
Activity and Function in Human Cells of the Evolutionary Conserved Exonuclease Polynucleotide Phosphorylase


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Organization and Regulation of the PNPT1 Gene
2.1. PNPT1 Gene and mRNA Features
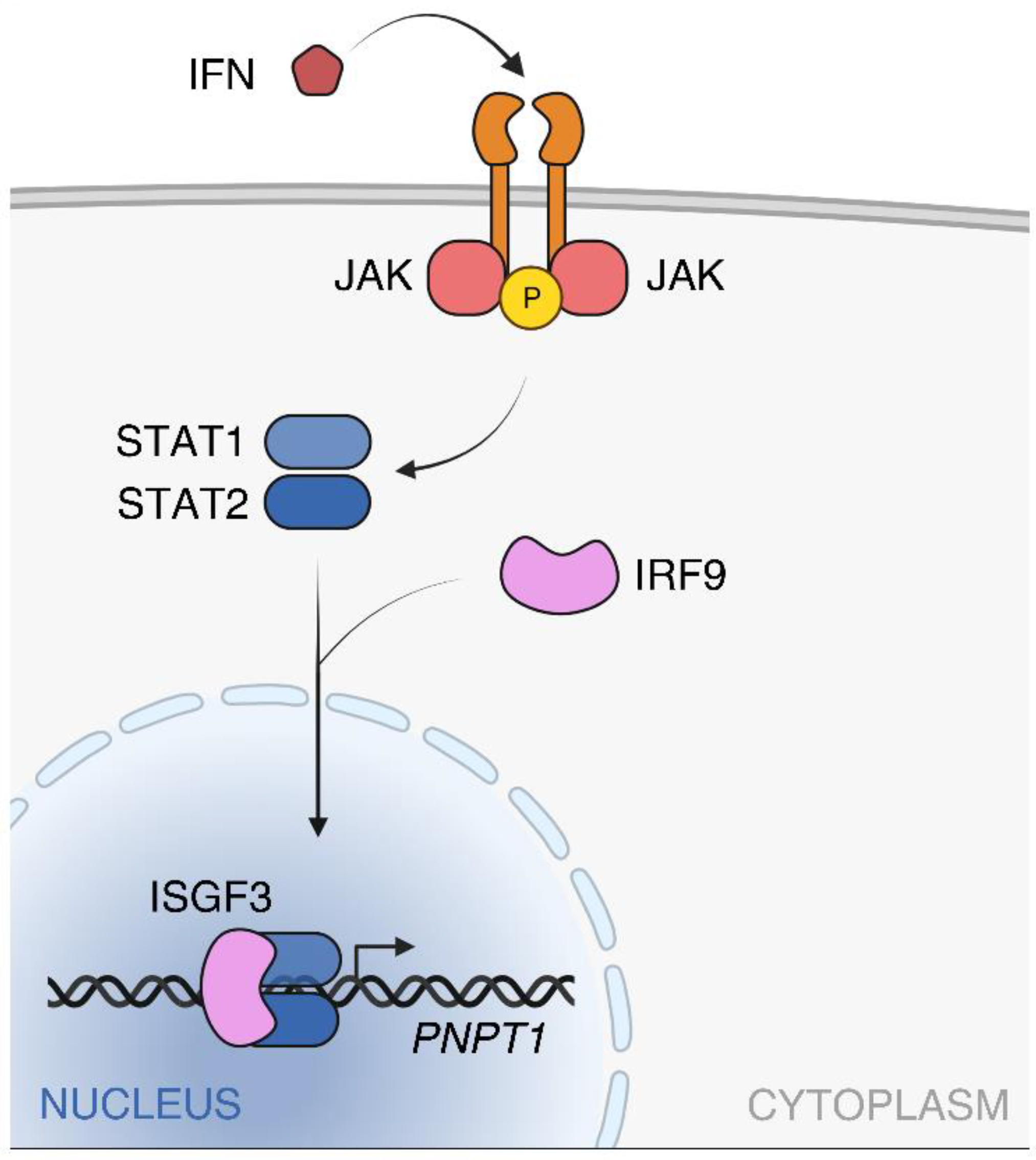
2.2. PNPT1 Regulation and Expression in Human Tissues
3. Structure of hPNPase: A Catalytic Core Capped by an RNA Binding Pore
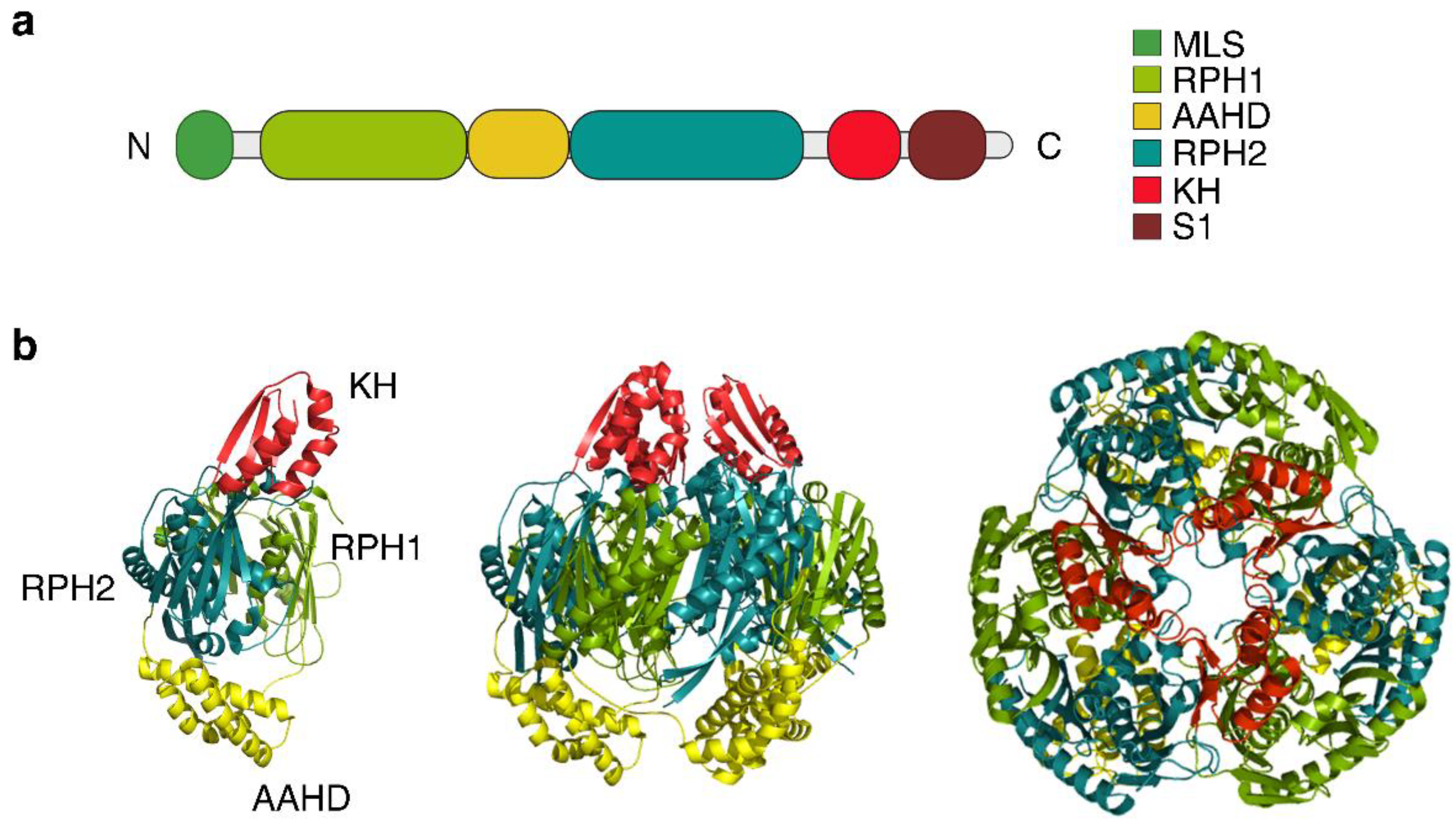
3.1. Protomer Domains and Quaternary Structure of hPNPase
3.2. Post-Translational Modifications and Interaction with Small Regulatory Molecules
4. hPNPase Catalytic and RNA Binding Activity
4.1. Catalytic Activity and Active Site Composition
4.2. Both the Enzyme Core and the KH-S1 Domains Contribute to RNA Binding
5. Function of hPNPase in Different Cell Compartments
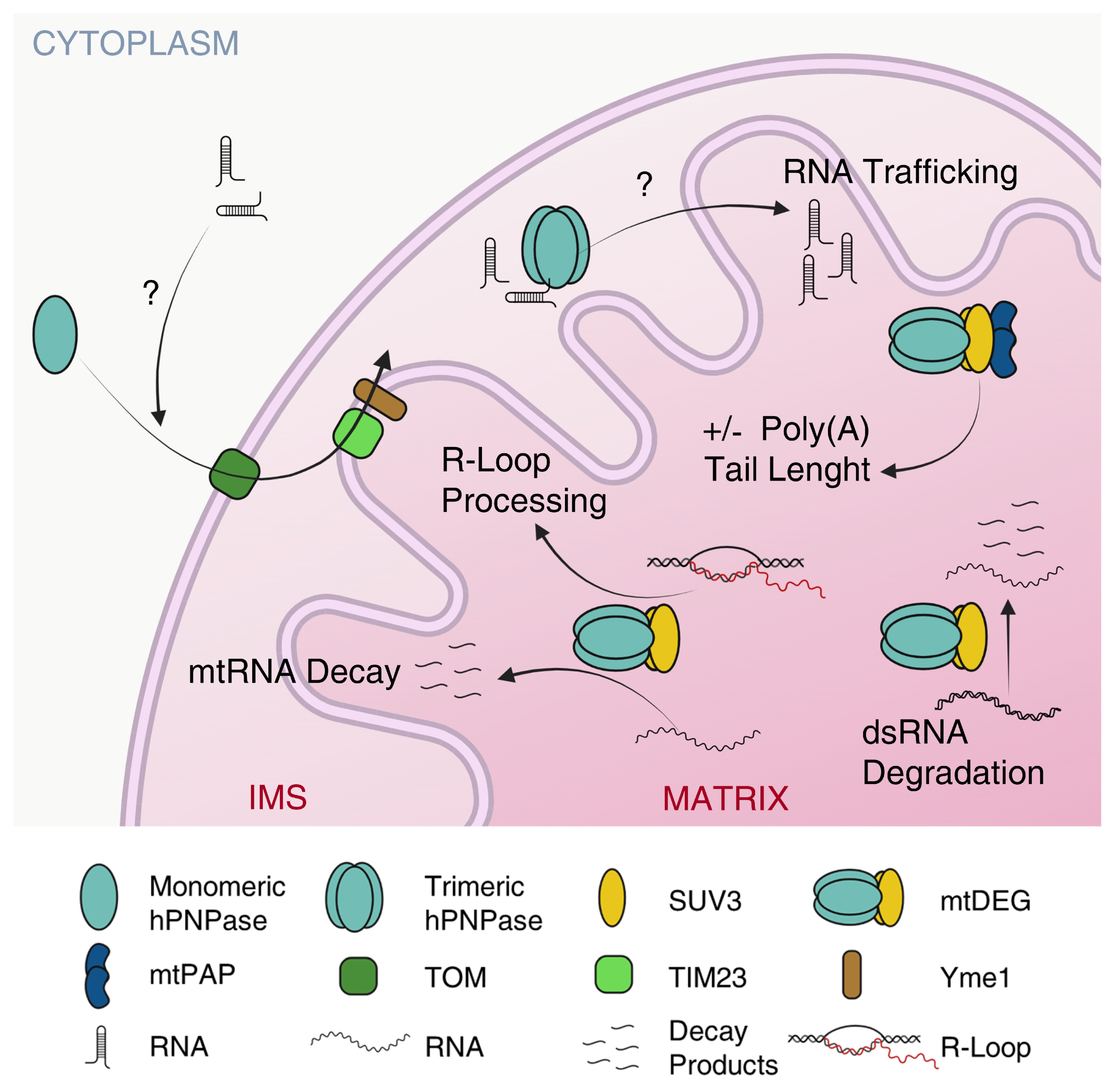
5.1. hPNPase Controls mtRNA Trafficking
5.2. hPNPase Controls mtRNA Decay and Prevents mtDNA Loss
5.3. Extra-Mitochondrial hPNPase Controls Cell Proliferation and Apoptosis
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Leszczyniecka, M.; DeSalle, R.; Kang, D.C.; Fisher, P.B. The origin of polynucleotide phosphorylase domains. Mol. Phylogenet. Evol. 2004, 31, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Briani, F.; Carzaniga, T.; Dehò, G. Regulation and functions of bacterial PNPase. Wiley Interdiscip. Rev. RNA 2016, 7, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Lin-Chao, S.; Chiou, N.T.; Schuster, G. The PNPase, exosome and RNA helicases as the building components of evolutionarily-conserved RNA degradation machines. J. Biomed. Sci. 2007, 14, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Grunberg-Manago, M.; Ortiz, P.J.; Ochoa, S. Enzymic synthesis of polynucleotides I. polynucleotide phosphorylase of Azotobacter vinelandii. Biochim. Biophys. Acta 1956, 20, 269–285. [Google Scholar] [CrossRef]
- Hakim, A.A. Synthetic activity of polynucleotide phosphorylase from sperm. Nature 1959, 183, 334. [Google Scholar] [CrossRef]
- Yagi, K.; Ozawa, T.; Konogi, H. Occurrence of Polynucleotide Phosphorylase in Atypical Epithelioma of Rat. Nature 1959, 184, 1939. [Google Scholar] [CrossRef]
- Leszczyniecka, M.; Kang, D.-C.; Sarkar, D.; Su, Z.-Z.; Holmes, M.; Valerie, K.; Fisher, P.B. Identification and cloning of human polynucleotide phosphorylase, hPNPase old-35, in the context of terminal differentiation and cellular senescence. Proc. Natl. Acad. Sci. USA 2002, 99, 16636–16641. [Google Scholar] [CrossRef]
- Piwowarski, J.; Grzechnik, P.; Dziembowski, A.; Dmochowska, A.; Minczuk, M.; Stepien, P.P. Human Polynucleotide Phosphorylase, hPNPase, is Localized in Mitochondria. J. Mol. Biol. 2003, 329, 853–857. [Google Scholar] [CrossRef]
- Eaton, A.; Bernier, F.P.; Goedhart, C.; Caluseriu, O.; Lamont, R.E.; Boycott, K.M.; Parboosingh, J.S.; Innes, A.M. Is PNPT1-related hearing loss ever non-syndromic? Whole exome sequencing of adult siblings expands the natural history of PNPT1-related disorders. Am. J. Med. Genet. Part A 2018, 176, 2487–2493. [Google Scholar] [CrossRef]
- Rius, R.; Van Bergen, N.J.; Compton, A.G.; Riley, L.G.; Kava, M.P.; Balasubramaniam, S.; Amor, D.J.; Fanjul-Fernandez, M.; Cowley, M.J.; Fahey, M.C.; et al. Clinical spectrum and functional consequences associated with bi-allelic pathogenic Pnpt1 variants. J. Clin. Med. 2019, 8, 2020. [Google Scholar] [CrossRef]
- Vedrenne, V.; Gowher, A.; De Lonlay, P.; Nitschke, P.; Serre, V.; Boddaert, N.; Altuzarra, C.; Mager-Heckel, A.M.; Chretien, F.; Entelis, N.; et al. Mutation in PNPT1, which encodes a polyribonucleotide nucleotidyltransferase, impairs RNA import into mitochondria and causes respiratory-chain deficiency. Am. J. Hum. Genet. 2012, 91, 912–918. [Google Scholar] [CrossRef]
- Leszczyniecka, M.; Su, Z.Z.; Kang, D.C.; Sarkar, D.; Fisher, P.B. Expression regulation and genomic organization of human polynucleotide phosphorylase, hPNPaseold-35, a Type I interferon inducible early response gene. Gene 2003, 316, 143–156. [Google Scholar] [CrossRef]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef]
