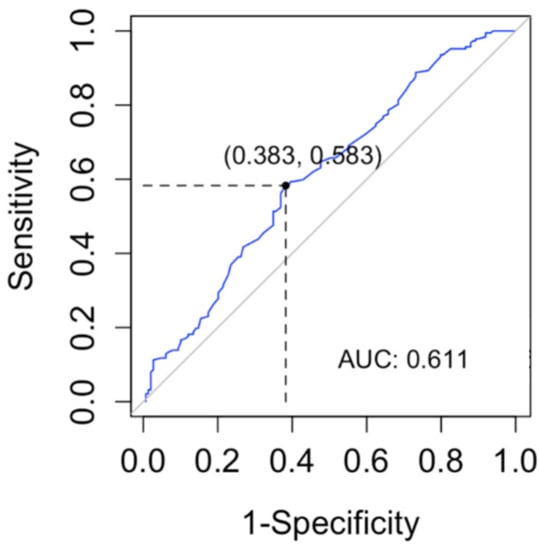
Figure 5.
Receiver operating characteristic (ROC) curve was used to evaluate the lyso-Gb1 ability for classifying carrier vs. wild-type GBA1. The area under the curve (AUC) was calculated.
3. Discussion
In our study, we confirmed the utility of DBS for diagnosing GD based on lyso-Gb1 levels and subsequent molecular analysis. All subjects with biallelic variants in GBA1, diagnosed as GD, had elevated lyso-Gb1, i.e., above 9 ng/mL. The combination of lyso-Gb1 and whole-gene sequence provided a 100% accurate diagnosis of GD, meaning that this can now become the new standard for screening of patients suspected to have GD. This recommendation updates the gold-standard diagnostic test (low activity of the glucocerebrosidase enzyme), which was defined by some of us in the two most recently published textbooks of hematology (see References [1,21]), as this current analysis had not been completed at the time those chapters were submitted. Severe cases of GD, all having significantly elevated lyso-Gb1 levels, would have been diagnosed definitively based on lyso-Gb1 testing alone. Lower lyso-Gb1 levels (less than 16 ng/mL) were also found in heterozygote GD carriers and individuals without relevant GBA1 variants, using state-of-the-art exon sequencing and copy-number analysis. The levels of lyso-Gb1 could not be used to differentiate between the heterozygous GBA1 carrier and wild type.
Early and accurate diagnosis of GD is essential to facilitate timely management decision-making, prevent unnecessary tests (some invasive), reduce anxiety induced by a lack of diagnosis, and importantly to prevent the birth of another sibling with potentially severe inherited disease [5]. Delayed diagnosis and misdiagnosis of GD, as for other rare diseases, remain major problems, due to nonspecific signs, symptoms, and laboratory findings; low awareness by healthcare professionals; and, in many cases, the lack of access to a simple diagnostic test. The inclusion of enzymatic GCase levels on fresh blood samples as the gold standard for the diagnosis of GD may be a major obstacle to the diagnosis of GD. Currently, very few laboratories are able to provide reliable enzymatic assays on blood samples. Most of them are research labs that cannot offer a clinical-scale activity or certification. In addition, the shipment of fresh blood samples is costly, and the number of samples sent at the same time is limited. The enzymatic GCase test needs a relatively large blood volume (6–10 mL) and preparation within 2 or 3 days. In contrast, the shipment of DBS cards is cheap, many cards can be sent at the same time, and the samples in the DBS remain stable for years. From a small volume of blood, both lyso-Gb1 levels and molecular analysis can be performed accurately.
The role of lyso-Gb1 in diagnosing GD is well established [19,22]. Our present study highlights the reliability of using lyso-Gb1 measured in DBS for diagnosing all types of GD in a large cohort of subjects who underwent testing for different reasons. Lyso-Gb1 measurement on DBS was also used in a case-control study conducted in the Russian Federation and was found to be highly sensitive and specific for GD [23]. In an Australian study, DBS measurements of lyso-Gb1 were also used for the prenatal diagnosis of GD [24]. The measurement of lyso-Gb1 levels from DBS has been used in newborn screening for GD [25].
