
Disruption of Kidney–Immune System Crosstalk in Sepsis with Acute Kidney Injury: Lessons Learned from Animal Models and Their Application to Human Health


Abstract
:1. Introduction
2. Pathophysiology of AKI in Sepsis
2.1. Introduction
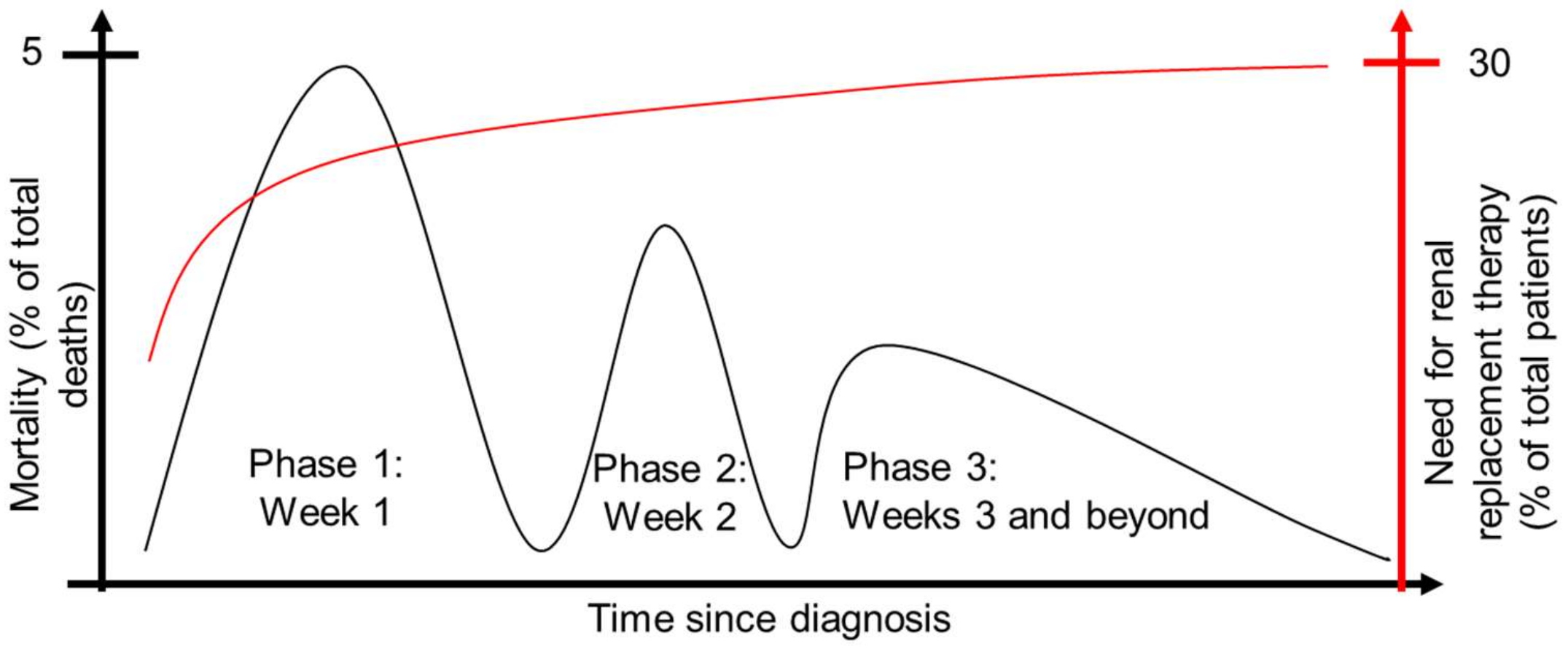
2.2. Pathophysiology of Lethal Human Sepsis
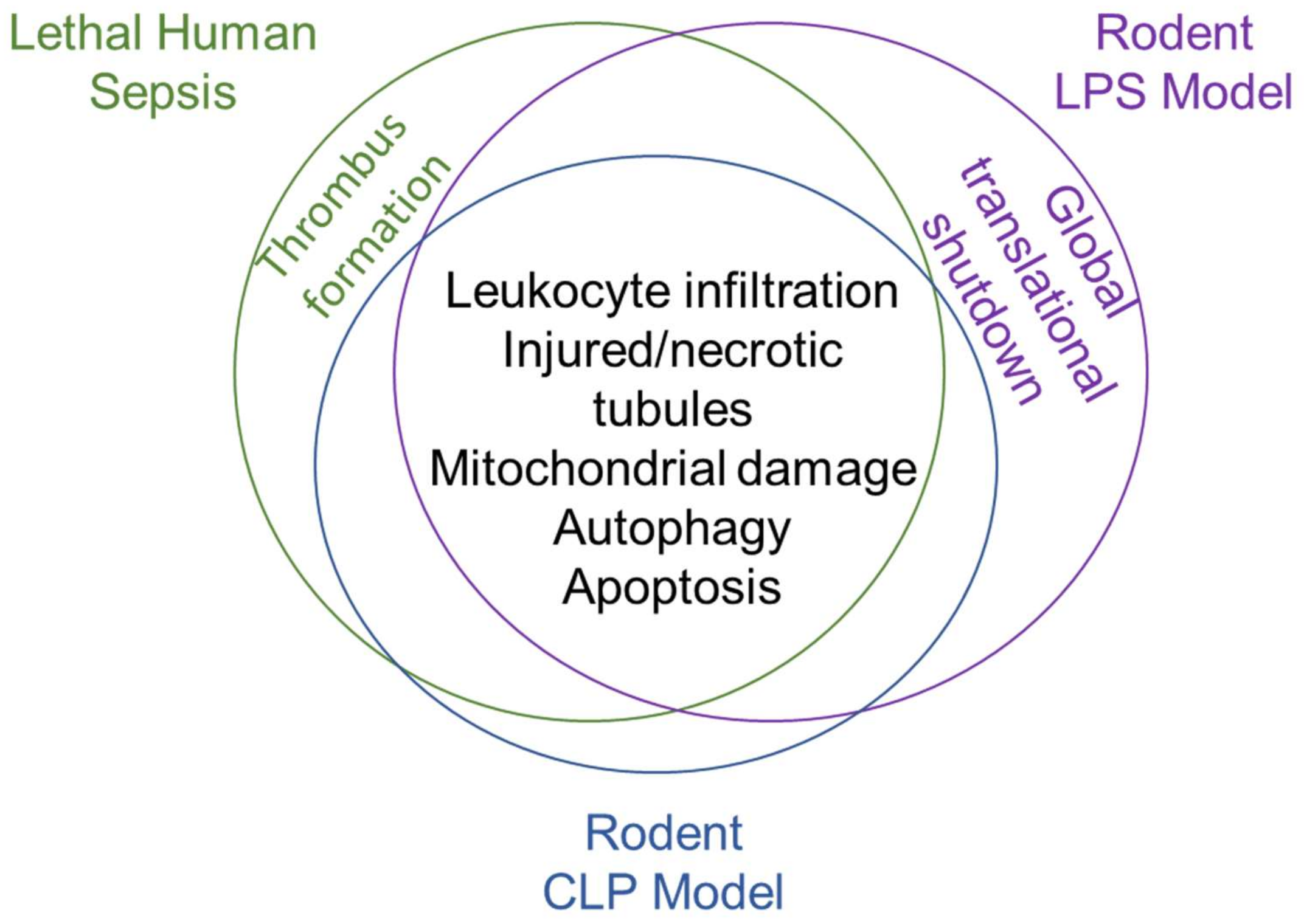
2.3. Pathophysiology of Preclinical Animal Models of Sepsis
2.3.1. Pathophysiology of the Lipopolysaccharide Model
2.3.2. Pathophysiology of the Cecal Ligation and Puncture Model
3. Immune Response in Sepsis
3.1. Activation of Inflammatory Signaling by Pathogen and Damage-Associated Molecular Patterns
3.2. Early Production of Anti-Inflammatory Cytokines
3.3. Long-Term Immune Derangement Following Acute Sepsis and Implications for Immunomodulatory Therapeutics
4. Macrophages as Drivers of Sepsis Progression
4.1. Macrophage Contribution to Sepsis and Septic Shock
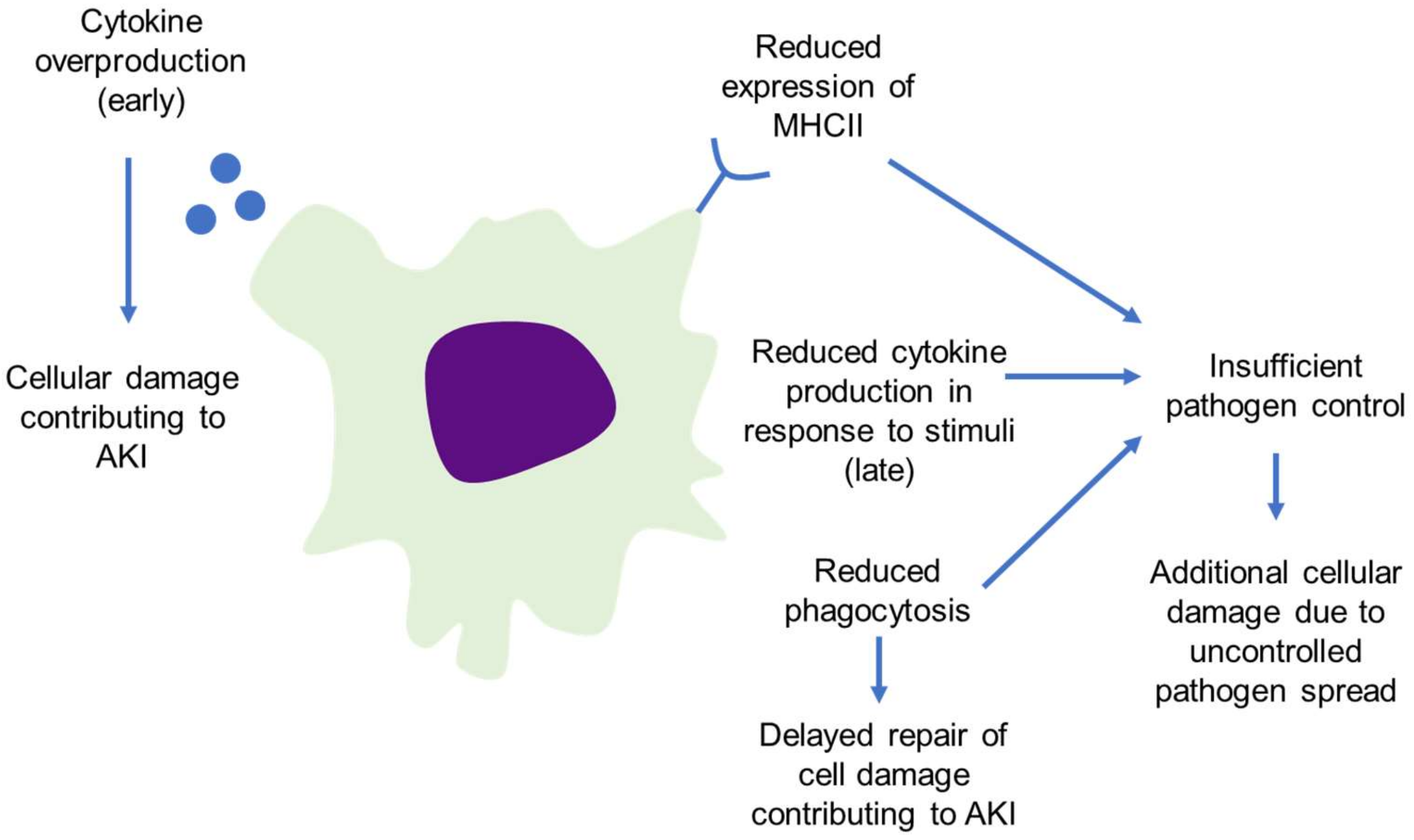
4.2. Loss of Macrophage Function in Sepsis Worsens Outcomes
4.3. Macrophage Infiltration Drives Both Damage and Repair in AKI
5. Kidney–Macrophage Crosstalk in Sepsis
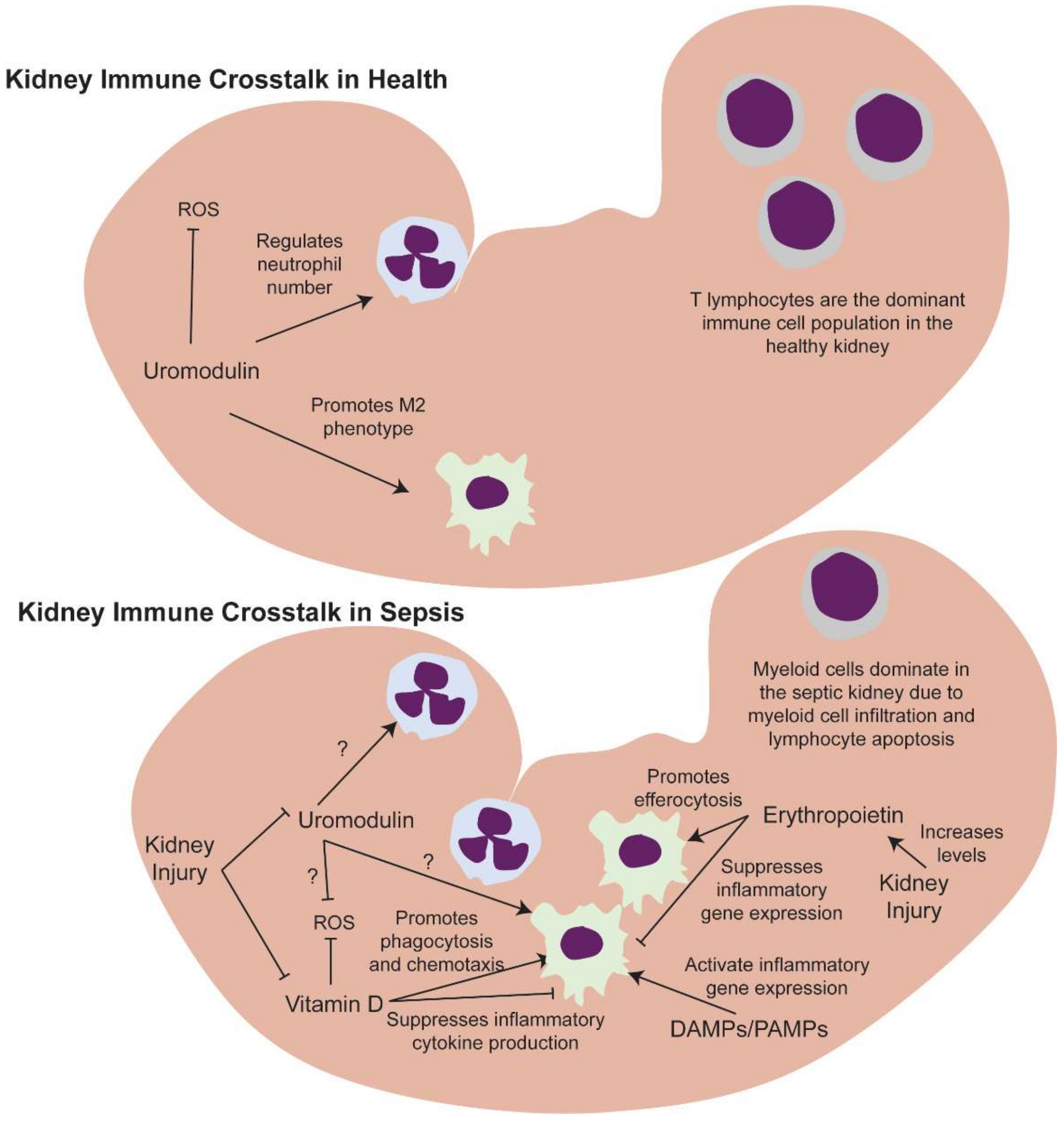
5.1. Kidney Resident and Infiltrating Immune Cells
5.2. Kidney-Derived Molecules That Regulate Macrophage Function Could Be Key Drivers of Sepsis Progression
5.2.1. Erythropoietin
5.2.2. Vitamin D
5.2.3. Uromodulin
6. Conclusions
6.1. Impact of Kidney–Immune Crosstalk in Sepsis
6.2. Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Heron, M. Deaths: Leading Causes for 2015. Natl. Vital. Stat. Rep. 2017, 66, 1–76. [Google Scholar]
- Hoste, E.A.; Lameire, N.H.; Vanholder, R.C.; Benoit, D.D.; Decruyenaere, J.M.; Colardyn, F.A. Acute renal failure in patients with sepsis in a surgical ICU: Predictive factors, incidence, comorbidity, and outcome. J. Am. Soc. Nephrol. 2003, 14, 1022–1030. [Google Scholar] [CrossRef] [Green Version]
- Bellomo, R.; Kellum, J.A.; Ronco, C.; Wald, R.; Martensson, J.; Maiden, M.; Bagshaw, S.M.; Glassford, N.J.; Lankadeva, Y.; Vaara, S.T.; et al. Acute kidney injury in sepsis. Intensive Care Med. 2017, 43, 816–828. [Google Scholar] [CrossRef] [Green Version]
- Lai, T.S.; Wang, C.Y.; Pan, S.C.; Huang, T.M.; Lin, M.C.; Lai, C.F.; Wu, C.H.; Wu, V.C.; Chien, K.L. Risk of developing severe sepsis after acute kidney injury: A population-based cohort study. Crit. Care 2013, 17, R231. [Google Scholar] [CrossRef] [Green Version]
- Wesche, D.E.; Lomas-Neira, J.L.; Perl, M.; Chung, C.S.; Ayala, A. Leukocyte apoptosis and its significance in sepsis and shock. J. Leukoc. Biol. 2005, 78, 325–337. [Google Scholar] [CrossRef]
- Peerapornratana, S.; Manrique-Caballero, C.L.; Gomez, H.; Kellum, J.A. Acute kidney injury from sepsis: Current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. 2019, 96, 1083–1099. [Google Scholar] [CrossRef]
- Luan, Y.Y.; Yao, Y.M.; Xiao, X.Z.; Sheng, Z.Y. Insights into the apoptotic death of immune cells in sepsis. J. Interferon Cytokine Res. 2015, 35, 17–22. [Google Scholar] [CrossRef] [Green Version]
- Otto, G.P.; Sossdorf, M.; Claus, R.A.; Rödel, J.; Menge, K.; Reinhart, K.; Bauer, M.; Riedemann, N.C. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit. Care 2011, 15, R183. [Google Scholar] [CrossRef] [Green Version]
- Lerolle, N.; Nochy, D.; Guérot, E.; Bruneval, P.; Fagon, J.Y.; Diehl, J.L.; Hill, G. Histopathology of septic shock induced acute kidney injury: Apoptosis and leukocytic infiltration. Intensive Care Med. 2010, 36, 471–478. [Google Scholar] [CrossRef] [Green Version]
