1. Introduction
Alzheimer’s disease is a devastating neurodegenerative disease affecting a large and growing number of individuals world-wide. Understanding the underlying molecular mechanisms is crucial to developing a targeted approach in the search for a cure. Fluorescence based super resolution microscopy methods such as direct stochastic optical reconstruction microscopy (dSTORM) or stimulated emission depletion (STED) can provide valuable structural information [
1,
2,
3,
4]. The efficient incorporation of fluorophores as molecular probes in the proteins of interest can open up avenues of additional optical methods such as for example the investigation of molecular size distributions such as fluorescence correlation spectroscopy (FCS) [
5,
6], cell biology using flow cytometry and fluorescence microscopy [
7], Western blotting and protein interaction screening using protein arrays combined with fluorescence scanners and microarray readers [
8], as well as determination of affinities and exchange rates using Förster resonance energy transfer (FRET) [
9,
10].
Of particular interest for neuronal function and dysfunction, fluorescence microscopy studies with fluorophore-labelled recombinant protein have provided key insights into the self assembly and membrane interaction of
-synuclein, the amyloid protein involved in Parkinson’s disease [
11,
12,
13]. Super-resolution fluorescence microscopy studies of A
42, the amyloid peptide involved in the pathology of Alzheimer’s disease, have provided valuable information on the in vitro fibril elongation [
14] as well as cellular uptake [
15] and aggregation inside cells [
16]. These studies have mainly employed synthetic fluorescent A
42 peptide due to challenges in successfully covalently attaching fluorophores to the peptide. Previous attempts to covalently label recombinant A
42 with Alexa fluorophores at the N-terminus have yielded low labeling efficiency and significantly retarded the aggregation process, which was remedied by using a low ratio of labelled to unlabelled peptide [
17]. The same approach permitted the identification of glycogen synthase 3
and
(also named tau kinase 1) as interaction partners of on-pathway A
42 oligomers [
18].