In our study, we identified 99 new patients with GD. As expected, children with a severe genotype, i.e., nGD and compound heterozygote GD1, were diagnosed at an earlier age [26]. Disease severity based on genotype was also reflected by higher lyso-Gb1 levels. Since lyso-Gb1 was shown to have a role in the follow-up of treated and untreated patients [27,28,29,30], we believe that the inclusion of lyso-Gb1 in the diagnostic process can help in the assessment of the patient baseline status and for follow-up. For example, in an algorithm for asymptomatic patients who initially do not need treatment, an increase of Lyso-Gb1 levels could trigger the initiation of GD-specific therapy [2].
Subjects with monoallelic variants in GBA1 have an increased risk of developing PD [31,32,33]. In a meta-analysis of patients with PD, it was found that carriers of the N370S mutation have a 3-fold higher risk for developing PD, while carriers of other GBA1 mutations have up to a 15-fold higher risk for developing PD [34]. Although considerable clinical variation is seen [33], GBA-associated PD is typically associated with an earlier age of onset, a faster deterioration of motor functions, and a higher frequency and faster progression of cognitive decline compared to non GBA-associated PD, thus highlighting the importance of detection of the GBA1 variant in patients with PD (as well as with other synucleinopathies; see Reference [32]). Although all subjects with heterozygous GBA1 carrier status had a lyso-Gb1 above 2.5 ng/mL, the specificity of this cutoff was too low to recommend using lyso-Gb1 for screening for a heterozygous GBA1 variant. Lyso-Gb1 levels in subjects with PD were similar to those screened for other reasons. Plasma lyso-Gb1 accumulation is, therefore, unlikely to be related to the higher risk of PD in monoallelic GBA1 variants [35,36].
4. Materials and Methods
All DBS samples sent for GBA1 molecular sequencing and lyso-Gb1 from the Gaucher Unit, Shaare Zedek Medical Center, from July 2014 to July 2021, were included in the study. The number of tests performed increased with time (Figure 6).
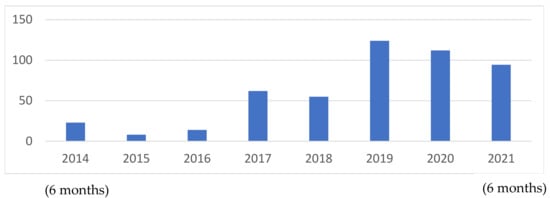
Figure 6.
Number of patients tested per year over the seven years of study.
All samples were analyzed in Centogene GmbH, Rostock, Germany. Genotyping was performed by whole GBA1 sequencing (exons and exon-intron boundaries) from DNA extracted from all DBS samples independent of the lyso-Gb1 levels. The sequencing approach would have detected all types of GBA1 aberrations, including GBAP-mediated recombination events. The definitions of mild versus severe genotypes were determined by N370S (c.1226A > G) homozygous and N370S/R496H (c.1604G) compound heterozygous were categorized as “mild”, whereas all other genotypes as “severe”. Lyso-Gb1 levels were performed according to the previously described method [19].
4.1. Biological Material
The material used was DBSs prepared by dropping 60 µL blood on CentoCard® filter paper (Centogene GmbH, Rostock, Germany); the spots were allowed to dry for 2–4 h at room temperature. The filter cards are the ideal material for shipping and long-term storage. Before analysis, 3.2 mm discs were cut from the homogeneous parts of the DBS, using a DBS puncher (Perkin Elmer LAS GmbH, Hamburg, Germany). Each disc contains approximately 3.1 µL blood.
4.2. Sample Preparation
For each subject, 3 DBS discs were cut and collected into a round-bottom 2 mL tube (Sarstedt AG & Co. KG, Nümbrecht, Germany). Then 50 µL extraction solution (DMSO/water, 1/1) and 100 µL internal standard solution (200 ng/mL Lyso-Gb2 (Biotrend Chemikalien GmbH, Köln, Germany) in ethanol, were added to the tube and incubated for 30 min, at 37 °C, under agitation at 700 rpm. The samples were briefly sonicated (1 min) before they were transferred to a PALL-8048 96-well filter plate with a PTFE membrane (WVR International GmbH, Dresden, Germany) on top of a 96-well V-shaped plate (WVR International GmbH, Dresden, Germany). The cellular and paper debris were filtered by centrifugation for 5 min, at 3500 rpm, in a Hermle Z300 plate centrifuge (Hermle Labortehnik GmbH, Wehingen, Germany). The V-shaped plate was covered with aluminum foil and inserted into the sample manager.