- Takasu, O.; Gaut, J.P.; Watanabe, E.; To, K.; Fagley, E.; Sato, B.; Jarman, S.; Efimov, I.R.; Janks, D.L.; Srivastava, A.; et al. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am. J. Respir. Crit. Care Med. 2013, 187, 509–517. [Google Scholar] [CrossRef]
- Aslan, A.; van den Heuvel, M.C.; Stegeman, C.A.; Popa, E.R.; Leliveld, A.M.; Molema, G.; Zijlstra, J.G.; Moser, J.; van Meurs, M. Kidney histopathology in lethal human sepsis. Crit. Care 2018, 22, 359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Lu, J.; Liao, Y.; Liu, S.; Chen, Y.; He, R.; Men, L.; Lu, C.; Chen, Z.; Li, S.; et al. Dihydroartemisinin attenuates lipopolysaccharide-induced acute kidney injury by inhibiting inflammation and oxidative stress. Biomed. Pharmacother. 2019, 117, 109070. [Google Scholar] [CrossRef]
- Kalakeche, R.; Hato, T.; Rhodes, G.; Dunn, K.W.; El-Achkar, T.M.; Plotkin, Z.; Sandoval, R.M.; Daghter, P.C. Endotoxin uptake by S1 proximal tubular segment causes oxidative stress in the downstream S2 segment. J. Am. Soc. Nephrol. 2011, 22, 1505–1516. [Google Scholar] [CrossRef]
- Hato, T.; Maier, B.; Syed, F.; Myslinski, J.; Zollman, A.; Plotkin, Z.; Eadon, M.T.; Dagher, P.C. Bacterial sepsis triggers an antiviral response that causes translation shutdown. J. Clin. Invest. 2019, 129, 296–309. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Zhu, J.; Liu, Y.; Dong, Z.; Liu, H.; Liu, Y.; Zhou, X.; Liu, F.; Chen, G. Lipopolysaccharide Induces Chronic Kidney Injury and Fibrosis through Activation of mTOR Signaling in Macrophages. Am. J. Nephrol. 2015, 42, 305–317. [Google Scholar] [CrossRef]
- Zanotti-Cavazzoni, S.L.; Goldfarb, R.D. Animal models of sepsis. Crit. Care Clin. 2009, 25, 703–719. [Google Scholar] [CrossRef]
- Toscano, M.G.; Ganea, D.; Gamero, A.M. Cecal ligation puncture procedure. J. Vis. Exp. 2011, 51, e2860. [Google Scholar] [CrossRef]
- Pan, T.; Jia, P.; Chen, N.; Fang, Y.; Liang, Y.; Guo, M.; Ding, X. Delayed Remote Ischemic Preconditioning Confers Renoprotection against Septic Acute Kidney Injury via Exosomal miR-21. Theranostics 2019, 9, 405–423. [Google Scholar] [CrossRef]
- Lin, Z.; Jin, J.; Shan, X. Fish oils protects against cecal ligation and puncture induced septic acute kidney injury via the regulation of inflammation, oxidative stress and apoptosis. Int. J. Mol. Med. 2019, 44, 1771–1780. [Google Scholar] [CrossRef]
- Gomez, H.; Ince, C.; De Backer, D.; Pickkers, P.; Payen, D.; Hotchkiss, J.; Kellum, J.A. A unified theory of sepsis-induced acute kidney injury: Inflammation, microcirculatory dysfunction, bioenergetics, and the tubular cell adaptation to injury. Shock 2014, 41, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Sunahara, S.; Watanabe, E.; Hatano, M.; Swanson, P.E.; Oami, T.; Fujimura, L.; Teratake, Y.; Shimazui, T.; Lee, C.; Oda, S. Influence of autophagy on acute kidney injury in a murine cecal ligation and puncture sepsis model. Sci Rep. 2018, 8, 1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiao, H.W.; Tsai, K.L.; Wang, L.F.; Chen, Y.H.; Chiang, P.C.; Chuang, S.M.; Hsu, C. The decline of autophagy contributes to proximal tubular dysfunction during sepsis. Shock 2012, 37, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Gunst, J.; Derese, I.; Aertgeerts, A.; Ververs, E.J.; Wauters, A.; Van den Berghe, G.; Vanhorebeek, I. Insufficient autophagy contributes to mitochondrial dysfunction, organ failure, and adverse