In the present study we optimize the position of covalent labelling of recombinant A
42 with fluorophores incorporated at different surface-exposed positions of the peptide in A
42 fibrils, as guided by recent high-resolution structures [
19,
20,
21]. To enable covalent labelling with thiol reactive fluorophores, we thus create five cysteine mutants of A
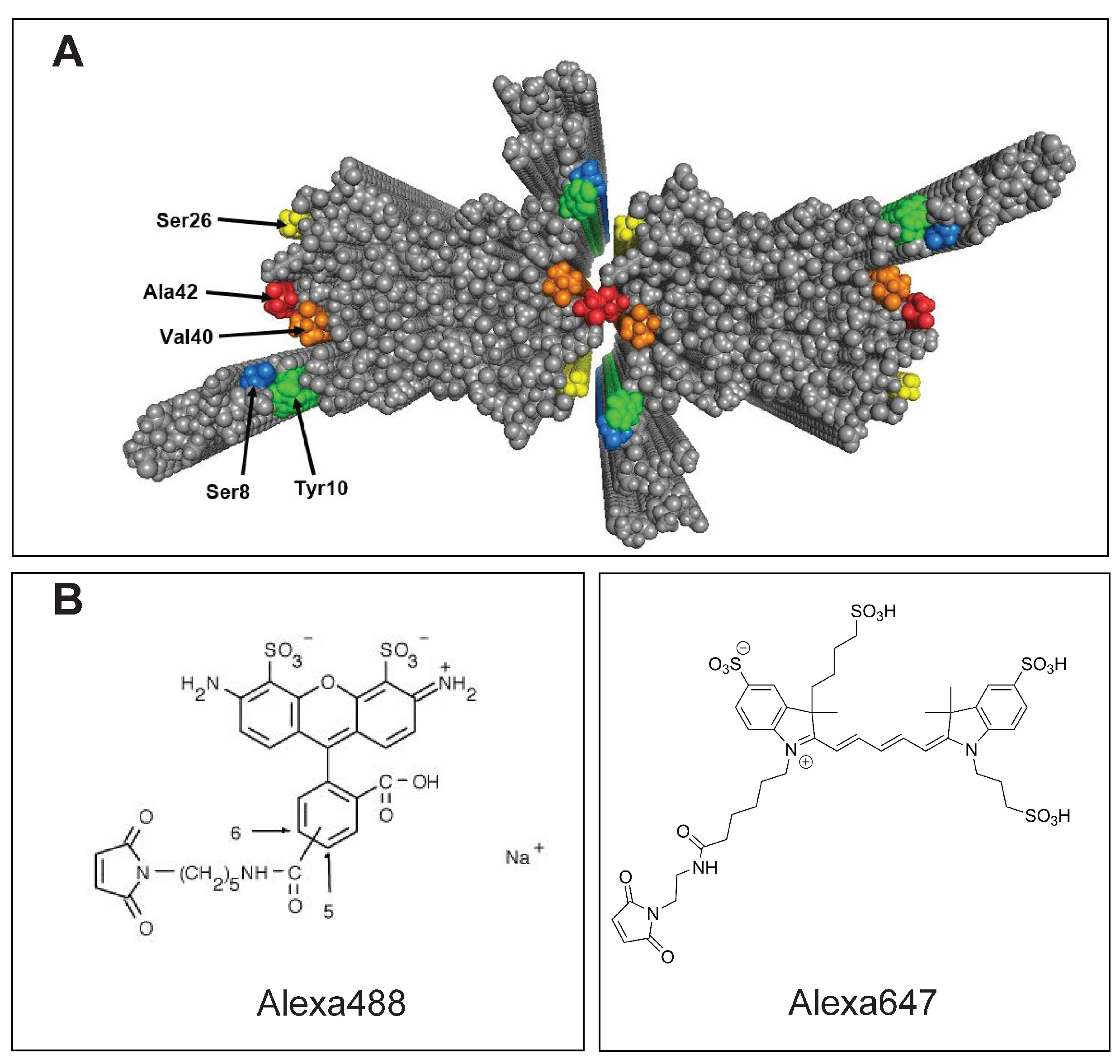
42: S8C and Y10C for labelling of the unstructured N-terminal region, S26C, V40C and A42C on the surface of the fibril core (
Figure 1). As a proof-of-concept, each mutant is labelled with Alexa488 and Alexa647, and the labelling efficiency is evaluated using size exclusion chromatography (SEC) and SDS PAGE. Aggregation assays of the labelled peptides, alone and in mixtures with unlabelled wild-type peptide, using thioflavin T (ThT) as well as Alexa probe fluorescence, were performed to study whether the perturbation of the fibril formation rate depends on the position of the label. Additionally, cryogenic transmission electron microscopy (cryoTEM) was used to determine the morphology of the labelled peptide fibrils compared to unlabelled WT A
42 fibrils.
2. Materials and Methods
2.1. Expression and Purification of Peptides
The plasmid carrying synthetic genes with
Escehrichia coli (E. coli) optimized codons for A
42 wild-type (PetSac, cloned by us [
22]) as well as S8C, Y10C, S26C, V40C, and A42C (Pet3a, purchased from Genscript) were transformed into Ca
competent cells of
E. coli strain BL21 DE3 pLysS star and the protein was expressed in auto-induction medium [
23]. The peptides were purified using ion exchange chromatography (IEX) as described with the minor change that lower salt concentration (50 mM NaCl, (Duchefa Biochemie, CAS no. 7647-14-5)) was used to elute the peptides, and size exclusion chromatography (SEC) on a 26 × 600 mm Superdex 75 column was used instead of spin filters for base on molecular size. The ion exchange and SEC buffers contained 1 mM dithiothreitol (DTT, PanReac Applichem, CAS no. 3483-12-3) to avoid dimerization of cysteine mutants. The final SEC was performed in buffer without DTT in order to isolate monomer and remove DTT from the sample prior to adding the label. The purified monomeric peptides were lyophilized as aliquots until further use.