4.3. LC/MS Method
The samples were separated by liquid chromatography on an ACE 3 C8, 50 × 2.1 mm column (MZ-Analysentechnik GmbH, Mainz, Germany), using a Waters I-Class UPLC (Waters GmbH, Eschborn, Germany). Solvents: 50 mM FA in water (A) and 50 mM FA in acetone/acetonitrile 1/1 (B). A flow rate of 0.9 mL/min preheated at 60 °C was used. The gradient was linear, and the analytes were eluted between 40% and 70% B solvent.
The UPLC was coupled with an AB-Sciex TQ-5500 (AB Sciex Germany GmbH, Darmstadt, Germany) mass spectrometer, using a 3:1 flow splitter. An MRM method was used for monitoring the analytes with the following settings: curtain gas, 40 psi; ion spray, 5500 V; desolvation temperature, 500 °C; declustering potential, 40 V; entrance potential, 10 V; collision energy, 30 V; and MRM transitions, Lyso-Gb1 (462.3–282.2) and Lyso-Gb2 internal standard (624.3–282.2).
4.4. Quantification
A 7-points calibration line was added to each plate before measurement. The preparation was similar to the samples, but the DBSs were replaced with standard solutions of increasing Lyso-Gb1 (Biotrend Chemikalien GmbH, Köln, Germany) concentrations: 0, 1, 5, 10, 50, 100, and 200 ng/mL. Analysis and quantification were performed by using the Analyst 1.6.2 software (AB Sciex Germany GmbH, Darmstadt, Germany).
4.5. Statistical Analysis
To report summary descriptive statistics, we used median (range) for continuous variables. For nominal data, we report the absolute and relative frequencies. The Kruskal–Wallis H test was used to determine statistically significant differences between two or more groups of an independent variable on a continuous or ordinal dependent variable. A chi-square test was used to compare categorical data. The optimal.cutpoints r program with maximum sensitivity analysis was used to calculate the optimal cut point for diagnosing GD and monoallelic GBA1 variant and plotting the ROC curves [37]. The criterion was based on the maximization of sensitivity [38,39,40]. If more than one cut point fulfills this condition, those that yield maximum specificity are chosen. A two-sided significance level of α = 0.05 was considered.
5. Conclusions
Based on our results and others, we call for a shift in the diagnostic approach to GD based on lyso-Gb1 measured in DBS accompanied with molecular analysis of GBA1, without the need to confirm the diagnosis with blood-based measurement of GCase enzyme activity.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/ijms23031627/s1.
Author Contributions
Conceptualization, T.D., A.R. and S.R.-V.; methodology and formal analysis, S.R.-V.; data curation, T.D., M.B.-C., M.I., P.B., C.B., G.K., C.C., M.-I.I. and A.R.; writing—original draft preparation, T.D. and S.R.-V.; writing—review and editing, P.B., C.B., A.R. and A.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Institutional Review Board approved the publication of anonymized data. Shaare Zedek Medical Center IRB number-(0291-18-SZMC).
Informed Consent Statement
Subjects or legal guardians, in the case of children, signed an informed consent form before sending the DBS sample.
Data Availability Statement
Data cannot be shared due to ethical and privacy issues.
Conflicts of Interest
The SZMC Gaucher Unit receives support from Sanofi/Genzyme for participation in the ICGG Registry, from Takeda for the GOS Registry, and Pfizer for TALIAS. The Unit also receives research grants from Takeda, Pfizer, Sanofi/Genzyme, and Centogene. T.D., M.B.-C. and M.I. have no conflict of interest to declare. P.B., C.B., G.K., C.C. and M-I.I. are employees of Centogene GmbH. A.R. is the founder and was the CEO of Centogene GmbH during the study. A.Z. receives honoraria from Takeda, Pfizer, and BioEvents and consultancy fees from Takeda, NLC Pharma, Insightec, and Prevail therapeutics. S.R.-V. receives grant/research support, honoraria, and advisory fee from Takeda, Pfizer, and Sanofi/Genzyme.