- Wang, G.; Chen, H.W.; Oktay, Y.; Zhang, J.; Allen, E.L.; Smith, G.M.; Fan, K.C.; Hong, J.S.; French, S.W.; McCaffery, J.M.; et al. PNPASE regulates RNA import into mitochondria. Cell 2010, 142, 456–467. [Google Scholar] [CrossRef]
- Shimada, E.; Ahsan, F.M.; Nili, M.; Huang, D.; Atamdede, S.; TeSlaa, T.; Case, D.; Yu, X.; Gregory, B.D.; Perrin, B.J.; et al. PNPase knockout results in mtDNA loss and an altered metabolic gene expression program. PLoS ONE 2018, 13, e0200925. [Google Scholar] [CrossRef]
- Chen, H.-W.; Rainey, R.N.; Balatoni, C.E.; Dawson, D.W.; Troke, J.J.; Wasiak, S.; Hong, J.S.; McBride, H.M.; Koehler, C.M.; Teitell, M.A.; et al. Mammalian Polynucleotide Phosphorylase Is an Intermembrane Space RNase That Maintains Mitochondrial Homeostasis. Mol. Cell. Biol. 2006, 26, 8475–8487. [Google Scholar] [CrossRef]
- Takahashi, H.; Furukawa, T.; Yano, T.; Sato, N.; Takizawa, J.; Kurasaki, T.; Abe, T.; Narita, M.; Masuko, M.; Koyama, S.; et al. Identification of an overexpressed gene, HSPA4L, the product of which can provoke prevalent humoral immune responses in leukemia patients. Exp. Hematol. 2007, 35, 1091–1099. [Google Scholar] [CrossRef]
- Gewartowski, K.; Tomecki, R.; Muchowski, L.; Dmochowska, A.; Dzwonek, A.; Malecki, M.; Skurzak, H.; Ostrowski, J.; Stepien, P.P. Up-regulation of human PNPase mRNA by β-interferon has no effect on protein level in melanoma cell lines. Acta Biochim. Pol. 2006, 53, 179–187. [Google Scholar] [CrossRef]
- Wang, G.; Shimada, E.; Koehler, C.M.; Teitell, M.A. PNPASE and RNA trafficking into mitochondria. Biochim. Biophys. Acta Gene Regul. Mech. 2012, 1819, 998–1007. [Google Scholar] [CrossRef]
- Feng, H.; Zhang, Y.B.; Gui, J.F.; Lemon, S.M.; Yamane, D. Interferon regulatory factor 1 (IRF1) and anti-pathogen innate immune responses. PLoS Pathog. 2021, 17, e1009220. [Google Scholar] [CrossRef]
- Michalska, A.; Blaszczyk, K.; Wesoly, J.; Bluyssen, H.A.R. A Positive Feedback Amplifier Circuit That Regulates Interferon (IFN)-Stimulated Gene Expression and Controls Type I and Type II IFN Responses. Front. Immunol. 2018, 9, 1135. [Google Scholar] [CrossRef] [PubMed]
- Pine, R.; Decker, T.; Kessler, D.S.; Levy, D.E.; Darnell, J.E. Purification and cloning of interferon-stimulated gene factor 2 (ISGF2): ISGF2 (IRF-1) can bind to the promoters of both beta interferon- and interferon-stimulated genes but is not a primary transcriptional activator of either. Mol. Cell. Biol. 1990, 10, 2448–2457. [Google Scholar] [CrossRef] [PubMed]
- Marchi, P.; Longhi, V.; Zangrossi, S.; Gaetani, E.; Briani, F.; Dehò, G. Autogenous regulation of Escherichia coli polynucleotide phosphorylase during cold acclimation by transcription termination and antitermination. Mol. Genet. Genom. 2007, 278, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Carzaniga, T.; Briani, F.; Zangrossi, S.; Merlino, G.; Marchi, P.; Dehò, G. Autogenous regulation of Escherichia coli polynucleotide phosphorylase expression revisited. J. Bacteriol. 2009, 191, 1738–1748. [Google Scholar] [CrossRef] [PubMed]
- Jarrige, A.C.; Mathy, N.; Portier, C. PNPase autocontrols its expression by degrading a double-stranded structure in the pnp mRNA leader. EMBO J. 2001, 20, 6845–6855. [Google Scholar] [CrossRef]
- Shaw, G.; Kamen, R. A conserved AU sequence from the 3’ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell 1986, 46, 659–667. [Google Scholar] [CrossRef]
- Chen, C.Y.A.; Shyu, A. Bin Mechanisms of deadenylation-dependent decay. Wiley Interdiscip. Rev. RNA 2011, 2, 167–183. [Google Scholar] [CrossRef]
- Savant-Bhonsale, S.; Cleveland, D.W. Evidence for instability of mRNAs containing AUUUA motifs mediated through translation-dependent assembly of a >20S degradation complex. Genes Dev. 1992, 6, 1927–1939. [Google Scholar] [CrossRef]
- Von Ameln, S.; Wang, G.; Boulouiz, R.; Rutherford, M.A.; Smith, G.M.; Li, Y.; Pogoda, H.M.; Nürnberg, G.; Stiller, B.; Volk, A.E.; et al. A mutation in PNPT1, encoding mitochondrial-RNA-import protein PNPase, causes hereditary hearing loss. Am. J. Hum. Genet. 2012, 91, 919–927. [Google Scholar] [CrossRef]
- Li, Z.; Deutscher, M.P. Exoribonucleases and Endoribonucleases. EcoSal Plus 2004, 1. [Google Scholar] [CrossRef]
