1. Introduction
Breast cancer (BC) is a heterogenous group of diseases with a high incidence rate worldwide, with 2.261.419 estimated new cases and 684.996 deaths in 2020, mostly women [
1]. Intrinsic subtypes such as hormone-receptor-dependent (progesterone receptor (PR) and oestrogen receptor (ER)) expression), human epidermal growth factor receptor 2 (HER2) positive, and triple negative breast cancer (TNBC), which is PR, ER and HER2 negative, are clinically relevant because their therapeutic stratification depends on molecular diagnosis [
2]. Surgery, in the setting of early BC, chemotherapy, endocrine and anti-HER2 therapy, and sometimes a combination of these, are the current therapeutic approaches [
3]. Hormone-receptor-positive (HR+) is the most favourable diagnosis (stands out for its favourable prognosis), however, there is a prolonged significant risk of recurrence [
4]. Endocrine therapy is established for these cases and the two most important drug categories in postmenopausal patients are tamoxifen and aromatase inhibitors [
5]. BC expressing HER2 comprises about 15% of all cases [
6], and treatment requires anti-HER2 monoclonal antibodies, namely, trastuzumab, sometimes associated with pertuzumab, as adjuvant or neoadjuvant therapy. More recently, lapatinib, a dual EGFR/HER2 reversible tyrosine kinase inhibitor, has been introduced as a potential option for adjuvant therapy. Chemotherapy plus trastuzumab combination and dual-targeted therapy with trastuzumab plus lapatinib in patients with locally advanced HER2-positive breast cancer shows increased complete pathologic response [
7]. Nevertheless, TNBC affects 15–20% of patients, and it is associated with a worse prognosis because targeted treatment is not available, and chemotherapy is the only option [
8]. Poly-ADP-ribose polymerase (PARP) inhibitors are best known as a targeted treatment for BRCA1 and BRCA2 genes, and they are in clinical trials combined with chemotherapy [
3,
9]. All these therapies are accompanied by unwanted side effects [
10]. Thus, the research on new therapies is crucial, and recently, cold plasma has emerged as a novel approach for anticancer therapy with a selective potential regarding phenotypically normal cells [
11,
12].
Plasma, commonly known as the fourth state of matter, has enough energy to ionize a significant amount of charged particles, being able to generate reactive oxygen and nitrogen species, and its future applications in field of biomedicine are promising [
13]. There are two types of plasma: atmospheric pressure and low pressure. One of the main differences between them is the mean free paths between electrons and heavy particles that are extremely short in atmospheric pressure plasma, promoting collision among particles [
14]. Within these, there are two distinct models: thermal and non-thermal plasmas. The core gas temperatures in thermal plasmas are above 10.000 K, and in non-thermal plasma, the sensible temperature remains at room temperature [
14]. Moreover, plasma has a wide range of applications such as the use as surface disinfectants in healthcare facilities, even though more tests are required in order to prove its safety and efficacy [
15].
Recent studies suggest an interesting potential of cold atmospheric plasma (CAP) in cancer therapy, as demonstrated by the selective eradication of cancer cells in vitro [
16,
17,
18,
19,
20]. Previously, we studied the effect of cold plasma in different human cancer cell lines including BC, melanoma, colon carcinoma and prostate cancer. The results showed a significant decrease in cell viability in most cell lines. Particularly, the viability of BC cells decreased following only 90 s of CAP exposure [
19]. Furthermore, a robust study involving direct and indirect CAP treatment of retinoblastoma cells and phenotypically normal counterparts suggests that this therapy has the potential to selectively ablate tumour cells [
20]. Thus, the main goal of this study was to assess the biological outcome and molecular mechanisms of action of CAP in BC cell lines, namely, hormonal-receptor-positive breast cancer cell line MCF7 and triple-negative breast cancer cell line HCC1806.
3. Discussion
Over the past decade, CAP studies have emerged as a new therapeutic approach in various types of cancer, highlighting its anti-proliferative effect [
21,
22,
23], and several authors also describe the selective potential of therapy for tumour cells, preserving adjacent phenotypically normal cells [
24,
25].