outcome in an animal model of critical illness. Crit. Care Med. 2013, 41, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Sursal, T.; Stearns-Kurosawa, D.J.; Itagaki, K.; Oh, S.Y.; Sun, S.; Kurosawa, S.; Jauser, C.J. Plasma bacterial and mitochondrial DNA distinguish bacterial sepsis from sterile systemic inflammatory response syndrome and quantify inflammatory tissue injury in nonhuman primates. Shock 2013, 39, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlen, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal. Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef]
- Denning, N.L.; Aziz, M.; Gurien, S.D.; Wang, P. DAMPs and NETs in Sepsis. Front. Immunol. 2019, 10, 2536. [Google Scholar] [CrossRef]
- Nedeva, C.; Menassa, J.; Puthalakath, H. Sepsis: Inflammation Is a Necessary Evil. Front. Cell Dev. Biol. 2019, 7, 108. [Google Scholar] [CrossRef] [Green Version]
- Gogos, C.A.; Drosou, E.; Bassaris, H.P.; Skoutelis, A. Pro- versus Anti-inflammatory Cytokine Profile in Patients with Severe Sepsis: A Marker for Prognosis and Future Therapeutic Options. J. Infect. Dis. 2000, 181, 176–180. [Google Scholar] [CrossRef]
- Boomer, J.S.; To, K.; Chang, K.C.; Takasu, O.; Osborne, D.F.; Walton, A.H.; Bricker, T.L.; Jarman, S.D.; Kreisel, D., 2nd; Krupnick, A.S.; et al. Immunosuppression in Patients Who Die of Sepsis and Multiple Organ Failure. JAMA 2011, 306, 2594–2605. [Google Scholar] [CrossRef]
- Delano, M.; Ward, P.A. Sepsis-induced immune dysfunction: Can immune therapies reduce mortality? J. Clin. Investig. 2016, 126, 23–31. [Google Scholar] [CrossRef] [PubMed]
- The Recovery Collaborative Group. Dexamethasone in Hospitalized Patients with Covid-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef]
- van Paassen, J.; Vos, J.S.; Hoekstra, E.M.; Neumann, K.M.I.; Boot, P.C.; Arbous, S.M. Corticosteroid use in COVID-19 patients: A systematic review and meta-analysis on clinical outcomes. Crit. Care 2020, 24, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V. Targeting macrophage immunometabolism: Dawn in the darkness of sepsis. Int. Immunopharmacol. 2018, 58, 173–185. [Google Scholar] [CrossRef]
- Sica, A.; Erreni, M.; Allavena, P.; Porta, C. Macrophage polarization in pathology. Exp. 2015, 72, 4111–4126. [Google Scholar] [CrossRef]
- Matsumoto, H.; Ogura, H.; Shimizu, K.; Ikeda, M.; Hirose, T.; Matsuura, H.; Kang, S.; Takahashi, K.; Tanaka, T.; Shimazu, T. The clinical importance of a cytokine network in the acute phase of sepsis. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Luan, L.; Patil, N.K.; Wang, J.; Bohannon, J.K.; Rabacal, W.; Fensterheim, B.A.; Hernandez, A.; Sherwood, E.R. IL-15 Enables Septic Shock by Maintaining NK Cell Integrity and Function. J. Immunol. 2017, 198, 1320–1333. [Google Scholar] [CrossRef] [Green Version]
- Friedman, G.; Jankowski, S.; Marchant, A.; Goldman, M.; Kahn, R.J.; Vincent, J.L. Blood interleukin 10 levels parallel the severity of septic shock. J. Crit. Care 1997, 12, 183–187. [Google Scholar] [CrossRef]
- Gårdlund, B.; Sjölin, J.; Nilsson, A.; Roll, M.; Wickerts, C.-J.; Wretlind, B. Plasma Levels of Cytokines in Primary Septic Shock in Humans: Correlation with Disease Severity. J. Infect. Dis. 1995, 172, 296–301. [Google Scholar] [CrossRef]
- Cauwels, A. Nitric oxide in shock. Kidney Int. 2007, 72, 557–565. [Google Scholar] [CrossRef] [Green Version]