- Bermüdez-Cruz, R.M.; Ramïrez, F.; Kameyama-Kawabe, L.; Montañez, C. Conserved domains in polynucleotide phosphorylase among eubacteria. Biochimie 2005, 87, 737–745. [Google Scholar] [CrossRef]
- Symmons, M.F.; Jones, G.H.; Luisi, B.F. A duplicated fold is the structural basis for polynucleotide phosphorylase catalytic activity, processivity, and regulation. Structure 2000, 8, 1215–1226. [Google Scholar] [CrossRef]
- Nurmohamed, S.; Vaidialingam, B.; Callaghan, A.J.; Luisi, B.F. Crystal Structure of Escherichia coli Polynucleotide Phosphorylase Core Bound to RNase E, RNA and Manganese: Implications for Catalytic Mechanism and RNA Degradosome Assembly. J. Mol. Biol. 2009, 389, 17–33. [Google Scholar] [CrossRef]
- Hardwick, S.W.; Gubbey, T.; Hug, I.; Jenal, U.; Luisi, B.F. Crystal structure of Caulobacter crescentus polynucleotide phosphorylase reveals a mechanism of RNA substrate channelling and RNA degradosome assembly. Open Biol. 2012, 2, 120028. [Google Scholar] [CrossRef]
- Lin, C.L.; Wang, Y.T.; Yang, W.Z.; Hsiao, Y.Y.; Yuan, H.S. Crystal structure of human polynucleotide phosphorylase: Insights into its domain function in RNA binding and degradation. Nucleic Acids Res. 2012, 40, 4146–4157. [Google Scholar] [CrossRef]
- Shi, Z.; Yang, W.Z.; Lin-Chao, S.; Chak, K.F.; Yuan, H.S. Crystal structure of Escherichia coli PNPase: Central channel residues are involved in processive RNA degradation. RNA 2008, 14, 2361–2371. [Google Scholar] [CrossRef]
- Matus-Ortega, M.E.; Regonesi, M.E.; Piña-Escobedo, A.; Tortora, P.; Dehò, G.; García-Mena, J. The KH and S1 domains of Escherichia coli polynucleotide phosphorylase are necessary for autoregulation and growth at low temperature. Biochim. Biophys. Acta 2007, 1769, 194–203. [Google Scholar] [CrossRef]
- García-Mena, J.; Das, A.; Sánchez-Trujillo, A.; Portier, C.; Montanez, C. A novel mutation in the KH domain of polynucleotide phosphorylase affects autoregulation and mRNA decay in Escherichia coli. Mol. Microbiol. 1999, 33, 235–248. [Google Scholar] [CrossRef]
- Jarrige, A.-C.; Bréchemier-Baey, D.; Mathy, N.; Duché, O.; Portier, C. Mutational Analysis of Polynucleotide Phosphorylase from Escherichia coli. J. Mol. Biol. 2002, 321, 397–409. [Google Scholar] [CrossRef]
- Symmons, M.F.; Williams, M.G.; Luisi, B.F.; Jones, G.H.; Carpousis, A.J. Running rings around RNA: A superfamily of phosphate-dependent RNases. Trends Biochem. Sci. 2002, 27, 11–18. [Google Scholar] [CrossRef]
- Dendooven, T.; Sinha, D.; Roeselová, A.; Cameron, T.A.; De Lay, N.R.; Luisi, B.F.; Bandyra, K.J. A cooperative PNPase-Hfq-RNA carrier complex facilitates bacterial riboregulation. Mol. Cell 2021, 81, 2901–2913.e5. [Google Scholar] [CrossRef] [PubMed]
- Carzaniga, T.; Mazzantini, E.; Nardini, M.; Regonesi, M.E.; Greco, C.; Briani, F.; De Gioia, L.; Dehò, G.; Tortora, P. A conserved loop in polynucleotide phosphorylase (PNPase) essential for both RNA and ADP/phosphate binding. Biochimie 2014, 97, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Januszyk, K.; Lima, C.D. Structural components and architectures of RNA exosomes. In RNA Exosomes; Springer: Berlin, Germany, 2010. [Google Scholar] [CrossRef]
- Raijmakers, R.; Vree Egberts, W.; Van Venrooij, W.J.; Pruijn, G.J.M. Protein-protein interactions between human exosome components support the assembly of RNase PH-type subunits into a six-membered PNPase-like ring. J. Mol. Biol. 2002, 323, 653–663. [Google Scholar] [CrossRef]
- Lorentzen, E.; Walter, P.; Fribourg, S.; Evguenieva-Hackenberg, E.; Klug, G.; Conti, E. The archaeal exosome core is a hexameric ring structure with three catalytic subunits. Nat. Struct. Mol. Biol. 2005, 12, 575–581. [Google Scholar] [CrossRef]
- Sikorska, N.; Zuber, H.; Gobert, A.; Lange, H.; Gagliardi, D. RNA degradation by the plant RNA exosome involves both phosphorolytic and hydrolytic activities. Nat. Commun. 2017, 8, 2162. [Google Scholar] [CrossRef]
- Dziembowski, A.; Lorentzen, E.; Conti, E.; Séraphin, B. A single subunit, Dis3, is essentially responsible for yeast exosome core activity. Nat. Struct. Mol. Biol. 2006, 14, 15–22. [Google Scholar] [CrossRef]