Our previous results have studied the effect of CAP exposure in metabolic activity and viability of MCF7 and HCC1806 cells [
19]. By comparing the metabolic activity of several cell lines after plasma exposure, we observed that longer CAP exposures were required to reduce metabolic activity in breast cancer cells than most of the cells tested [
19]. However, the effect of plasma treatment in the cells’ viability differed between triple-negative (MCF7) and hormonal-receptor-positive (HCC1806) breast cancer cell lines. While only longer exposures decreased the viability of HCC1806 cells, parallel to the previous results, a short plasma exposure caused a marked and disproportional reduction in the MCF7 cell number. Thus, MCF7 cells were assumed to be stressed. Since plasma reduced the proliferation rate but failed to produce an equivalent reduction in metabolic activity under these conditions, stressed MCF7 cells displayed an increased metabolic rate per cell. The selection of a highly metabolic subpopulation of MCF7 cells or adaptation of remaining cells with increased mitochondrial biogenesis was hypothesized. Whichever the mechanism explaining the increase in metabolic index, plasma exposure to longer duration abolished this response. This work further explores the possible mechanisms of action of new plasma-based therapies in these breast cancer cell lines [
19].
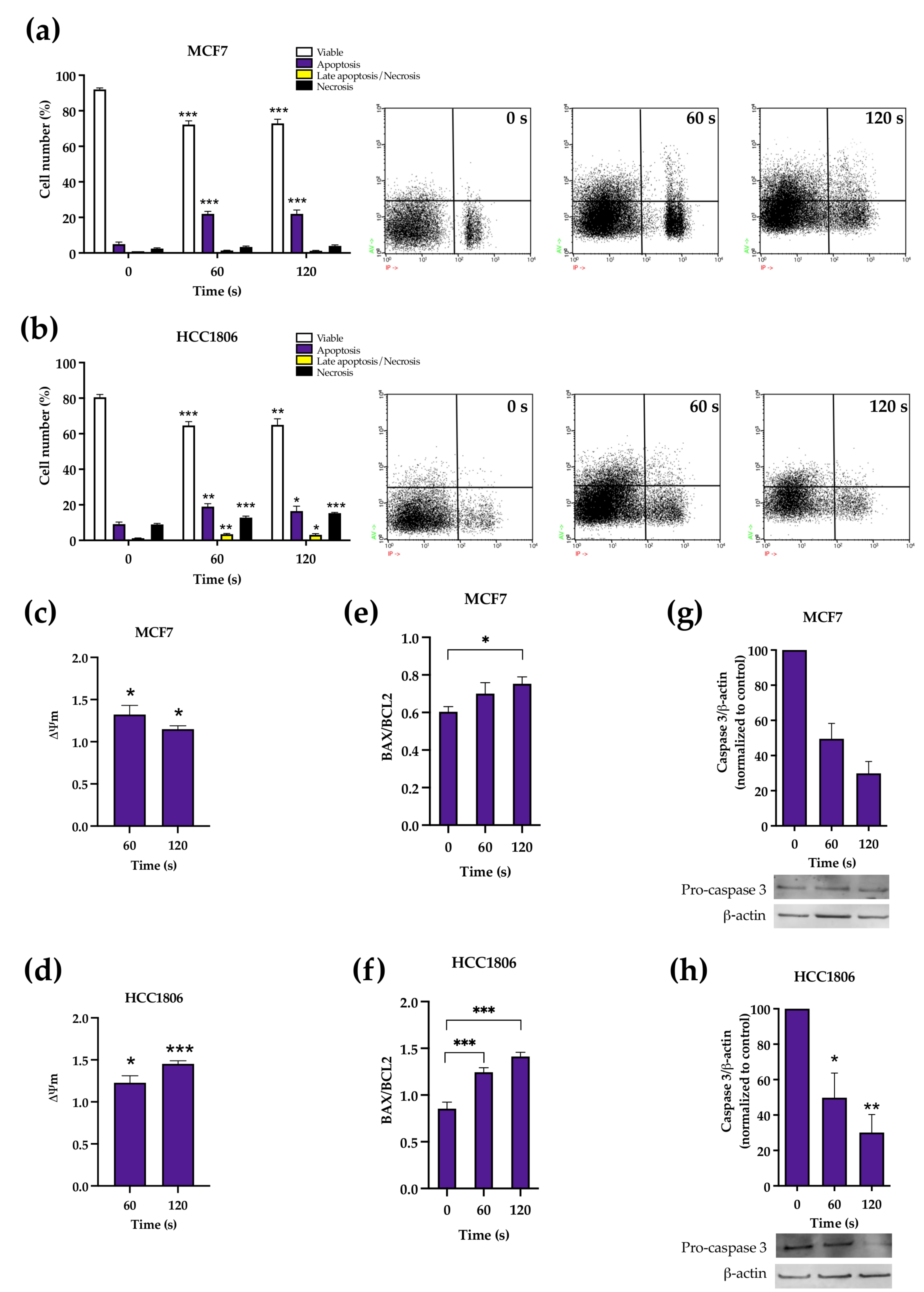
We have selected referred time exposures because we previously documented this differential response at 60 and 120 s. The presented results confirm the previously described reduction in cell viability after CAP exposure [
19]. Firstly, we analysed the type of cell death in each cell line. For both cell lines and therapy durations, the proportion of viable cells in both cell lines were reduced. However, while apoptosis was the main type of death in MCF7 cells, a combination of apoptosis and necrosis was seen in HCC1806 cells. The evaluation of mitochondrial potential showed that at least some portion of treated cells lost their normal mitochondrial inner membrane electrical potential, a feature of apoptosis, consistent with the increased proportion of this type of death in both cell lines. An abrupt modification of MMP, as seen particularly in HCC1806, can drop adenosine triphosphate (ATP) concentration to levels insufficient for cell survival, not allowing the apoptotic process to end [
