
Type 1 Diabetes Impairs Cardiomyocyte Contractility in the Left and Right Ventricular Free Walls but Preserves It in the Interventricular Septum

 , , , ,
, , , ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Model of Type 1 Diabetes
2.2. ECG Recording and Blood Pressure Measurement
2.3. Laboratory Blood Tests
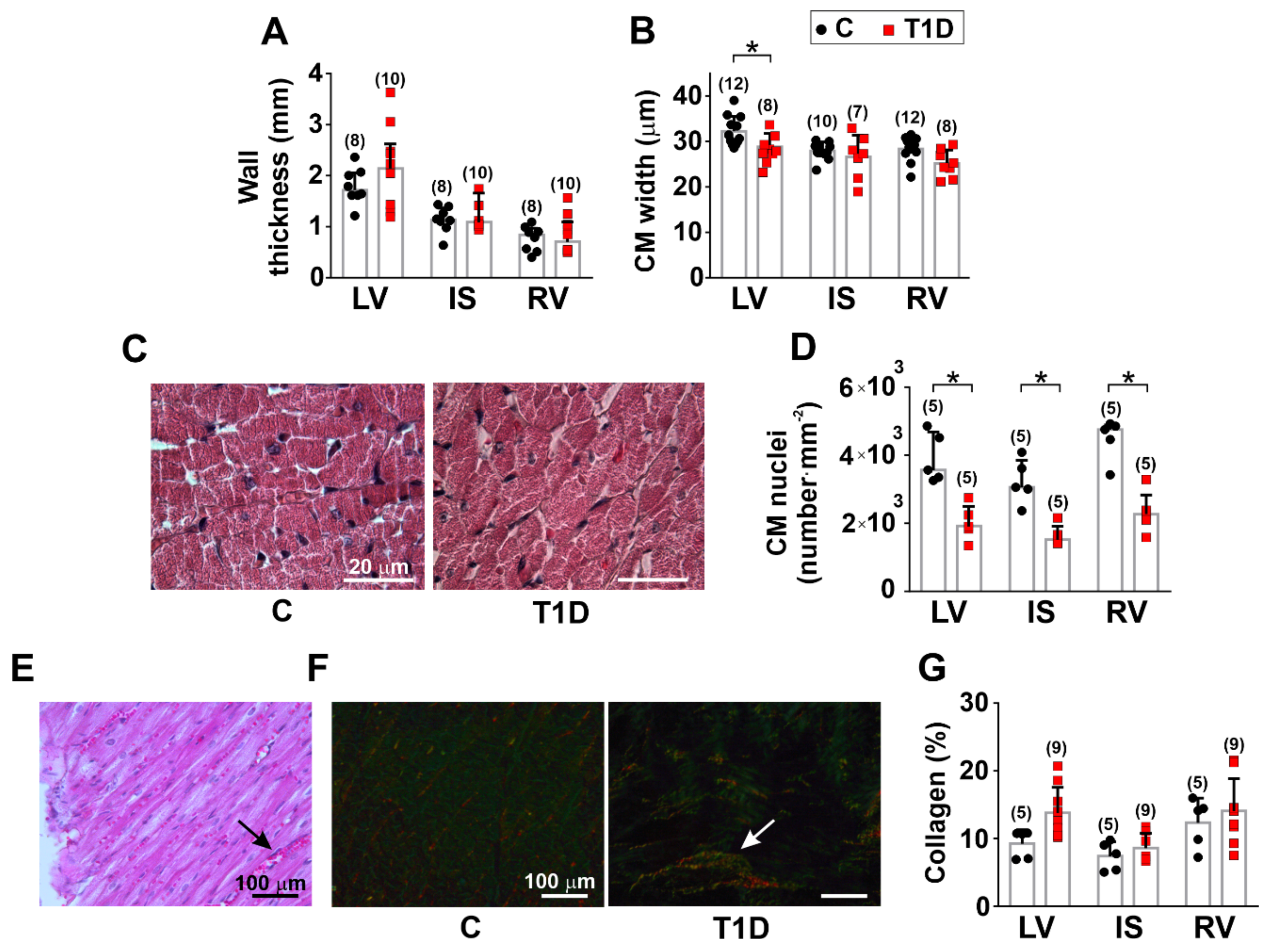
2.4. Histological and Immunohistochemical Studies
2.5. Ventricular Myocyte Isolation
2.6. Measurements of Myocyte Size, [Ca2+]i Transients, and