- Liu, Q.; Greimann, J.C.; Lima, C.D. Reconstitution, activities, and structure of the eukaryotic RNA exosome. Cell 2006, 127, 1223–1237. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef]
- Yu, Y.L.; Chou, R.H.; Wu, C.H.; Wang, Y.N.; Chang, W.J.; Tseng, Y.J.; Chang, W.C.; Lai, C.C.; Lee, H.J.; Huo, L.; et al. Nuclear EGFR Suppresses Ribonuclease Activity of Polynucleotide Phosphorylase through DNAPK-mediated Phosphorylation at Serine 776. J. Biol. Chem. 2012, 287, 31015. [Google Scholar] [CrossRef]
- Tuckerman, J.R.; Gonzalez, G.; Gilles-Gonzalez, M.A. Cyclic di-GMP Activation of Polynucleotide Phosphorylase Signal-Dependent RNA Processing. J. Mol. Biol. 2011, 407, 633–639. [Google Scholar] [CrossRef]
- Siculella, L.; Damiano, F.; Di Summa, R.; Tredici, S.M.; Alduina, R.; Gnoni, G.V.; Alifano, P. Guanosine 5’-diphosphate 3’-diphosphate (ppGpp) as a negative modulator of polynucleotide phosphorylase activity in a “rare” actinomycete. Mol. Microbiol. 2010, 77, 716–729. [Google Scholar] [CrossRef]
- Gatewood, M.L.; Jones, G.H. (p)ppGpp inhibits polynucleotide phosphorylase from Streptomyces but not from Escherichia coli and increases the stability of bulk mRNA in Streptomyces coelicolor. J. Bacteriol. 2010, 192, 4275–4280. [Google Scholar] [CrossRef]
- Del Favero, M.; Mazzantini, E.; Briani, F.; Zangrossi, S.; Tortora, P.; Dehò, G. Regulation of Escherichia coli polynucleotide phosphorylase by ATP. J. Biol. Chem. 2008, 283, 27355–27359. [Google Scholar] [CrossRef]
- Nurmohamed, S.; Vincent, H.A.; Titman, C.M.; Chandran, V.; Pears, M.R.; Du, D.; Griffin, J.L.; Callaghan, A.J.; Luisi, B.F. Polynucleotide phosphorylase activity may be modulated by metabolites in Escherichia coli. J. Biol. Chem. 2011, 286, 14315–14323. [Google Scholar] [CrossRef]
- Stone, C.M.; Butt, L.E.; Bufton, J.C.; Lourenco, D.C.; Gowers, D.M.; Pickford, A.R.; Cox, P.A.; Vincent, H.A.; Callaghan, A.J. Inhibition of homologous phosphorolytic ribonucleases by citrate may represent an evolutionarily conserved communicative link between RNA degradation and central metabolism. Nucleic Acids Res. 2017, 45, 4655–4666. [Google Scholar] [CrossRef]
- Icard, P.; Poulain, L.; Lincet, H. Understanding the central role of citrate in the metabolism of cancer cells. Biochim. Biophys. Acta 2012, 1825, 111–116. [Google Scholar] [CrossRef]
- Golzarroshan, B.; Lin, C.-L.; Li, C.-L.; Yang, W.-Z.; Chu, L.-Y.; Agrawal, S.; Yuan, H.S. Crystal structure of dimeric human PNPase reveals why disease-linked mutants suffer from low RNA import and degradation activities. Nucleic Acids Res. 2018, 46, 8630–8640. [Google Scholar] [CrossRef]
- Portnoy, V.; Palnizky, G.; Yehudai-Resheff, S.; Glaser, F.; Schuster, G. Analysis of the human polynucleotide phosphorylase (PNPase) reveals differences in RNA binding and response to phosphate compared to its bacterial and chloroplast counterparts. Rna 2008, 14, 297–309. [Google Scholar] [CrossRef]
- Cheng, Z.F.; Deutscher, M.P. An important role for RNase R in mRNA decay. Mol. Cell 2005, 17, 313–318. [Google Scholar] [CrossRef]
- Spickler, C.; Mackie, G.A. Action of RNase II and polynucleotide phosphorylase against RNAs containing stem-loops of defined structure. J. Bacteriol. 2000, 182, 2422–2427. [Google Scholar] [CrossRef]
- Andrade, J.M.; Pobre, V.; Silva, I.J.; Domingues, S.; Arraiano, C.M. The Role of 3′–5′ Exoribonucleases in RNA Degradation. Prog. Mol. Biol. Transl. Sci. 2009, 85, 187–229. [Google Scholar] [CrossRef]
- Viegas, S.C.; Matos, R.G.; Arraiano, C.M. The Bacterial Counterparts of the Eukaryotic Exosome: An Evolutionary Perspective. Methods Mol. Biol. 2020, 2062, 37–46. [Google Scholar] [CrossRef]
- Wang, D.D.H.; Shu, Z.; Lieser, S.A.; Chen, P.L.; Lee, W.H. Human mitochondrial SUV3 and polynucleotide phosphorylase form a 330-kDa heteropentamer to cooperatively degrade double-stranded RNA with a 3′-to-5′ directionality. J. Biol. Chem. 2009, 284, 20812–20821. [Google Scholar] [CrossRef]
- Liu, X.; Fu, R.; Pan, Y.; Meza-Sosa, K.F.; Zhang, Z.; Lieberman, J. PNPT1 Release from Mitochondria during Apoptosis Triggers Decay of Poly(A) RNAs. Cell 2018, 174, 187–201.e12. [Google Scholar] [CrossRef]