26] and leading to necrosis. Apoptosis in response to plasma was also supported by the documentation of the overexpression of proapoptotic protein BAX when compared to antiapoptotic Bcl-2 protein and a reduction in procaspase-3 expression, a surrogate marker of its cleavage and conversion into the executioner caspase-3, the main effector of apoptosis [
27]. Interestingly, the baseline BAX/Bcl2 ratio was intrinsically lower in MCF7 cells and did not significantly change when these cells were exposed to the shortest exposure.
Since the main hypothesized mechanism of plasma therapy involves reactive oxidative and nitroxidative species (RONS), we determined the intracellular concentrations of these species shortly (2 h) and 24 h after exposure. Our previous works [
19,
20] suggests that H
2O
2 and NO
2 are generated inside the plasma plume or in the interface between plasma and liquid and then solubilized in culture media. Then, we assume that RONS concentrations inside cells can be increased by (1) scavenging reactive species from media briefly after plasma irradiation or (2) mitochondria release by a mitochondrial-induced mitochondrial release mechanism later [
28]. Conversely, intracellular RONS concentrations can be decreased by (1) consumption through conversion in other species shortly after treatment or (2) resetting the equilibrium following alterations between production and removal rate of RONS.
We detected distinct patterns in nitroxidative states between cell lines. Shortly after irradiation, HCC1806′s intracellular NO concentrations increased and O2− concentrations decreased, while the inverse pattern occurred 24 h after treatment coupled with an increase in GSH levels. Perhaps the only noticeable difference between HCC1806 cells irradiated during 60 and 120 s was peroxide concentration, which was only slightly decreased in cells exposed longer to plasma. Differently, shortly after irradiation, MCF7 intracellular peroxide concentration and glutathione levels were equally decreased in cells exposed to 60 and 120 s. However, 24 h later, MCF7 cells behaved in different ways, with increased peroxide concentration seen in cells exposed to 60 s and decreased NO and O2− concentrations in those exposed to the highest treatment duration.