References
- Revel-Vilk, S.; Szer, J.; Zimran, A. Gaucher disease and related lysosomal storage diseases. In Williams Hematology, 10th ed.; Kaushansky, K., Lichtman, M., Prchal, J., Levi, M., Press, O., Burns, L., Caligiuri, M., Eds.; McGraw-Hill: New York, NY, USA, 2021; pp. 1189–1202. [Google Scholar]
- Revel-Vilk, S.; Szer, J.; Mehta, A.; Zimran, A. How we manage Gaucher Disease in the era of choices. Br. J. Haematol. 2018, 182, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, S.; Lahad, A.; Shmueli, A.; Zimran, A.; Peleg, L.; Orr-Urtreger, A.; Levy-Lahad, E.; Sagi, M. Carrier screening for Gaucher disease: Lessons for low-penetrance, treatable diseases. JAMA 2007, 298, 1281–1290. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Grabowski, G.A.; Zimran, A.; Ida, H. Gaucher disease types 1 and 3: Phenotypic characterization of large populations from the ICGG Gaucher Registry. Am. J. Hematol. 2015, 90 (Suppl. 1), S12–S18. [Google Scholar] [CrossRef] [PubMed]
- Revel-Vilk, S.; Szer, J.; Zimran, A. Hematological manifestations and complications of Gaucher disease. Expert Rev. Hematol. 2021, 14, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.K.; Lopez, G.; Schiffmann, R.; Barton, N.W.; Weinreb, N.J.; Sidransky, E. Gaucher disease: Progress and ongoing challenges. Mol. Genet. Metab. 2017, 120, 8–21. [Google Scholar] [CrossRef]
- Bobillo Lobato, J.; Jiménez Hidalgo, M.; Jiménez Jiménez, L.M. Biomarkers in lysosomal storage diseases. Diseases 2016, 4, 40. [Google Scholar] [CrossRef]
- Ferraz, M.J.; Kallemeijn, W.W.; Mirzaian, M.; Herrera Moro, D.; Marques, A.; Wisse, P.; Boot, R.G.; Willems, L.I.; Overkleeft, H.S.; Aerts, J.M. Gaucher disease and Fabry disease: New markers and insights in pathophysiology for two distinct glycosphingolipidoses. Biochim. Biophys. Acta 2014, 1841, 811–825. [Google Scholar] [CrossRef]
- Stein, P.; Yang, R.; Liu, J.; Pastores, G.M.; Mistry, P.K. Evaluation of high density lipoprotein as a circulating biomarker of Gaucher disease activity. J Inherit Metab Dis. 2011, 34, 429–437. [Google Scholar] [CrossRef]
- Stein, P.; Yu, H.; Jain, D.; Mistry, P.K. Hyperferritinemia and iron overload in type 1 Gaucher disease. Am. J. Hematol. 2010, 85, 472–476. [Google Scholar] [CrossRef]
- Koppe, T.; Doneda, D.; Siebert, M.; Paskulin, L.; Camargo, M.; Tirelli, K.M.; Vairo, F.; Daudt, L.; Schwartz, I.V.D. The prognostic value of the serum ferritin in a southern Brazilian cohort of patients with Gaucher disease. Genet. Mol. Biol. 2016, 39, 30–34. [Google Scholar] [CrossRef]
- Stirnemann, J.; Boutten, A.; Vincent, C.; Mekinian, A.; Heraoui, D.; Fantin, B.; Fain, O.; Mentré, F.; Belmatoug, N. Impact of imiglucerase on the serum glycosylated-ferritin level in Gaucher disease. Blood Cells Mol. Dis. 2011, 46, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Salazar, M.A.; O’Rourke, E.; Henderson, N.; Wessel, H.; Barranger, J.A. Correlation of surrogate markers of Gaucher disease. Implications for long-term follow up of enzyme replacement therapy. Clin. Chim. Acta Int. J. Clin. Chem. 2004, 344, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Boot, R.G.; Verhoek, M.; de Fost, M.; Hollak, C.E.M.; Maas, M.; Bleijlevens, B.; van Breemen, M.J.; van Meurs, M.; Boven, L.A.; Laman, J.D.; et al. Marked elevation of the chemokine CCL18/PARC in Gaucher disease: A novel surrogate marker for assessing therapeutic intervention. Blood 2004, 103, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Hollak, C.E.; van Weely, S.; van Oers, M.H.; Aerts, J.M. Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher disease. J. Clin. Investig. 1994, 93, 1288–1292. [Google Scholar] [CrossRef] [PubMed]