- Soboll, S.; Sholz, R.; Heldt, H.W. Subcellular Metabolite Concentrations: Dependence of Mitochondrial and Cytosolic ATP Systems on the Metabolic State of Perfuse Rat Liver. Eur. J. Biochem. 1978, 87, 377–390. [Google Scholar] [CrossRef]
- Amin, N.; Peterkofsky, A. A dual mechanism for regulating cAMP levels in Escherichia coli. J. Biol. Chem. 1995, 270, 11803–11805. [Google Scholar] [CrossRef]
- Sarpel, G.; Barp, A.N.; Lubansky, H.J.; Omachi, A. Erythocyte phosphate content in Huntington’s disease. Neurosci. Lett. 1982, 31, 91–96. [Google Scholar] [CrossRef]
- Nesmeyanova, M.A. Polyphosphates and enzymes of polyphosphate metabolism in Escherichia coli. Biochemistry 2000, 65, 309–314. [Google Scholar]
- Unciuleac, M.C.; Ghosh, S.; de la Cruz, M.J.; Goldgur, Y.; Shuman, S. Structure and mechanism of Mycobacterium smegmatis polynucleotide phosphorylase. RNA 2021, 27, 959–969. [Google Scholar] [CrossRef]
- Briani, F.; Del Favero, M.; Capizzuto, R.; Consonni, C.; Zangrossi, S.; Greco, C.; De Gioia, L.; Tortora, P.; Dehò, G. Genetic analysis of polynucleotide phosphorylase structure and functions. Biochimie 2007, 89, 145–157. [Google Scholar] [CrossRef]
- Matilainen, S.; Carroll, C.J.; Richter, U.; Euro, L.; Pohjanpelto, M.; Paetau, A.; Isohanni, P.; Suomalainen, A. Defective mitochondrial RNA processing due to PNPT1 variants causes Leigh syndrome. Hum. Mol. Genet. 2017, 26, 3352–3361. [Google Scholar] [CrossRef] [PubMed]
- Dhir, A.; Dhir, S.; Borowski, L.S.; Jimenez, L.; Teitell, M.; Rötig, A.; Crow, Y.J.; Rice, G.I.; Duffy, D.; Tamby, C.; et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 2018, 560, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, G.; Littauer, U.Z. Deoxyadenosine diphosphate as substrate for polynucleotide phosphorylase from Escherichia coli. FEBS Lett. 1969, 4, 79–83. [Google Scholar] [CrossRef]
- Gillam, S.; Waterman, K.; Doel, M.; Smith, M. Enzymatic synthesis of deoxyribo-oiigonucleotides of defined sequence. Deoxyribo-oligonucleotide synthesis. Nucleic Acids Res. 1974, 1, 1649–1664. [Google Scholar] [CrossRef]
- Cardenas, P.P.; Carrasco, B.; Sanchez, H.; Deikus, G.; Bechhofer, D.H.; Alonso, J.C. Bacillus subtilis polynucleotide phosphorylase 3’-to-5’ DNase activity is involved in DNA repair. Nucleic Acids Res. 2009, 37, 4157–4169. [Google Scholar] [CrossRef] [PubMed]
- Carzaniga, T.; Sbarufatti, G.; Briani, F.; Dehò, G. Polynucleotide phosphorylase is implicated in homologous recombination and DNA repair in Escherichia coli. BMC Microbiol. 2017, 17, 81. [Google Scholar] [CrossRef]
- Cardenas, P.P.; Carzaniga, T.; Zangrossi, S.; Briani, F.; Garcia-Tirado, E.; Dehò, G.; Alonso, J.C. Polynucleotide phosphorylase exonuclease and polymerase activities on single-stranded DNA ends are modulated by RecN, SsbA and RecA proteins. Nucleic Acids Res. 2011, 39, 9250–9261. [Google Scholar] [CrossRef]
- Becket, E.; Tse, L.; Yung, M.; Cosico, A.; Miller, J.H. Polynucleotide phosphorylase plays an important role in the generation of spontaneous mutations in Escherichia coli. J. Bacteriol. 2012, 194, 5613–5620. [Google Scholar] [CrossRef][Green Version]
- Rath, D.; Mangoli, S.H.; Pagedar, A.R.; Jawali, N. Involvement of pnp in survival of UV radiation in Escherichia coli K-12. Microbiology 2012, 158, 1196–1205. [Google Scholar] [CrossRef]
- Bermúdez-Cruz, R.M.; García-Mena, J.; Montaez, C. Polynucleotide phosphorylase binds to ssRNA with same affinity as to ssDNA. Biochimie 2002, 84, 321–328. [Google Scholar] [CrossRef]
- Lisitsky, I.; Schuster, G. Preferential degradation of polyadenylated and polyuridinylated RNAs by the bacterial exoribonuclease polynucleotide phosphorylase. Eur. J. Biochem. 1999, 261, 468–474. [Google Scholar] [CrossRef]
- Wong, A.G.; McBurney, K.L.; Thompson, K.J.; Stickney, L.M.; Mackie, G.A. S1 and KH domains of polynucleotide phosphorylase determine the efficiency of RNA binding and autoregulation. J. Bacteriol. 2013, 195, 2021–2031. [Google Scholar] [CrossRef]
- Valverde, R.; Edwards, L.; Regan, L. Structure and function of KH domains. FEBS J. 2008, 275, 2712–2726. [Google Scholar] [CrossRef]
- Sarkar, D.; Park, E.S.; Emdad, L.; Randolph, A.; Valerie, K.; Fisher, P.B. Defining the domains of human polynucleotide phosphorylase (hPNPaseOLD-35) mediating cellular senescence. Mol. Cell. Biol. 2005, 25, 7333–7343. [Google Scholar] [CrossRef]