- Hershman, M.J.; Cheadle, W.G.; Wellhausen, S.R.; Davidson, P.F.; Polk, H.C. Monocyte HLA-DR antigen expression characterizes clinical outcome in the trauma patient. Br. J. Surg. 1990, 77, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Cajander, S.; Tina, E.; Bäckman, A.; Magnuson, A.; Strålin, K.; Söderquist, B.; Källman, J. Quantitative Real-Time Polymerase Chain Reaction Measurement of HLA-DRA Gene Expression in Whole Blood Is Highly Reproducible and Shows Changes That Reflect Dynamic Shifts in Monocyte Surface HLA-DR Expression during the Course of Sepsis. PLoS ONE 2016, 11, e0154690. [Google Scholar] [CrossRef] [PubMed]
- Cazalis, M.-A.; Friggeri, A.; Cavé, L.; Demaret, J.; Barbalat, V.; Cerrato, E.; Lepape, A.; Pachot, A.; Monneret, G.; Venet, F. Decreased HLA-DR antigen-associated invariant chain (CD74) mRNA expression predicts mortality after septic shock. Crit. Care 2013, 17, R287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cajander, S.; Bäckman, A.; Tina, E.; Strålin, K.; Söderquist, B.; Källman, J. Preliminary results in quantitation of HLA-DRA by real-time PCR: A promising approach to identify immunosuppression in sepsis. Crit. Care 2013, 17, R223. [Google Scholar] [CrossRef] [Green Version]
- Munoz, C.; Carlet, J.; Fitting, C.; Misset, B.; Blériot, J.P.; Cavaillon, J.M. Dysregulation of in vitro cytokine production by monocytes during sepsis. J. Clin. Investig. 1991, 88, 1747–1754. [Google Scholar] [CrossRef] [Green Version]
- Danikas, D.D.; Karakantza, M.; Theodorou, G.L.; Sakellaropoulos, G.C.; Gogos, C.A. Prognostic value of phagocytic activity of neutrophils and monocytes in sepsis. Correlation to CD64 and CD14 antigen expression. Clin. Exp. Immunol. 2008, 154, 87–97. [Google Scholar] [CrossRef]
- Xing, L.; Zhongqian, L.; Chunmei, S.; Pinfa, C.; Lei, H.; Qin, J.; Genhua, M.; Yijun, D. Activation of M1 macrophages in sepsis-induced acute kidney injury in response to heparin-binding protein. PLoS ONE 2017, 13, e0196423. [Google Scholar] [CrossRef] [Green Version]
- Liangliang, Z.; Mu, G.; Song, C.; Zhou, L.; He, L.; Jin, Q.; Lu, Z. Role of M2 Macrophages in Sepsis-Induced Acute Kidney Injury. Shock 2018, 50, 233–239. [Google Scholar] [CrossRef]
- Shalova, I.N.; Lim, J.Y.; Chittezhath, M.; Zinkernagel, A.S.; Beasley, F.; Hernández-Jiménez, E.; Toledano, V.; Cubillos-Zapata, C.; Rapisarda, A.; Chen, J.; et al. Human Monocytes Undergo Functional Re-programming during Sepsis Mediated by Hypoxia-Inducible Factor-1α. Immunity 2015, 42, 484–498. [Google Scholar] [CrossRef] [Green Version]
- White, L.E.; Chaudhary, R.; Moore, L.J.; Moore, F.A.; Hassoun, H.T. Surgical Sepsis and Organ Crosstalk: The Role of the Kidney. J. Surg. Res. 2011, 167, 306–315. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-G.; Na, M.; Kim, M.-G.; Park, S.H.; Lee, H.J.; Kim, D.K.; Kwak, C.; Kim, Y.S.; Chang, S.; Moon, K.C.; et al. Immune cell composition in normal human kidneys. Sci. Rep. 2020, 10, 15678. [Google Scholar] [CrossRef] [PubMed]
- Stewart, B.J.; Ferdinand, J.R.; Young, M.D.; Mitchell, T.J.; Loudon, K.W.; Riding, A.M.; Richoz, N.; Frazer, G.L.; Staniforth, J.U.L.; Braga, F.A.V.; et al. Spatiotemporal immune zonation of the human kidney. Science 2019, 365, 1461–1466. [Google Scholar] [CrossRef] [PubMed]