2.2. Labelling of Purified Peptides with Alexa Fluor
Lyophilized fractions were dissolved in 50 L milliQ water yielding a peptide concentration of ∼14 M. Alexa fluor 488 or 647 at a concentration of 3–4 mM in 20 L dimethyl sulfoxide (DMSO) (Sigma, CAS no. 67-68-5) was added to the dissolved peptide in order to have access dye in the labelling mixture, which was kept overnight at 4 C. The mixture was then added to 1 mL of 6 M GuHCl (Sigma, CAS no. 50-01-1), 20 mM sodium phosphate, 0.2 mM ethylenediaminetetraacetic acid (EDTA; J. T Baker, CAS no. 6381-92-6), pH 8.5, and subjected to SEC on a Superdex 75 10/300 column in 20 mM sodium phosphate (Merck, CAS no. 6381-92-6) buffer pH 8.0, with 0.2 mM EDTA. The absorbance at 280 nm as well as 488 nm or 647 nm was monitored using a Quadtech detector to follow the elution of the labelled peptide, excess dye as well as any unlabelled peptide, if present. The aliquots collected from the SEC were analysed by SDS PAGE, and stored at −80 C until further use. The same peptide concentration and conditions were used in the labelling of all mutants. Note: In case of the Y10C mutant, the absorbance at 214 nm was monitored instead of the absorbance at 280 nm. Since the A42 sequence has only one Tyr residue contributing to A, the Y10C mutant gives no absorbance at 280 nm wavelength. Thus, we monitor the absorbance at 214 nm, which is contributed to by all peptide bonds.
Alexa488 and Alexa647 were chosen for labelling to test labelling with chemically different probes with widely different excitation wavelengths. The peptides S8C, Y10C, and V40C were labelled with both Alexa488 and Alexa647 to enable future experiments requiring peptides labelled with two different fluorophores.
The concentration of the labelled peptides was determined using the correction factor for Alexa488 and Alexa647 in the following formula:
where,
A is the absorbance at 488 or 647 nm,
c.f. is a correction factor, which is 0.11 for Alexa488, and 0.03 for Alexa647, and
is the extinction coefficient for A
42 which is 1400 L mol
cm
.
2.3. Preparation of Samples for Kinetic Experiments