- Chen, H.W.; Koehler, C.M.; Teitell, M.A. Human polynucleotide phosphorylase: Location matters. Trends Cell Biol. 2007, 17, 600–608. [Google Scholar] [CrossRef]
- Rainey, R.N.; Glavin, J.D.; Chen, H.-W.; French, S.W.; Teitell, M.A.; Koehler, C.M. A New Function in Translocation for the Mitochondrial i-AAA Protease Yme1: Import of Polynucleotide Phosphorylase into the Intermembrane Space. Mol. Cell. Biol. 2006, 26, 8488–8497. [Google Scholar] [CrossRef]
- French, S.W.; Dawson, D.W.; Chen, H.W.; Rainey, R.N.; Sievers, S.A.; Balatoni, C.E.; Wong, L.; Troke, J.J.; Nguyen, M.T.N.; Koehler, C.M.; et al. The TCL1 oncoprotein binds the RNase PH domains of the PNPase exoribonuclease without affecting its RNA degrading activity. Cancer Lett. 2007, 248, 198–210. [Google Scholar] [CrossRef]
- Khaw, S.L.; Min-Wen, C.; Koh, C.G.; Lim, B.; Shyh-Chang, N. Oocyte Factors Suppress Mitochondrial Polynucleotide Phosphorylase to Remodel the Metabolome and Enhance Reprogramming. Cell Rep. 2015, 12, 1080–1088. [Google Scholar] [CrossRef]
- Shyh-Chang, N.; Daley, G.Q.; Cantley, L.C. Stem cell metabolism in tissue development and aging. Development 2013, 140, 2535–2547. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Hallas, C.; Pekarsky, Y.; Itoyama, T.; Varnum, J.; Bichi, R.; Rothstein, J.L.; Croce, C.M. Genomic analysis of human and mouse TCL1 loci reveals a complex of tightly clustered genes. Proc. Natl. Acad. Sci. USA 1999, 96, 14418–14423. [Google Scholar] [CrossRef]
- Narducci, M.G.; Fiorenza, M.T.; Kang, S.M.; Bevilacqua, A.; Di Giacomo, M.; Remotti, D.; Picchio, M.C.; Fidanza, V.; Cooper, M.D.; Croce, C.M.; et al. TCL1 participates in early embryonic development and is overexpressed in human seminomas. Proc. Natl. Acad. Sci. USA 2002, 99, 11712–11717. [Google Scholar] [CrossRef]
- Gammage, P.A.; Moraes, C.T.; Minczuk, M. Mitochondrial Genome Engineering: The Revolution May Not Be CRISPR-Ized. Trends Genet. 2018, 34, 101–110. [Google Scholar] [CrossRef]
- Wang, G.; Shimada, E.; Zhang, J.; Hong, J.S.; Smith, G.M.; Teitell, M.A.; Koehler, C.M. Correcting human mitochondrial mutations with targeted RNA import. Proc. Natl. Acad. Sci. USA 2012, 109, 4840–4845. [Google Scholar] [CrossRef]
- Cheng, Y.; Liu, P.; Zheng, Q.; Gao, G.; Yuan, J.; Wang, P.; Huang, J.; Xie, L.; Lu, X.; Tong, T.; et al. Mitochondrial Trafficking and Processing of Telomerase RNA TERC. Cell Rep. 2018, 24, 2589–2595. [Google Scholar] [CrossRef]
- Jeandard, D.; Smirnova, A.; Tarassov, I.; Barrey, E.; Smirnov, A.; Entelis, N. Import of Non-Coding RNAs into Human Mitochondria: A Critical Review and Emerging Approaches. Cells 2019, 8, 286. [Google Scholar] [CrossRef]
- Smirnov, A.; Comte, C.; Mager-Heckel, A.M.; Addis, V.; Krasheninnikov, I.A.; Martin, R.P.; Entelis, N.; Tarassov, I. Mitochondrial enzyme rhodanese is essential for 5S ribosomal RNA import into human mitochondria. J. Biol. Chem. 2010, 285, 30792–30803. [Google Scholar] [CrossRef]
- Borowski, L.S.; Dziembowski, A.; Hejnowicz, M.S.; Stepien, P.P.; Szczesny, R.J. Human mitochondrial RNA decay mediated by PNPase-hSuv3 complex takes place in distinct foci. Nucleic Acids Res. 2013, 41, 1223–1240. [Google Scholar] [CrossRef]
- Chujo, T.; Ohira, T.; Sakaguchi, Y.; Goshima, N.; Nomura, N.; Nagao, A.; Suzuki, T. LRPPRC/SLIRP suppresses PNPase-mediated mRNA decay and promotes polyadenylation in human mitochondria. Nucleic Acids Res. 2012, 40, 8033–8047. [Google Scholar] [CrossRef]
- Rhee, H.W.; Zou, P.; Udeshi, N.D.; Martell, J.D.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 2013, 339, 1328–1331. [Google Scholar] [CrossRef]
- Szczesny, R.J.; Borowski, L.S.; Brzezniak, L.K.; Dmochowska, A.; Gewartowski, K.; Bartnik, E.; Stepien, P.P. Human mitochondrial RNA turnover caught in flagranti: Involvement of hSuv3p helicase in RNA surveillance. Nucleic Acids Res. 2009, 38, 279–298. [Google Scholar] [CrossRef] [PubMed]