Several RONS have been identified in cell media exposed to plasma, but most are short-lived. NO
2− and H
2O
2 are long-lived species and are thought to be the major effectors of plasma effects [
29]. We previously determined peroxide concentrations in irradiated cell media of about 1–4 mM with a single exposure of 120 s [
20]. However, intracellular H
2O
2 concentrations were not increased, contrary to what we have seen in previous works with retinoblastoma cells [
20]. These paradoxical results were also reported by other authors, reinforcing the idea that some cancer cells are more resistant to oxidative species, thus suggesting a highly effective antioxidative system [
30]. We therefore measured GSH levels and did find a decrease in MCF7 cells paralleling a decrease in intracellular peroxides, suggesting a mutual consumption reaction. HCC1806 cells increased GSH levels 24 h after plasma exposition, compatible with a reactive production of these proteins capable of detoxifying peroxides. However, these variations in GSH levels, albeit statistically significant, were marginal and directed our attention to other antioxidative defences that could better explain the antioxidative capacity of these breast cancer cell lines. Based on previous reports [
30], we tested the role of mitochondrial cytochrome c oxidase. Biochemical inhibition of cytochrome c oxidase activity was performed with NaN
3 in a non-cytotoxic concentration. Incubation with NaN
3 and while irradiating with plasma was able to achieve an accentuated reduction in metabolic activity following a 60 s exposure. This was particularly relevant since plasma per se did not achieve a significant reduction in metabolic activity of breast cancer cells [
19]. Thus, plasma resistance was reduced to levels comparable to cell lines regarded as susceptible. While NaN
3 may also quench singlet oxygen derived from photodynamic reactions between endogenous photosensitizers and UV light photons emitted by plasma [
20,
31], this mechanism would not increase the enhanced cell killing capacity of plasma therapy as seen in our data since it would abrogate a generation of oxidative damage. An interesting observation was an increase in peroxides 24 h after plasma exposure in MCF7 cells and following restauration of GSH levels, which may represent a new balance in the redox state of this cells, which can be advantageous in highly metabolically active cells, since oxidized cytochrome c oxidase can bind hydrogen peroxide and use it as an electron acceptor in mitochondrial electronic chain [
32,
33].
While there is NO in the plasma plume, it is not soluble in medium, and therefore it is not scavenged by cells. NO is thought to be produced inside cells from the conversion of NO
2 produced in the plasma plume and solubilized in medium. Thus, our results suggest that HCC1806 cells scavenge and metabolize NO
2, pointing out that these cells, but not MCF7 cells, have NO
2− reductase activity, which can be used to enhance plasma toxicity. Superoxide anions seem to inversely correlate with NO, which suggests that it might be consumed in a reaction with NO to form peroxynitrite (ONOO
−). Note that NO is the only molecule produced by biological systems in high enough concentrations to out-compete superoxide dismutase for superoxide [