- Schutyser, E.; Richmond, A.; Van Damme, J. Involvement of CC chemokine ligand 18 (CCL18) in normal and pathological processes. J. Leukoc. Biol. 2005, 78, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Van Dussen, L.; Hendriks, E.J.; Groener, J.E.M.; Boot, R.G.; Hollak, C.E.M.; Aerts, J.M.F.G. Value of plasma chitotriosidase to assess non-neuronopathic Gaucher disease severity and progression in the era of enzyme replacement therapy. J. Inherit. Metab. Dis. 2014, 37, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Dekker, N.; van Dussen, L.; Hollak, C.E.; Overkleeft, H.; Scheij, S.; Ghauharali, K.; van Breemen, M.J.; Ferraz, M.J.; Groener, J.E.; Maas, M.; et al. Elevated plasma glucosylsphingosine in Gaucher disease: Relation to phenotype, storage cell markers, and therapeutic response. Blood 2011, 118, e118–e127. [Google Scholar] [CrossRef] [PubMed]
- Rolfs, A.; Giese, A.K.; Grittner, U.; Mascher, D.; Elstein, D.; Zimran, A.; Bottcher, T.; Lukas, J.; Hubner, R.; Golnitz, U.; et al. Glucosylsphingosine is a highly sensitive and specific biomarker for primary diagnostic and follow-up monitoring in Gaucher disease in a non-Jewish, Caucasian cohort of Gaucher disease patients. PLoS ONE 2013, 8, e79732. [Google Scholar] [CrossRef]
- Moat, S.J.; George, R.S.; Carling, R.S. Use of Dried Blood Spot Specimens to Monitor Patients with Inherited Metabolic Disorders. Int. J. Neonatal Screen 2020, 6, 26. [Google Scholar] [CrossRef]
- Mehta, A.; Horowitz, M.; Carrillo-Farga, J.; Zimran, A. Lysosomal Storage Diseases—Focusing on Gaucher disease: Perspectives and Principles. In Hematology: Basic Principles and Practice, 8th ed.; Hoffman, R., Benz, E., Silberstein, L., Heslop, H., Weitz, J., Salama, M., Abutalib, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2022; In press. [Google Scholar]
- Revel-Vilk, S.; Fuller, M.; Zimran, A. Value of Glucosylsphingosine (Lyso-Gb1) as a Biomarker in Gaucher Disease: A Systematic Literature Review. Int. J. Mol. Sci. 2020, 21, 7159. [Google Scholar] [CrossRef]
- Savostyanova, K.; Pushkova, A.; Mura’vovaa, L.; Movsisyana, G.; Rykunovaa, A.; Ponomarevb, R.; Lukinab, K.; Lukinab, E.; Namazova-Baranovaa, L. Glucosylfingosine (lyso-GL1) may be the primary biomarker for screening Gaucher disease in Russian patients [Abstract number 318]. Mol. Genet. Metab. 2019, 126, S17–S156. [Google Scholar]
- Saville, J.T.; McDermott, B.K.; Chin, S.J.; Fletcher, J.M.; Fuller, M. Expanding the clinical utility of glucosylsphingosine for Gaucher disease. J. Inherit. Metab. Dis. 2020, 43, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Zhan, X.; Gu, X.; Zhang, H. Successful newborn screening for Gaucher disease using fluorometric assay in China. J. Hum. Genet. 2017, 62, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Gary, S.E.; Ryan, E.; Steward, A.M.; Sidransky, E. Recent advances in the diagnosis and management of Gaucher disease. Expert Rev. Endocrinol. Metab. 2018, 13, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Cozma, C.; Cullufi, P.; Kramp, G.; Hovakimyan, M.; Velmishi, V.; Gjikopulli, A.; Tomori, S.; Fischer, S.; Oppermann, S.; Grittner, U.; et al. Treatment Efficiency in Gaucher Patients Can Reliably Be Monitored by Quantification of Lyso-Gb1 Concentrations in Dried Blood Spots. Int. J. Mol. Sci. 2020, 21, 4577. [Google Scholar] [CrossRef]