- Venø, S.T.; Kulikowicz, T.; Pestana, C.; Stepien, P.P.; Stevnsner, T.; Bohr, V.A. The human Suv3 helicase interacts with replication protein A and flap endonuclease 1 in the nucleus. Biochem. J. 2011, 440, 293. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.A.; Vicente, A.M.; Hardwick, S.W.; Alvelos, D.M.; Mazzon, R.R.; Luisi, B.F.; Marques, M.V. Association of the Cold Shock DEAD-Box RNA Helicase RhlE to the RNA Degradosome in Caulobacter crescentus. J. Bacteriol. 2017, 199, e00135-17. [Google Scholar] [CrossRef] [PubMed]
- Regonesi, M.E.; Del Favero, M.; Basilico, F.; Briani, F.; Benazzi, L.; Tortora, P.; Mauri, P.; Dehò, G. Analysis of the Escherichia coli RNA degradosome composition by a proteomic approach. Biochimie 2006, 88, 151–161. [Google Scholar] [CrossRef]
- Khemici, V.; Toesca, I.; Poljak, L.; Vanzo, N.F.; Carpousis, A.J. The RNase E of Escherichia coli has at least two binding sites for DEAD-box RNA helicases: Functional replacement of RhlB by RhlE. Mol. Microbiol. 2004, 54, 1422–1430. [Google Scholar] [CrossRef]
- Prud’homme-Géńreux, A.; Beran, R.K.; Iost, I.; Ramey, C.S.; Mackie, G.A.; Simons, R.W. Physical and functional interactions among RNase E, polynucleotide phosphorylase and the cold-shock protein, CsdA: Evidence for a “cold shock degradosome”. Mol. Microbiol. 2004, 54, 1409–1421. [Google Scholar] [CrossRef]
- Tejada-Arranz, A.; de Crécy-Lagard, V.; de Reuse, H. Bacterial RNA degradosomes: Molecular machines under tight control. Trends Biochem. Sci. 2020, 45, 42. [Google Scholar] [CrossRef]
- Py, B.; Higgins, C.F.; Krisch, H.M.; Carpousis, A.J. A DEAD-box RNA helicase in the Escherichia coli RNA degradosome. Nature 1996, 381, 169–172. [Google Scholar] [CrossRef]
- Lin, P.H.; Lin-Chao, S. RhlB helicase rather than enolase is the β-subunit of the Escherichia coli polynucleotide phosphorylase (PNPase)–exoribonucleolytic complex. Proc. Natl. Acad. Sci. USA 2005, 102, 16590–16595. [Google Scholar] [CrossRef]
- Dziembowski, A.; Piwowarski, J.; Hoser, R.; Minczuk, M.; Dmochowska, A.; Siep, M.; Van der Spek, H.; Grivell, L.; Stepien, P.P. The yeast mitochondrial degradosome. Its composition, interplay between RNA helicase and RNase activities and the role in mitochondrial RNA metabolism. J. Biol. Chem. 2003, 278, 1603–1611. [Google Scholar] [CrossRef]
- Jedynak-Slyvka, M.; Jabczynska, A.; Szczesny, R.J. Human Mitochondrial RNA Processing and Modifications: Overview. Int. J. Mol. Sci. 2021, 22, 7999. [Google Scholar] [CrossRef]
- Pietras, Z.; Wojcik, M.A.; Borowski, L.S.; Szewczyk, M.; Kulinski, T.M.; Cysewski, D.; Stepien, P.P.; Dziembowski, A.; Szczesny, R.J. Dedicated surveillance mechanism controls G-quadruplex forming non-coding RNAs in human mitochondria. Nat. Commun. 2018, 9, 2558. [Google Scholar] [CrossRef]
- Pietras, Z.; Wojcik, M.A.; Borowski, L.S.; Szewczyk, M.; Kulinski, T.M.; Cysewski, D.; Stepien, P.P.; Dziembowski, A.; Szczesny, R.J. Controlling the mitochondrial antisense–role of the SUV3-PNPase complex and its co-factor GRSF1 in mitochondrial RNA surveillance. Mol. Cell. Oncol. 2018, 5, e1516452. [Google Scholar] [CrossRef]
- Silva, S.; Camino, L.P.; Aguilera, A. Human mitochondrial degradosome prevents harmful mitochondrial R loops and mitochondrial genome instability. Proc. Natl. Acad. Sci. USA 2018, 115, 11024–11029. [Google Scholar] [CrossRef]
- Wang, D.D.H.; Guo, X.E.; Modrek, A.S.; Chen, C.F.; Chen, P.L.; Lee, W.H. Helicase SUV3, polynucleotide phosphorylase, and mitochondrial polyadenylation polymerase form a transient complex to modulate mitochondrial mRNA polyadenylated tail lengths in response to energetic changes. J. Biol. Chem. 2014, 289, 16727–16735. [Google Scholar] [CrossRef]
- Mildenhall, K.B.; Wiese, N.; Chung, D.; Maples, V.F.; Mohanty, B.K.; Kushner, S.R. RNase E-based degradosome modulates polyadenylation of mRNAs after Rho-independent transcription terminators in Escherichia coli. Mol. Microbiol. 2016, 101, 645–655. [Google Scholar] [CrossRef]
- Mohanty, B.K.; Maples, V.F.; Kushner, S.R. The Sm-like protein Hfq regulates polyadenylation dependent mRNA decay in Escherichia coli. Mol. Microbiol. 2004, 54, 905–920. [Google Scholar] [CrossRef]
- Yagi, M.; Uchiumi, T.; Takazaki, S.; Okuno, B.; Nomura, M.; Yoshida, S.I.; Kanki, T.; Kang, D. p32/gC1qR is indispensable for fetal development and mitochondrial translation: Importance of its RNA-binding ability. Nucleic Acids Res. 2012, 40, 9717–9737. [Google Scholar] [CrossRef]