34], possibly explaining why SOD levels do not change in HCC1806 cells.
These results combined suggest that both cell lines are relatively resistant to high concentrations of peroxides produced inside plasma due to the peroxidase activity of cytochrome c oxidase and can be made susceptible by addition of NaN3. Furthermore, optimal midrange peroxide concentration can help to boost the metabolic index of viable MCF7 as an electron acceptor in the mitochondrial electron transport chain. At last, the nitrate reductase activity of HCC1806 cells may partially explain the effects of CAP in this cell line.
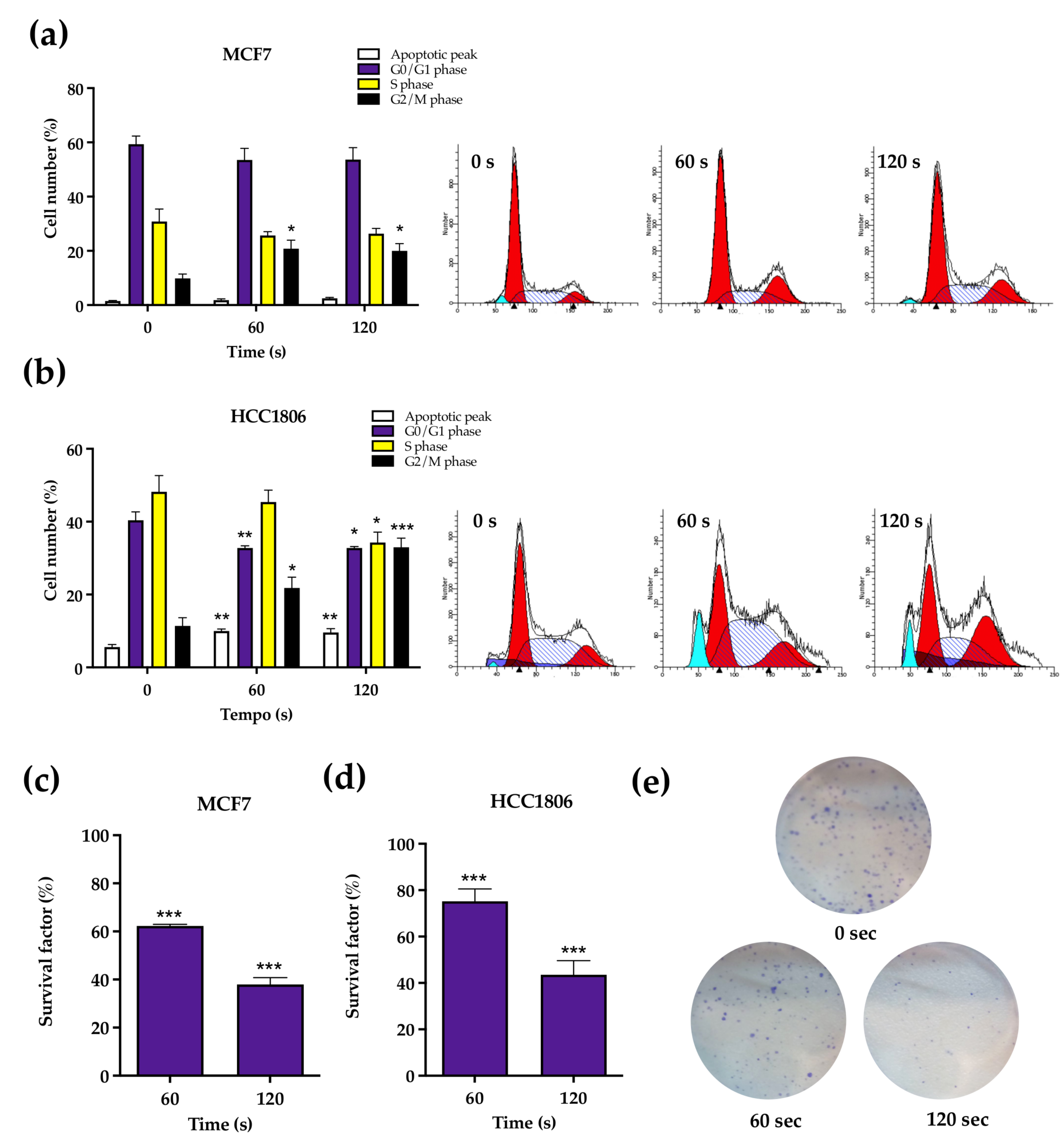
We next explored the cell cycle distribution of both cell lines, which revealed subtle differences between the two. Both cell lines display an increase in G2/M, which can be explained by an arrest in this phase following the activation of cell cycle checkpoints after the detection of damages in DNA replication possibly induced by RONS.
At last, clonogenic assay results, the gold standard for long time survival, expanded the observed cytotoxic results of atmospheric plasma to the long term. These results include MCF7 cells exposed to CAP for 60 s, thus suggesting that their enhanced metabolic index does not translate into enhanced survival.
4. Materials and Methods
4.1. Cell Culture
In this study, we used two adherent human cancer cell lines representative of two distinct subtypes of BC, namely, the hormonal-receptor-positive breast cancer cell line MCF7) and triple-negative breast cancer cell line HCC1806. These cell lines were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and were cultured under standard cell culture conditions (37 °C, 5% CO2). MCF7 cells were cultured with Dulbecco’s Modified Eagle Medium (Sigma Aldrich, St. Louis, MO, USA) medium, and HCC1806 cells were maintained in Roswell Park Memorial Institute (Sigma Aldrich, St. Louis, MO, USA) medium, according to suppliers’ recommendations. Media were supplemented with 5% foetal bovine serum (Sigma Aldrich, St. Louis, MO, USA) and 1% of penicillin-streptomycin solution (10,000 U/mL penicillin, 10 mg/mL streptomycin, and 25 μg/mL amphotericin B; Sigma Aldrich, St. Louis, MO, USA).
4.2. CAP Treatment
An electronic device capable generating CAP into cell culture plates was developed at the Institute of Biophysics, Faculty of Medicine, University of Coimbra, as described in detail elsewhere [
19]. Briefly, this device generates high voltage (4 kV) pulses with a frequency of 1 KHz through a sterilized needle with 0.9 mm of radius and 40mm of length (Microlance 3, Becton Dickinson, Franklin Lakes, NJ, USA). When charged, the needle works as an open-air single electrode CAP jet. An electrically grounded needle was submerged in the culture media. In this design, the cultures act both as target and grounded electrode, enabling plasma generation. The high voltage needle was placed 2mm above the surface of the cell culture’s medium. Cells were platted in 24-well plates (Sarstedt, Nümbrecht, Germany) at a density of 500,000 cells/mL in a volume of 500 μL per well and submitted to CAP treatment for short periods of time: 60 and 120 s. Further assays were performed 2 or 24 h after the exposure.