The lyophilized aliquots of the purified WT A42 were dissolved in 1 mL of 6 M GuHCl, 20 mM sodium phosphate, 0.2 mM EDTA, pH 8.5, and subjected to SEC on a Superdex 75 10/300 column in 20 mM sodium phosphate buffer pH 8.0, with 0.2 mM EDTA. The middle part of monomer peak was collected in a low-binding tube (Axygen, MCT-150-L-C) on ice, and was typically found to have a concentration in the range 20–80 M (determined by absorbance of the collected fraction using = 1400 L molcm). Frozen aliquots of the Alexa-labelled peptides were kept on ice for thawing.
2.4. Aggregation Kinetics by Thioflavin T Fluorescence
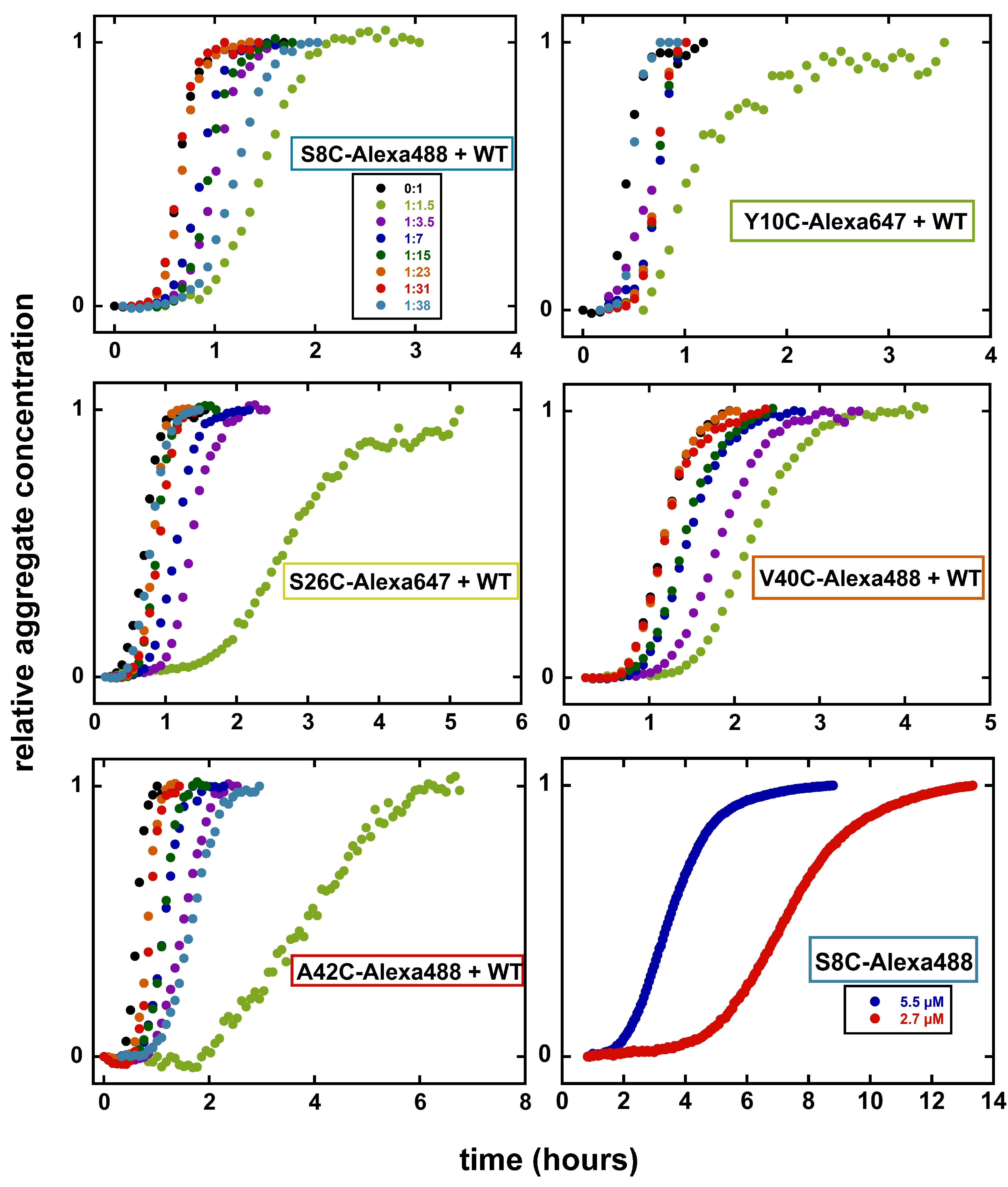
Aggregation kinetics experiments were performed for samples with different ratios of Alexa-labelled peptide to WT A42, as follows, 1:1.5, 1:3.5, 1:7, 1:15, 1:23, 1:31 and 1:38 in 20 mM sodium phosphate and 0.2 mM EDTA, pH 8.0 buffer. The total monomer concentration was close to 5 M. The samples were pipetted as multiple replicates into a 96-well plate (Corning 3881), 100 L per well. The experiments were initiated by placing the 96-well plate at 37 C in a plate reader (Fluostar Omega). The ThT fluorescence was measured through the bottom of the plate every 120 s using an excitation filter at 440 nm and an emission filter at 480 nm.
2.5. Cryo-TEM
For all peptides, fibrils were prepared from samples with monomer close to 5 M total concentration, which were incubated at 37 C in PEGylated plates (Corning 3881) in a plate reader and collected after reaching the plateau in ThT fluorescence. Specimens for cryo-TEM were prepared in an automatic plunge freezer system (Leica EM GP). The climate chamber temperature was kept at 21 C, and relative humidity was ≥90% to minimise loss of solution during sample preparation. The specimens were prepared by placing 4 L solution on glow discharged lacey formvar carbon coated copper grids (Ted Pella) and blotted with filter paper before being plunged into liquid ethane at −183 C. This leads to vitrified specimens, avoiding component segmentation and rearrangement, and the formation of water crystals, thereby preserving original microstructures. The vitrified specimens were stored under liquid nitrogen until measured. A Fischione Model 2550 cryo transfer tomography holder was used to transfer the specimen into the electron microscope, JEM 2200FS, equipped with an in-column energy filter (Omega filter), which allows zero-loss imaging. The acceleration voltage was 200 kV and zero-loss images were recorded digitally with a TVIPS F416 camera using SerialEM under low dose conditions with a 10 eV energy selecting slit in place.