- Dominguez, C.; Fisette, J.F.; Chabot, B.; Allain, F.H.T. Structural basis of G-tract recognition and encaging by hnRNP F quasi-RRMs. Nat. Struct. Mol. Biol. 2010, 17, 853–861. [Google Scholar] [CrossRef]
- Mustafi, S.B.; Aznar, N.; Dwivedi, S.K.D.; Chakraborty, P.K.; Basak, R.; Mukherjee, P.; Ghosh, P.; Bhattacharya, R. Mitochondrial BMI1 maintains bioenergetic homeostasis in cells. FASEB J. 2016, 30, 4042–4055. [Google Scholar] [CrossRef]
- Sarkar, D.; Leszczyniecka, M.; Kang, D.C.; Lebedeva, I.V.; Valerie, K.; Dhar, S.; Pandita, T.K.; Fisher, P.B. Down-regulation of Myc as a Potential Target for Growth Arrest Induced by Human Polynucleotide Phosphorylase (hPNPaseold-35) in Human Melanoma Cells. J. Biol. Chem. 2003, 278, 24542–24551. [Google Scholar] [CrossRef]
- Sarkar, D.; Park, E.S.; Fisher, P.B. Defining the mechanism by which IFN-β dowregulates c-myc expression in human melanoma cells: Pivotal role for human polynucleotide phosphorylase (hPNPaseold-35). Cell Death Differ. 2006, 13, 1541–1553. [Google Scholar] [CrossRef]
- Das, S.K.; Bhutia, S.K.; Sokhi, U.K.; Dash, R.; Azab, B.; Sarkar, D.; Fisher, P.B. Human polynucleotide phosphorylase (hPNPase old-35): An evolutionary conserved gene with an expanding repertoire of RNA degradation functions. Oncogene 2011, 30, 1733–1743. [Google Scholar] [CrossRef] [PubMed]
- Fornari, F.; Gramantieri, L.; Ferracin, M.; Veronese, A.; Sabbioni, S.; Calin, G.A.; Grazi, G.L.; Giovannini, C.; Croce, C.M.; Bolondi, L.; et al. MiR-221 controls CDKN1C/p57 and CDKN1B/p27 expression in human hepatocellular carcinoma. Oncogene 2008, 27, 5651–5661. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, D.; Park, E.S.; Barber, G.N.; Fisher, P.B. Activation of double-stranded RNA-dependent protein kinase, a new pathway by which human polynucleotide phosphorylase (hPNPaseold-35) induces apoptosis. Cancer Res. 2007, 67, 7333–7343. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ghosh, S.; Guimaraes, J.C.; Lanzafame, M.; Schmidt, A.; Syed, A.P.; Dimitriades, B.; Börsch, A.; Ghosh, S.; Mittal, N.; Montavon, T.; et al. Prevention of dsRNA-induced interferon signaling by AGO1x is linked to breast cancer cell proliferation. EMBO J. 2020, 39, e103922. [Google Scholar] [CrossRef]
- Cailleau, R.; Olivé, M.; Cruciger, Q.V.J. Long-term human breast carcinoma cell lines of metastatic origin: Preliminary characterization. In Vitro 1978, 14, 911–915. [Google Scholar] [CrossRef]
- Hayakawa, H.; Sekiguchi, M. Human Polynucleotide Phosphorylase Protein in Response to Oxidative Stress. Biochemistry 2006, 45, 6749–6755. [Google Scholar] [CrossRef]
- Hayakawa, H.; Kuwano, M.; Sekiguchi, M. Specific Binding of 8-Oxoguanine-Containing RNA to Polynucleotide Phosphorylase Protein. Biochemistry 2001, 40, 9977–9982. [Google Scholar] [CrossRef]
- Wu, J.; Jiang, Z.; Liu, M.; Gong, X.; Wu, S.; Burns, C.M.; Li, Z. Polynucleotide phosphorylase protects Escherichia coli against oxidative stress. Biochemistry 2009, 48, 2012–2020. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falchi, F.A.; Pizzoccheri, R.; Briani, F. Activity and Function in Human Cells of the Evolutionary Conserved Exonuclease Polynucleotide Phosphorylase. Int. J. Mol. Sci. 2022, 23, 1652. https://doi.org/10.3390/ijms23031652
Falchi FA, Pizzoccheri R, Briani F. Activity and Function in Human Cells of the Evolutionary Conserved Exonuclease Polynucleotide Phosphorylase. International Journal of Molecular Sciences. 2022; 23(3):1652. https://doi.org/10.3390/ijms23031652
Chicago/Turabian StyleFalchi, Federica A., Roberto Pizzoccheri, and Federica Briani. 2022. "Activity and Function in Human Cells of the Evolutionary Conserved Exonuclease Polynucleotide Phosphorylase" International Journal of Molecular Sciences 23, no. 3: 1652. https://doi.org/10.3390/ijms23031652
APA StyleFalchi, F. A., Pizzoccheri, R., & Briani, F. (2022). Activity and Function in Human Cells of the Evolutionary Conserved Exonuclease Polynucleotide Phosphorylase. International Journal of Molecular Sciences, 23(3), 1652. https://doi.org/10.3390/ijms23031652