4.3. Cell Viability
Twenty-four hours after CAP treatment, samples with 106 cells were centrifuged for 5 min at 1300× g. The pellet was suspended in 100 µL of binding buffer, constituted by 0.01M Hepes (Sigma Aldrich, St. Louis, MO, USA), 0.14M NaCl (Sigma Aldrich, St. Louis, MO, USA) and 0.2 mM CaCl2 (Sigma Aldrich, St. Louis, MO, USA). Then, cells were labelled with annexin-V-fluorescein isothiocyanate (AnV-FITC) and propidium iodide (IP) as recommended by the kit supplier (Immunotech, Marseille, France). Samples were analysed in a four-color flow cytometer FACSCalibur (Becton Dickinson, New Jersey, NY, USA).
4.4. Mitochondrial Membrane Potential
Twenty-four hours after CAP exposure, 106 cells were centrifuged for 5 min at 1300× g, and the samples were incubated with 40 µL of fluorescent probe JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolocarbocyanine iodide; Invitrogen®, Waltham, MA, USA) for 15 min at 37 °C in the dark. After washing, cells were evaluated in the flow cytometer. For monomers, the fluorescence was determined at 485/20 nm excitation and 528/20 nm emission wavelengths, being the aggregates fluorescence determined at 530/25.5 nm excitation and 590/35 nm emission wavelengths. The results are expressed as the variation of the monomers/aggregates (M/A) ratio regarding each experimental control.
4.5. BAX and BCL2 Proteins Expression
Twenty-four hours after CAP treatment, samples with 106 cells were centrifuged for 5 min at 1300× g and cells were fixed and permeabilized with the Intracell Kit (Immunostep Biotech, Salamanca, Spain), as recommended by the supplier. Samples were incubated with 0.6 µg of anti-BAX-PE antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and 0.6 µg of anti-BCL2-FITC (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 15 min at room temperature. Detection was performed by flow cytometry.
4.6. Caspase 3
Protein extracts were prepared using an appropriate lysing buffer and the protein concentration was determined by BCA method (Pierce™ BCA Protein Assay Kit, ThermoFisher Scientific, Porto Salvo, Portugal). Denaturated protein samples were run on electrophoresis gel during 15 min at 80 V and 1 h 15 min at 150 V, followed by the electrotransfer to PVDF membranes (Immun-Blot® PVDF Membrane, Bio-Rad, Amadora, Portugal). Caspase 3 was detected with a primary monoclonal antibody anti-caspase 3 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and succeeding a suitable secondary antibody anti-mouse. Finally, membranes were incubated with ECF substrate (GE Healthcare, Chicago, IL, USA) and revealed using a fluorescence reader Typhoon FLA 9000 (GE Healthcare, Chicago, IL, USA).
4.7. Reactive Oxygen and Nitrogen Species (RONS)
Two and 24 h after CAP therapy, 106 cells were centrifuged for 5 min at 1300× g and washed with phosphate saline buffer (PBS), constituted of 137 mM NaCl (Sigma Aldrich, St. Louis, MO, USA), 2.7 mM KCl (Sigma Aldrich, St. Louis, MO, USA), 10 mM Na2HPO4.2H2O (Sigma Aldrich, St. Louis, MO, USA), 2.0 mM KH2PO4 (Sigma Aldrich, St. Louis, MO, USA). The tests were performed by fluorescence in a multiwell plate spectrophotometer (Synergy HT, Winooski, VT, USA).
4.7.1. Intracellular peroxides
Cells were incubated with 5 μM of 2′,7′-dichlorodihydrofluorescein diacetate (Invitrogen®, Waltham, MA, USA) probe for 45 min in the dark at 37 °C. After washing, samples were analysed with the excitation and emission wavelengths of 485 and 520 nm, respectively.
4.7.2. Intracellular Superoxide Anion
Cells were incubated with 2 μM of dihydroethidium (Sigma Aldrich, St. Louis, MO, USA) probe for 15 min in the dark at 37 °C. After washing, detection was performed using the excitation and emission wavelengths of 500 and 645 nm, respectively.