- anosevic, D.; Myslinski, J.; McCarthy, T.W.; Zollman, A.; Syed, F.; Xuei, X.; Gao, H.; Liu, Y.; Collins, K.S.; Cheng, Y.-H.; et al. The orchestrated cellular and molecular responses of the kidney to endotoxin define a precise sepsis timeline. eLife 2021, 10, e62270. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.M.; Sabo, A.R.; Winfree, S.; Collins, K.S.; Janosevic, D.; Gulbronson, C.J.; Cheng, Y.-H.; Casbon, L.; Barwinska, D.; Ferkowicz, M.J.; et al. Integration of spatial and single-cell transcriptomics localizes epithelial cell–immune cross-talk in kidney injury. JCI Insight 2021, 6, e147703. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Lysak, N.; Adhikari, L.; Velez, L.M.; Sautina, L.; Mohandas, R.; Lopez, M.-C.; Ungaro, R.; Peng, Y.-C.; Kadri, F.; et al. Discovery and Validation of Urinary Molecular Signature of Early Sepsis. Crit. Care Explor. 2020, 2, e0195. [Google Scholar] [CrossRef]
- Luo, B.; Gan, W.; Liu, Z.; Shen, Z.; Wang, J.; Shi, R.; Liu, Y.; Liu, Y.; Jiang, M.; Zhang, Z.; et al. Erythropoeitin Signaling in Macrophages Promotes Dying Cell Clearance and Immune Tolerance. Immunity 2016, 44, 287–302. [Google Scholar] [CrossRef] [Green Version]
- Krafte-Jacobs, B.; Bock, G.H. Circulating erythropoietin and interleukin-6 concentrations increase in critically ill children with sepsis and septic shock. Crit. Care Med. 1996, 24, 1455–1459. [Google Scholar] [CrossRef]
- Tamion, F.; Le Cam-Duchez, V.; Menard, J.F.; Girault, C.; Coquerel, A.; Bonmarchand, G. Serum Erythropoietin Levels in Septic Shock. Anaesth. Intensiv. Care 2005, 33, 578–584. [Google Scholar] [CrossRef]
- Toro, L.; Barrientos, V.; León, P.; Rojas, M.; Gonzalez, M.; González-Ibáñez, A.; Illanes, S.; Sugikawa, K.; Abarzúa, N.; Bascuñán, C.; et al. Erythropoietin induces bone marrow and plasma fibroblast growth factor 23 during acute kidney injury. Kidney Int. 2018, 93, 1131–1141. [Google Scholar] [CrossRef]
- Coldewey, S.M.; Khan, A.I.; Kapoor, A.; Collino, M.; Rogazzo, M.; Brines, M.; Cerami, A.; Hall, P.; Sheaff, M.; Kieswich, J.E.; et al. Erythropoietin attenuates acute kidney dysfunction in murine experimental sepsis by activation of the β-common receptor. Kidney Int. 2013, 84, 482–490. [Google Scholar] [CrossRef] [Green Version]
- Heitrich, M.; de los Ángeles García, D.M.; Stoyanoff, T.R.; Rodríguez, J.P.; Todaro, J.S.; Aguirre, M.V. Erythropoietin attenuates renal and pulmonary injury in polymicrobial induced-sepsis through EPO-R, VEGF and VEGF-R2 modulation. Biomed. Pharmacother. 2016, 82, 606–613. [Google Scholar] [CrossRef]
- Qu, Y.; Sun, Q.; Song, X.; Jiang, Y.; Dong, H.; Zhao, W.; Li, C. Helix B surface peptide reduces sepsis-induced kidney injury via PI3K/Akt pathway. Nephrology 2020, 25, 527–534. [Google Scholar] [CrossRef]
- De Souza, A.C.C.P.; Volpini, R.A.; Shimizu, M.H.M.; Sanches, T.R.C.; Camara, N.; Semedo-Kuriki, P.; Rodrigues, C.; Seguro, A.C.; Andrade, L. Erythropoietin prevents sepsis-related acute kidney injury in rats by inhibiting NF-κB and upregulating endothelial nitric oxide synthase. Am. J. Physiol. Physiol. 2012, 302, F1045–F1054. [Google Scholar] [CrossRef] [Green Version]
- Stoyanoff, T.R.; Rodríguez, J.P.; Todaro, J.S.; Colavita, J.P.M.; Torres, A.M.; Aguirre, M.V. Erythropoietin attenuates LPS-induced microvascular damage in a murine model of septic acute kidney injury. Biomed. Pharmacother. 2018, 107, 1046–1055. [Google Scholar] [CrossRef]
- Stoyanoff, T.R.; Todaro, J.S.; Aguirre, M.V.; Zimmermann, M.C.; Brandan, N.C. Amelioration of lipopolysaccharide-induced acute kidney injury by erythropoietin: Involvement of mitochondria-regulated apoptosis. Toxicology 2014, 318, 13–21. [Google Scholar] [CrossRef]