2.6. dSTORM
dSTORM samples were prepared by depositing 200 L of 0.7 M fibrils on a poly-Lysine coated # 1.5 (0.17 mm) glass-bottom dish (WillCo., Well, Germany). The fibrils were allowed to settle for 20 min and washed with 200 L 20 mM sodium phosphate, 0.2 mM EDTA, pH 8.0 before loading the glass-bottom dish on the microscope. Samples were kept on ice and in the dark at all times.
dSTORM imaging was performed on the ELYRA P1 imaging system (Zeiss, Germany) which included an inverted microscope with 100X oil immerse objective lens (1.46 NA). The samples were mounted in the ZEISS level adjustable insert holder and placed on the PIEZO stage with Auto-focus adjusted. The fluorescence dyes were excited by the selected three laser lines, 488 nm, 543 nm and 633 nm respectively. Accordingly, the filter sets #4 for collection of the emission light were chosen dependent on the fluorescent dyes. For Alexa 488, the 488 nm laser line was used for excitation and the emission light filter was BP 495–550; for Alexa 546, a 543 nm laser line was used for excitation and the emission light filter was BP 570–620; for Alexa 647, a 633 nm laser line was used for excitation and the emission light filter was LP 655. The images were acquired onto a 256 × 256 pixel frame of an electron multiplying charge coupled device (EMCCD) camera (iXon DU897, Andor).
To generate the dSTORM images, the PALM processing function in the ZEN software was applied. First, the overlapping signals were discarded by using a multi-emitter model for the whole image sequences. Second, to distinguish the real signal peak, the mask size was set to 7 pixels and the ratio of signal/noise was set to 7. After the filtration, the lateral and axial drift during acquisition was corrected after reconstruction of dSTORM images by using the Drift function that was tested by a fluorescent beads as fiducial marker prior to the analysis. After drift correction, the images were proceeded by grouping function. Finally, the present dSTORM images were corrected in according to the distributions of followed parameters: photon number, precision size and first frame. To remove the unspecific background, the molecules were filtered out if the number of molecules was less than 10 in an area of 100 nm perimeter.
4. Discussion
The covalent labelling of fluorophores to A
42 is challenging because both aggregation kinetics and fibril structure are sensitive to small changes in peptide composition and fluorophores typically have the size comparable to 7–10 amino acid residues and may be both hydrophobic and charged. Previous studies have used synthetic A
42 peptide for fluorescence based studies; however, it has been shown that sequence purity is of great importance in the mechanistic analysis of protein aggregation. Synthetic peptides may contain mismatches and racemates, and indeed synthetic A
42 is shown to aggregate significantly slower than recombinant peptide, and at the same time is unable to mimic the neurotoxic behavior of the recombinant peptide [
24]. Strategic positioning of the fluorophore based on the fibril structure of A
42 may be one route to minimize the influence on the self-assembly of the peptide. Previous attempts to label A
42 with Alexa fluor at a cysteine residue added at the N-terminus resulted low labelling efficiency and caused great perturbation of aggregation kinetics. An labelled:unlabelled peptide ratio of 1:170 was required to achieve an aggregation profile similar to WT A
42 [
17]. This might be due to the general low reactivity close to the Asp1 residue of A
42 peptide, as previously noted when attempting to cleave the Met0-Asp1 peptide bond [
22].
In this study, we set out to find if there are more optimal fluorophore labelling positions in A
42 by covalently attaching Alexa fluorophores at different positions of the peptide based on their location in the fibril structure. Through site directed mutagenesis we introduce cysteine residues at five different positions of A
42 for fluorophore labelling using maleimide chemistry. We thus created the mutants S8C and Y10C at the N-terminal region of A
42, and mutants S26C, V40C, and A42C on the surface of the fibril core (
Figure 1). We tested the labelling efficiency for all of these mutants, and performed time dependent aggregation kinetics for all labelled mutants in presence of WT A
42 in different ratios to test the extent of perturbation due to the presence of the fluorophore on the peptide. We also studied the fibril morphology and fluorophore inclusion for the end-stage fibrils of these peptides using cryoTEM and dSTORM. Whether oligomers of the labelled peptides can successfully mimic the neurotoxicity of WT A
42 remains to be addressed.