- Dinur, T.; Grittner, U.; Revel-Vilk, S.; Becker-Cohen, M.; Istaiti, M.; Cozma, C.; Rolfs, A.; Zimran, A. Impact of Long-Term Enzyme Replacement Therapy on Glucosylsphingosine (Lyso-Gb1) Values in Patients with Type 1 Gaucher Disease: Statistical Models for Comparing Three Enzymatic Formulations. Int. J. Mol. Sci. 2021, 22, 7699. [Google Scholar] [CrossRef]
- Hurvitz, N.; Dinur, T.; Becker-Cohen, M.; Cozma, C.; Hovakimyan, M.; Oppermann, S.; Demuth, L.; Rolfs, A.; Abramov, A.; Zimran, A.; et al. Glucosylsphingosine (lyso-Gb1) as a Biomarker for Monitoring Treated and Untreated Children with Gaucher Disease. Int. J. Mol. Sci. 2019, 20, 3033. [Google Scholar] [CrossRef]
- Dinur, T.; Zimran, A.; Becker-Cohen, M.; Arkadir, D.; Cozma, C.; Hovakimyan, M.; Oppermann, S.; Demuth, L.; Rolfs, A.; Revel-Vilk, S. Long Term Follow-Up of 103 Untreated Adult Patients with Type 1 Gaucher Disease. J. Clin. Med. 2019, 8, 1662. [Google Scholar] [CrossRef]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Liong, C.; Alcalay, R.N. GBA-Associated Parkinson’s Disease and Other Synucleinopathies. Curr. Neurol. Neurosci. Rep. 2018, 18, 44. [Google Scholar] [CrossRef]
- Aharon-Peretz, J.; Rosenbaum, H.; Gershoni-Baruch, R. Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N. Engl. J. Med. 2004, 351, 1972–1977. [Google Scholar] [CrossRef] [PubMed]
- Gan-Or, Z.; Amshalom, I.; Kilarski, L.L.; Bar-Shira, A.; Gana-Weisz, M.; Mirelman, A.; Marder, K.; Bressman, S.; Giladi, N.; Orr-Urtreger, A. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 2015, 84, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Surface, M.; Balwani, M.; Waters, C.; Haimovich, A.; Gan-Or, Z.; Marder, K.S.; Hsieh, T.; Song, L.; Padmanabhan, S.; Hsieh, F.; et al. Plasma Glucosylsphingosine in GBA1 Mutation Carriers with and without Parkinson’s Disease. Mov. Disord. 2021. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Arkadir, D.; Bauer, P.; Dinur, T.; Lopez, G.; Rolfs, A.; Zimran, A. Substrate reduction therapy for GBA1-associated Parkinsonism: Are we betting on the wrong mouse? Mov. Disord. 2020, 35, 228–230. [Google Scholar] [CrossRef]
- López-Ratón, M.; Rodríguez-Álvarez, M.; Cadarso-Suárez, C.; Gude, F. OptimalCutpoints: An R Package for Selecting Optimal Cutpoints in Diagnostic Tests. J. Stat. Softw. 2014, 61, 1–36. [Google Scholar] [CrossRef]
- Alvarez-Garcia, G.; Collantes-Fernandez, E.; Costas, E.; Rebordosa, X.; Ortega-Mora, L.M. Influence of age and purpose for testing on the cut-off selection of serological methods in bovine neosporosis. Vet. Res. 2003, 34, 341–352. [Google Scholar] [CrossRef]
- Filella, X.; Alcover, J.; Molina, R.; Gimenez, N.; Rodriguez, A.; Jo, J.; Carretero, P.; Ballesta, A.M. Clinical usefulness of free PSA fraction as an indicator of prostate cancer. Int. J. Cancer 1995, 63, 780–784. [Google Scholar] [CrossRef]
- Hoffman, R.M.; Clanon, D.L.; Littenberg, B.; Frank, J.J.; Peirce, J.C. Using the free-to-total prostate-specific antigen ratio to detect prostate cancer in men with nonspecific elevations of prostate-specific antigen levels. J. Gen. Intern. Med. 2000, 15, 739–748. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).