- Song, Y.R.; Lee, T.; You, S.J.; Chin, H.J.; Chae, D.-W.; Lim, C.; Park, K.-H.; Han, S.; Kim, J.-H.; Na, K.Y. Prevention of Acute Kidney Injury by Erythropoietin in Patients Undergoing Coronary Artery Bypass Grafting: A Pilot Study. Am. J. Nephrol. 2009, 30, 253–260. [Google Scholar] [CrossRef]
- De Seigneux, S.; Ponte, B.; Weiss, L.; Pugin, J.; Romand, J.A.; Martin, P.-Y.; Saudan, P. Epoetin administrated after cardiac surgery: Effects on renal function and inflammation in a randomized controlled study. BMC Nephrol. 2012, 13, 132. [Google Scholar] [CrossRef]
- Oh, S.W.; Chin, H.J.; Chae, D.W.; Na, K.Y. Erythropoietin Improves Long-Term Outcomes in Patients with Acute Kidney Injury after Coronary Artery Bypass Grafting. J. Korean Med Sci. 2012, 27, 506–511. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-H.; Shim, J.-K.; Song, J.-W.; Song, Y.; Kim, H.-B.; Kwak, Y.-L. Effect of erythropoietin on the incidence of acute kidney injury following complex valvular heart surgery: A double blind, randomized clinical trial of efficacy and safety. Crit. Care 2013, 17, R254. [Google Scholar] [CrossRef] [Green Version]
- Tasanarong, A.; Duangchana, S.; Sumransurp, S.; Homvises, B.; Satdhabudha, O. Prophylaxis with erythropoietin versus placebo reduces acute kidney injury and neutrophil gelatinase-associated lipocalin in patients undergoing cardiac surgery: A randomized, double-blind controlled trial. BMC Nephrol. 2013, 14, 136. [Google Scholar] [CrossRef] [Green Version]
- Skrifvars, M.B.; Moore, E.; Mårtensson, J.; Bailey, M.; French, C.; Presneill, J.; Nichol, A.; Little, L.; Duranteau, J.; Huet, O.; et al. Erythropoietin in traumatic brain injury associated acute kidney injury: A randomized controlled trial. Acta Anaesthesiol. Scand. 2019, 63, 200–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R. Crosstalk between Vitamin D Metabolism, VDR Signalling, and Innate Immunity. Biomed. Res. Int 2016, 2016, 1375858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graidis, S.; Papavramidis, T.S.; Papaioannou, M. Vitamin D and Acute Kidney Injury: A Two-Way Causality Relation and a Predictive, Prognostic, and Therapeutic Role of Vitamin D. Front. Nutr. 2020, 7, 630951. [Google Scholar] [CrossRef]
- Xu, S.; Chen, Y.-H.; Tan, Z.-X.; Xie, D.-D.; Zhang, C.; Xia, M.-Z.; Wang, H.; Zhao, H.; Xu, D.-X.; Yu, D.-X. Vitamin D3 pretreatment alleviates renal oxidative stress in lipopolysaccharide-induced acute kidney injury. J. Steroid Biochem. Mol. Biol. 2015, 152, 133–141. [Google Scholar] [CrossRef]
- Du, J.; Jiang, S.; Hu, Z.; Tang, S.; Sun, Y.; He, J.; Li, Z.; Yi, B.; Wang, J.; Zhang, H.; et al. Vitamin D receptor activation protects against lipopolysaccharide-induced acute kidney injury through suppression of tubular cell apoptosis. Am. J. Physiol. Physiol. 2019, 316, F1068–F1077. [Google Scholar] [CrossRef]
- Micanovic, R.; LaFavers, K.; Garimella, P.S.; Wu, X.-R.; El-Achkar, T.M. Uromodulin (Tamm–Horsfall protein): Guardian of urinary and systemic homeostasis. Nephrol. Dial. Transplant. 2020, 35, 33–43. [Google Scholar] [CrossRef]
- Muchmore, A.V.; Decker, J.M. Uromodulin: A Unique 85-Kilodalton Immunosuppressive Glycoprotein Isolated from Urine of Pregnant Women. Science 1985, 229, 479–481. [Google Scholar] [CrossRef] [Green Version]
- Micanovic, R.; Khan, S.; Janosevic, D.; Lee, M.E.; Hato, T.; Srour, E.F.; Winfree, S.; Ghosh, J.; Tong, Y.; Rice, S.E.; et al. Tamm-Horsfall Protein Regulates Mononuclear Phagocytes in the Kidney. J. Am. Soc. Nephrol. 2018, 29, 841–856. [Google Scholar] [CrossRef]