The eighth residue of A
42 is a serine, a polar amino acid on the flexible N-terminal region, which is solvent exposed in all four monomers of a fibril plane (
Figure 1). We introduce cysteine, another polar amino acid, in its place through site directed mutagenesis in order to covalently attach Alex fluorophores using maleimide chemistry. The S8C mutant yields high labelling efficiency of Alexa488 as shown in
Figure 1. Aggregation kinetics for S8C in presence of different ratios of WT A
42 show an aggregation profile very similar to WT. Even with a high ratio of labelled:unlabelled peptide (1:1.5), the lag phase is not extended significantly compared to WT alone. The fibril morphology for S8C-Alexa488+WT (1:3.5) appears to be very similar to that of typical WT A
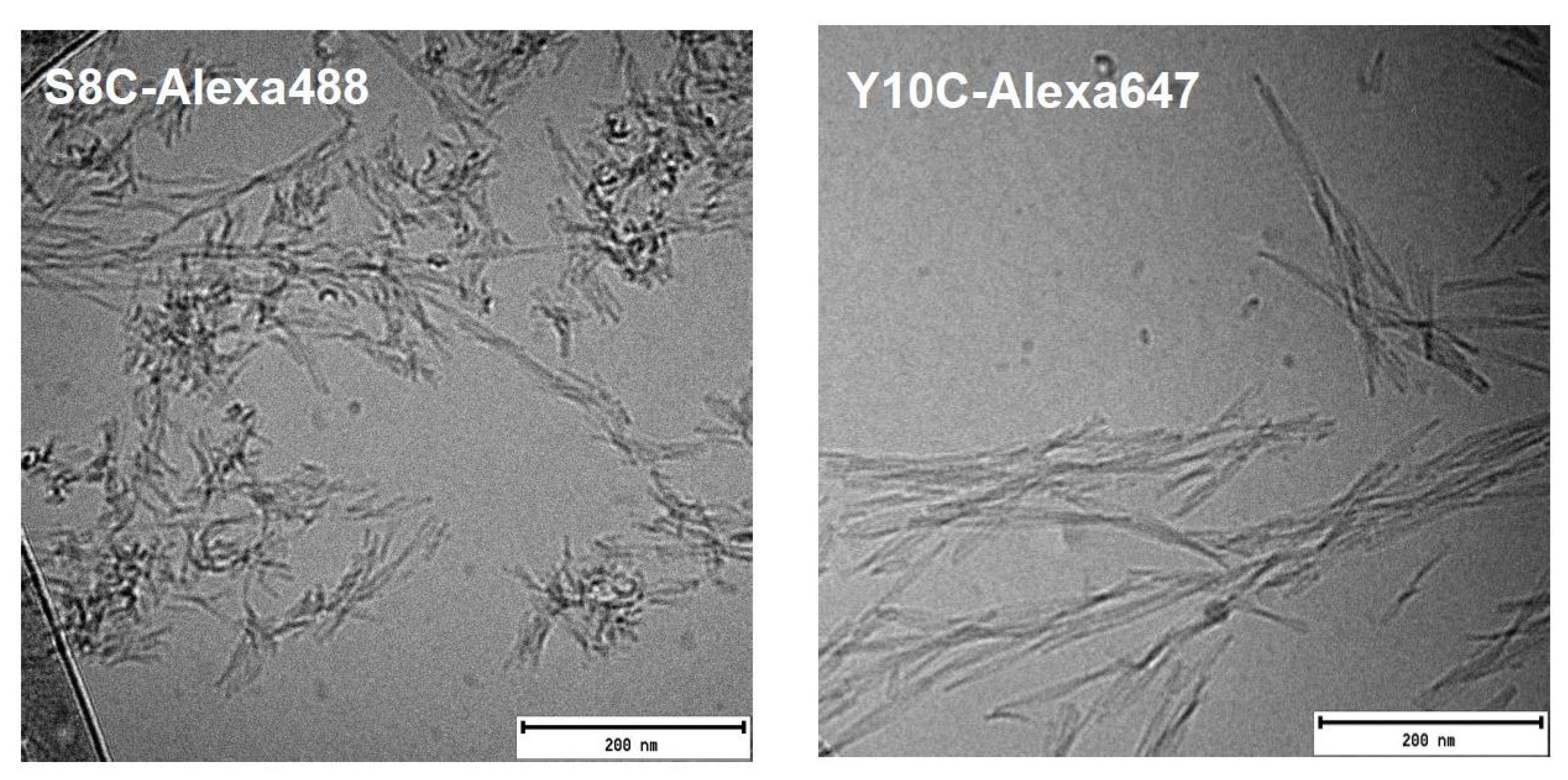
42. The fibrils are straight, with two filaments twisting around each other at regular intervals creating “nodes”. Labelling Alexa-fluor at this position thus does not cause any significant perturbations of the aggregation kinetics nor fibril morphology when mixed with WT.