4.7.3. Intracellular Nitric Oxide
Cultures were incubated with 1 μM of 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (Invitrogen®, Waltham, MA, USA) for 1 h in the dark at 37 °C. After washing, reading was performed with excitation and emission wavelengths of 495 and 515 nm, respectively.
4.8. Cytochrome c Oxidase Inhibition
Cells were incubated with 1 mM sodium azide (Sigma Aldrich, St. Louis, MO, USA) two hours prior CAP exposure. After CAP treatment, cells were incubated for 30 min and cell media renewed. Twenty-four hours after CAP exposure, cell cultures were washed with PBS, and 0.5 mg/mL of MTT (Sigma Aldrich, St. Louis, MO, USA) was added in each well. Cells were incubated at 37 °C, at least 4 h in dark, followed by gentle agitation with an isopropanol solution (Sigma Aldrich, St. Louis, MO, USA). Absorbance was read at wavelengths of 570 and 620 nm (Sigma Aldrich, St. Louis, MO, USA). It is important to note that previous studies were carried out in order to assess the non-toxic concentration of sodium azide for each cell line. A concentration of 1 mM of sodium azide solution was selected based on results displaying preserved metabolic activity of cells exposed to this solution (96.27 ± 9.06% in HCC1806 cells and 81.54 ± 4.24% in hormone-dependent HCC1806 cells).
4.9. Anti-Oxidative Defences
The evaluation of anti-oxidative defences was performed 2 and 24 h after CAP treatment. Samples with 106 cells were centrifuged for 5 min at 1300 × g and washed with PBS.
4.9.1. GSH
Cells were incubated with 10 μM of mercury orange (Sigma Aldrich, St. Louis, MO, USA) for 15 min in the dark at 37 °C. Samples were read with excitation and emission wavelengths of 485 and 590 nm, respectively, in the multi-well plate spectrophotometer.
4.9.2. SOD
Cell lyses was promoted with RIPA buffer, constituted of 25 mM Tris-HCl (Sigma Aldrich, St. Louis, MO, USA), 150 mM NaCl (Sigma Aldrich, St. Louis, MO, USA), 1% NP-40 (Sigma Aldrich, St. Louis, MO, USA), 1% sodium deoxycholate (Sigma Aldrich, St. Louis, MO, USA), 0.1% SDS (Sigma Aldrich, St. Louis, MO, USA) was added to cells. Cells were sonicated for 10 cycles of 3 s, each with an amplitude of 40%, centrifuged for 18 min at 14,000× g and the supernatant stored at −80 °C. Protein concentration was determined as already described. SOD activity was measured using the SOD Assay Kit-WST (Sigma Aldrich, St. Louis, MO, USA) according to suppliers’ protocol.
4.10. Cell Cycle
Twenty-four hours after CAP exposure, samples with 106 cells were centrifuged for 5 min at 1300× g and fixed in 70% ethanol (Sigma Aldrich, St. Louis, MO, USA) for 30 min at 4 °C. After washing, a propidium iodide PI/RNase solution (Immunostep, Salamanca, Spain) was added and incubated during 15 min. Samples were evaluated in the flow cytometer.
4.11. Clonogenic Assay
After CAP treatment, cells were incubated for 2 h. Then, 2000 cells were platted in 6-well plates (Sarstedt, Nümbrecht, Germany) with 3 mL of medium. After 15 days in MCF7 cell line and after 18 days in HCC1806 cell line, cells were fixed with methanol and wells were stained with 2 mL of crystal violet dye (Sigma Aldrich, St. Louis, MO, USA). Individual colonies were counted for each well and survival factor calculated according to Equation (1):
4.12. Statistical Analysis
Statistical analysis was performed using Prism 9 (San Diego, CA, USA). The normality of the distribution of quantitative variables was assessed according to the Shapiro–Wilk test. Parametric tests were used in case of a normal distribution, and nonparametric tests were used otherwise. The comparison of results with a hypothetical value (experiments where control was normalized to 1 or 100) using the one sample t test if the sample assumed a normal distribution and the Wilcoxon test if the last condition was not met. The comparison of quantitative variables between more than two groups was obtained with the ANOVA 1 Fixed Factor test (parametric test) or the Kruskal–Wallis test (non-parametric test). Multiple comparisons were corrected with Holm–Šídák’s or Dunn’s multiple comparisons test, as applicable. A significance value of 0.05 was considered for all comparisons.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