- Micanovic, R.; Chitteti, B.R.; Dagher, P.C.; Srour, E.F.; Khan, S.; Hato, T.; Lyle, A.; Tong, Y.; Wu, X.-R.; El-Achkar, T.M. Tamm-Horsfall Protein Regulates Granulopoiesis and Systemic Neutrophil Homeostasis. J. Am. Soc. Nephrol. 2015, 26, 2172–2182. [Google Scholar] [CrossRef] [Green Version]
- Mo, L.; Zhu, X.-H.; Huang, H.-Y.; Shapiro, E.; Hasty, D.L.; Wu, X.-R. Ablation of the Tamm-Horsfall protein gene increases susceptibility of mice to bladder colonization by type 1-fimbriatedEscherichia coli. Am. J. Physiol. Physiol. 2004, 286, F795–F802. [Google Scholar] [CrossRef] [Green Version]
- Bates, J.M.; Raffi, H.M.; Prasadan, K.; Mascarenhas, R.; Laszik, Z.; Maeda, N.; Hultgren, S.J.; Kumar, S. Tamm-Horsfall protein knockout mice are more prone to urinary tract infection Rapid Communication. Kidney Int. 2004, 65, 791–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raffi, H.S.; Bates, J.M., Jr.; Laszik, Z.; Kumar, S. Tamm-Horsfall Protein Protects Against Urinary Tract Infection by Proteus Mirabilis. J. Urol. 2009, 181, 2332–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coady, A.; Ramos, A.R.; Olson, J.; Nizet, V.; Patras, K.A. Tamm-Horsfall Protein Protects the Urinary Tract against Candida albicans. Infect. Immun. 2018, 86, e00451-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludes, P.-O.; de Roquetaillade, C.; Chousterman, B.G.; Pottecher, J.; Mebazaa, A. Role of Damage-Associated Molecular Patterns in Septic Acute Kidney Injury, From Injury to Recovery. Front. Immunol. 2021, 12, 606622. [Google Scholar] [CrossRef] [PubMed]
- Säemann, M.D.; Weichart, T.; Zeyda, M.; Staffler, G.; Schunn, M.; Stuhlmeier, K.M.; Sobanov, Y.; Stulnig, T.M.; Akira, S.; von Gabain, A.; et al. Tamm-Horsfall glycoprotein links innate immune cell activation with adaptive immunity via a Toll-like receptor-4-dependent mechanism. J. Clin. Invest. 2005, 115, 468–475. [Google Scholar] [CrossRef] [Green Version]
- Garimella, P.S.; Sarnak, M.J. Uromodulin in kidney health and disease. Curr. Opin. Nephrol. Hypertens. 2016, 26, 136–142. [Google Scholar] [CrossRef]
- Garimella, P.S.; Biggs, M.L.; Katz, R.; Ix, J.H.; Bennett, M.R.; Devarajan, P.; Kestenbaum, B.R.; Siscovick, D.S.; Jensen, M.K.; Shlipak, M.G.; et al. Urinary uromodulin, kidney function, and cardiovascular disease in elderly adults. Kidney Int. 2015, 88, 1126–1134. [Google Scholar] [CrossRef] [Green Version]
- Delgado, G.E.; Kleber, M.E.; Scharnagl, H.; Krämer, B.K.; März, W.; Scherberich, J.E. Serum Uromodulin and Mortality Risk in Patients Undergoing Coronary Angiography. J. Am. Soc. Nephrol. 2017, 28, 2201–2210. [Google Scholar] [CrossRef]
- Garimella, P.S.; Katz, R.; Ix, J.H.; Fried, L.F.; Kritchevsky, S.B.; Devarajan, P.; Bennett, M.R.; Parikh, C.; Shlipak, M.G.; Harris, T.B.; et al. Association of urinary uromodulin with kidney function decline and mortality: The health ABC study. Clin. Nephrol. 2017, 87, 278–286. [Google Scholar] [CrossRef]
- Leiherer, A.; Muendlein, A.; Saely, C.H.; Ebner, J.; Brandtner, E.M.; Fraunberger, P.; Drexel, H. Serum uromodulin is a predictive biomarker for cardiovascular events and overall mortality in coronary patients. Int. J. Cardiol. 2017, 231, 6–12. [Google Scholar] [CrossRef]
- El-Achkar, T.M.; Huang, X.; Plotkin, Z.; Sandoval, R.M.; Rhodes, G.J.; Dagher, P.C. Sepsis induces changes in the expression and distribution of Toll-like receptor 4 in the rat kidney. Am. J. Physiol. Physiol. 2005, 290, F1034–F1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaFavers, K.A.; Macedo, E.; Garimella, P.S.; Lima, C.; Khan, S.; Myslinski, J.; McClintick, J.; Witzmann, F.A.; Winfree, S.; Phillips, C.L.; et al. Circulating uromodulin inhibits systemic oxidative stress by inactivating the TRPM2 channel. Sci. Transl. Med. 2019, 11, eaaw3639. [Google Scholar] [CrossRef] [PubMed]