S8C-Alexa488 also aggregates in the absence of unlabelled WT, which was studied by following the fluorescence quenching of Alexa488 upon aggregate formation. In this case, the lag phase is considerably extended, when the aggregation of 2.7 and 5.5
M S8C-Alexa488 is compared to 5
M WT (
Figure 3). In this case, the fibrils are much shorter than those formed in presence of an equimolar amount of WT A
42.
Y10C is the second mutant we created on the N-terminal region of A42. We achieve high labelling efficiency in this case as well, and the aggregation kinetics don’t show any significant perturbation even at high ratio of labelled:unlabelled peptide. However, when co-aggregating with WT A42, we observe that the fibril morphology is perturbed for Y10C-Alexa647. Typical WT A42 fibrils appear straight and rigid. However, Y10C-Alexa647+WT (1:3.5) fibrils appear flexible and appear as short filaments uncharacteristic of mature end-stage fibrils. Y10C-Alexa647 also aggregates in the absence of unlabelled WT A42. The fibrils formed from the labelled peptide alone display longer node-to-node distances but are so short that the periodical twists around the fibril length are few. Labelling Alexa-flour at this position thus seems to cause perturbation in fibril structure and morphology.
Ser26 is positioned on the surface of the fibril core of the WT A
42 fibril. In a fibril plane of four monomers, two Ser26 are located where the two individual filaments meet and two Ser26 appear fully exposed to water [
21] (
Figure 1). Replacing a hydrophilic serine residue with a large moiety such as an Alexa fluorophor could thus potentially impose steric clashes in between the filaments leading to the formation of an altered fibril morphology. At high molar ratio, 1:1.5 S26C-Alexa647:WT, this variant displays a significantly extended lag phase compared to WT alone. The fibril morphology of S26C-Alexa647+WT (1:3.5) appears to be different than that of the typical WT A
42 fibrils. The fibrils are longer, indicating higher elongation rate and lowered rate of secondary nucleation. The node-to-node distance is longer and the fibril diameter is larger, indicating a change in the assembly of the two filaments in the fibril. This position of labelling thus serves as an example of significant perturbation caused by the Alexa fluorophores as regards both aggregation kinetics and fibril morphology.
It has been shown that site directed mutagenesis of Val40 and Ala42 to polar serine residues does not cause any major perturbations of the aggregation mechanism and fibril morphology [
25]. Hence we created mutants V40C and A42C to attach Alexa fluorophores at the respective positions. Val40 and Ala42 are also located where the two individual filaments meet (
Figure 1), however unlike S26C, fluorophore-labelled V40C and A42C do not show significant perturbations of fibril morphology. As for S26C, the aggregation kinetics at high molar ratio, 1:1.5 variant:WT, is significantly perturbed, but less so or not at all at lower ratio. A possible explanation for the maintained fibril morphology at 1:3.5 molar ratio, is the possible segregation in the plane of four monomers in a fibril; one Alexa-labelled monomer may be in the positions where the labelled residues are facing the solvent, while the unlabelled WT monomers may be incorporated at the positions where Val40 and Ala42 are buried in the interface where the two filaments meet.
It is interesting to note that the labelled S26C, V40C or A42C do not seem to aggregate in the absence of unlabelled WT A42. Most likely, the labelled fibrils are unable to form fibrils of an alternative fold that contains both a stable hydrophobic core and exposed fluorophores at all four positions in a fibril plane.
Finally, we note that the structures of the dye molecules Alexa488 or Alexa647 are very different, but this does not seem to affect fibril formation, which can be seen from V40C-Alexa488 and V40C-Alexa647 showing similar aggregation kinetics and fibril morphology (
Supplementary Materials Figures S2 and S3).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}