- El-Achkar, T.M.; McCracken, R.; Liu, Y.; Heitmeier, M.R.; Bourgeois, S.; Ryerse, J.; Wu, X.-R. Tamm-Horsfall protein translocates to the basolateral domain of thick ascending limbs, interstitium, and circulation during recovery from acute kidney injury. Am. J. Physiol. Physiol. 2013, 304, F1066–F1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule of Interest | Lessons Learned from Animal Models | Results from Human Sepsis | Future Work Needed |
|---|---|---|---|
| Erythropoietin | Primes macrophages for efferocytosis of apoptotic cells and suppresses macrophage expression of inflammatory genes [56] Levels increase in AKI and sepsis [59] Treatment with erythropoietin is protective [60,61,62,63,64,65] | Levels increase in sepsis [57,58] | Identification of sepsis patients who would benefit from erythropoietin supplementation Clinical trials of erythropoietin supplementation in sepsis |
| Vitamin D | Enhances macrophage phagocytosis and chemotaxis and suppresses inflammatory cytokine production [72] Treatment with vitamin D mitigates renal oxidative stress [74] and decreases expression of inflammatory cytokines in the kidney [75] in LPS | Systemic levels are predictors of sepsis survival [72] | Vitamin D treatment in other preclinical animal models of sepsis Identification of patients who would benefit from vitamin D Clinical trials of vitamin D supplementation in sepsis |
| Uromodulin (Tamm–Horsfall protein) | Regulates macrophage number and phagocytic function [78] Regulates neutrophil production [79] Protective against bladder and urinary tract infection [80] Depleted in ischemic AKI [92,93], but elevated in CLP sepsis [91] Treatment with uromodulin mitigates kidney injury in ischemic AKI [50] | Levels decrease in AKI [93] | Characterization of uromodulin and uromodulin treatment in animal models of sepsis Characterization of uromodulin in sepsis patient populations Identification of sepsis patients who would benefit from uromodulin supplementation Clinical trials of uromodulin supplementation in sepsis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
LaFavers, K. Disruption of Kidney–Immune System Crosstalk in Sepsis with Acute Kidney Injury: Lessons Learned from Animal Models and Their Application to Human Health. Int. J. Mol. Sci. 2022, 23, 1702. https://doi.org/10.3390/ijms23031702
LaFavers K. Disruption of Kidney–Immune System Crosstalk in Sepsis with Acute Kidney Injury: Lessons Learned from Animal Models and Their Application to Human Health. International Journal of Molecular Sciences. 2022; 23(3):1702. https://doi.org/10.3390/ijms23031702
Chicago/Turabian StyleLaFavers, Kaice. 2022. "Disruption of Kidney–Immune System Crosstalk in Sepsis with Acute Kidney Injury: Lessons Learned from Animal Models and Their Application to Human Health" International Journal of Molecular Sciences 23, no. 3: 1702. https://doi.org/10.3390/ijms23031702
APA StyleLaFavers, K. (2022). Disruption of Kidney–Immune System Crosstalk in Sepsis with Acute Kidney Injury: Lessons Learned from Animal Models and Their Application to Human Health. International Journal of Molecular Sciences, 23(3), 1702. https://doi.org/10.3390/ijms23031702