Mechanically Non-Loaded Sarcomere Shortening
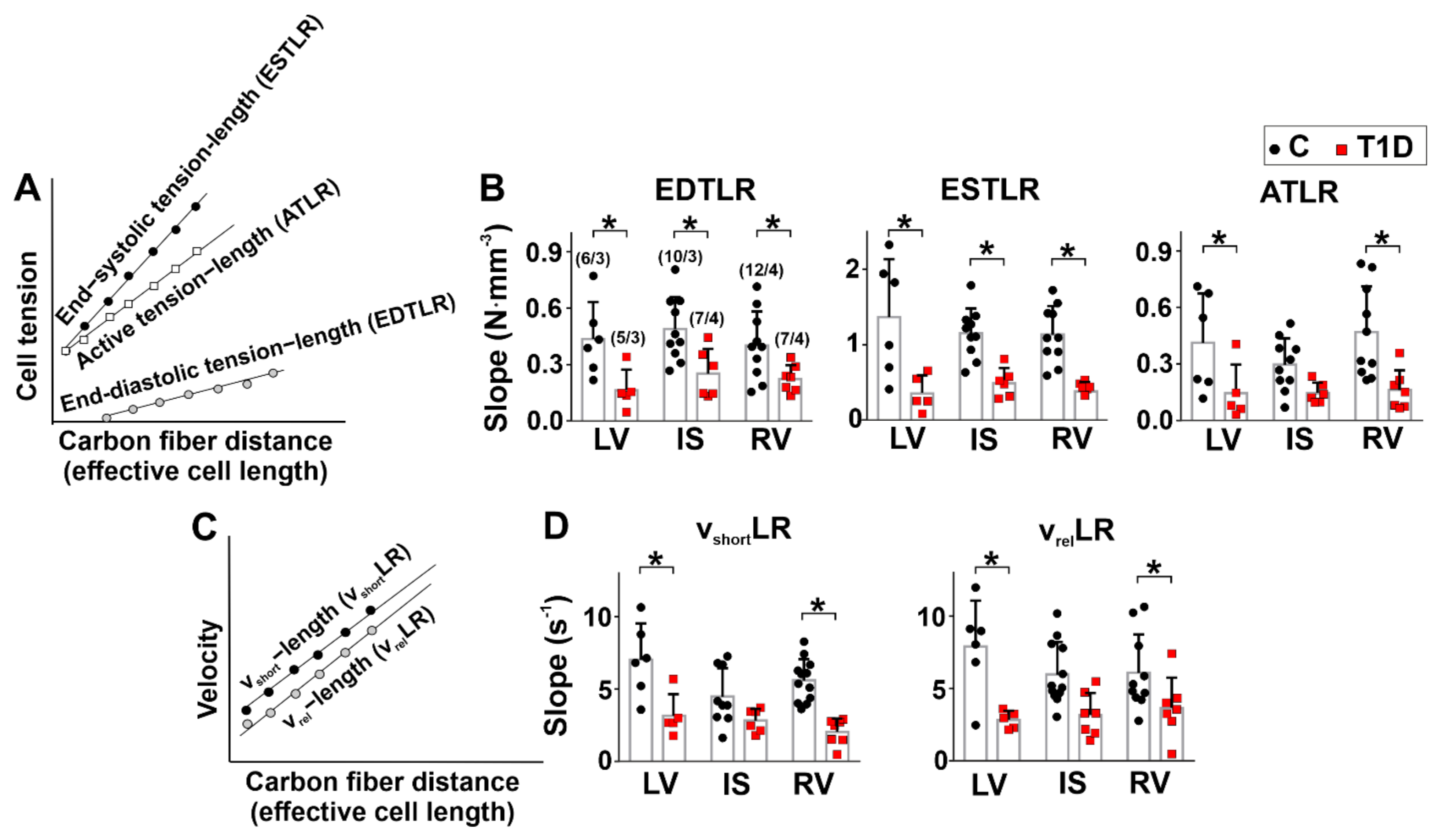
2.7. Measurements Tension-Length Dependence in Single Afterload Cardiomyocytes
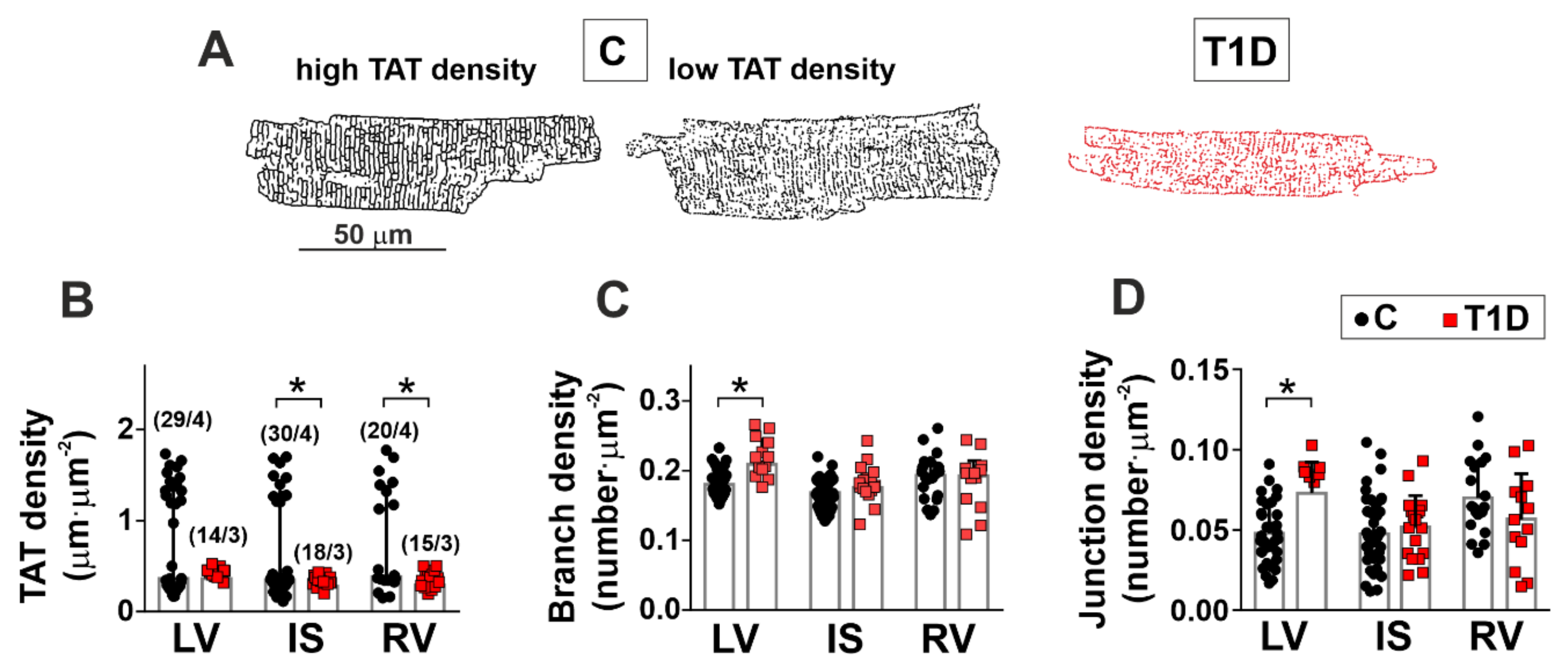
2.8. Transverse-Axial Tubule Network Analysis
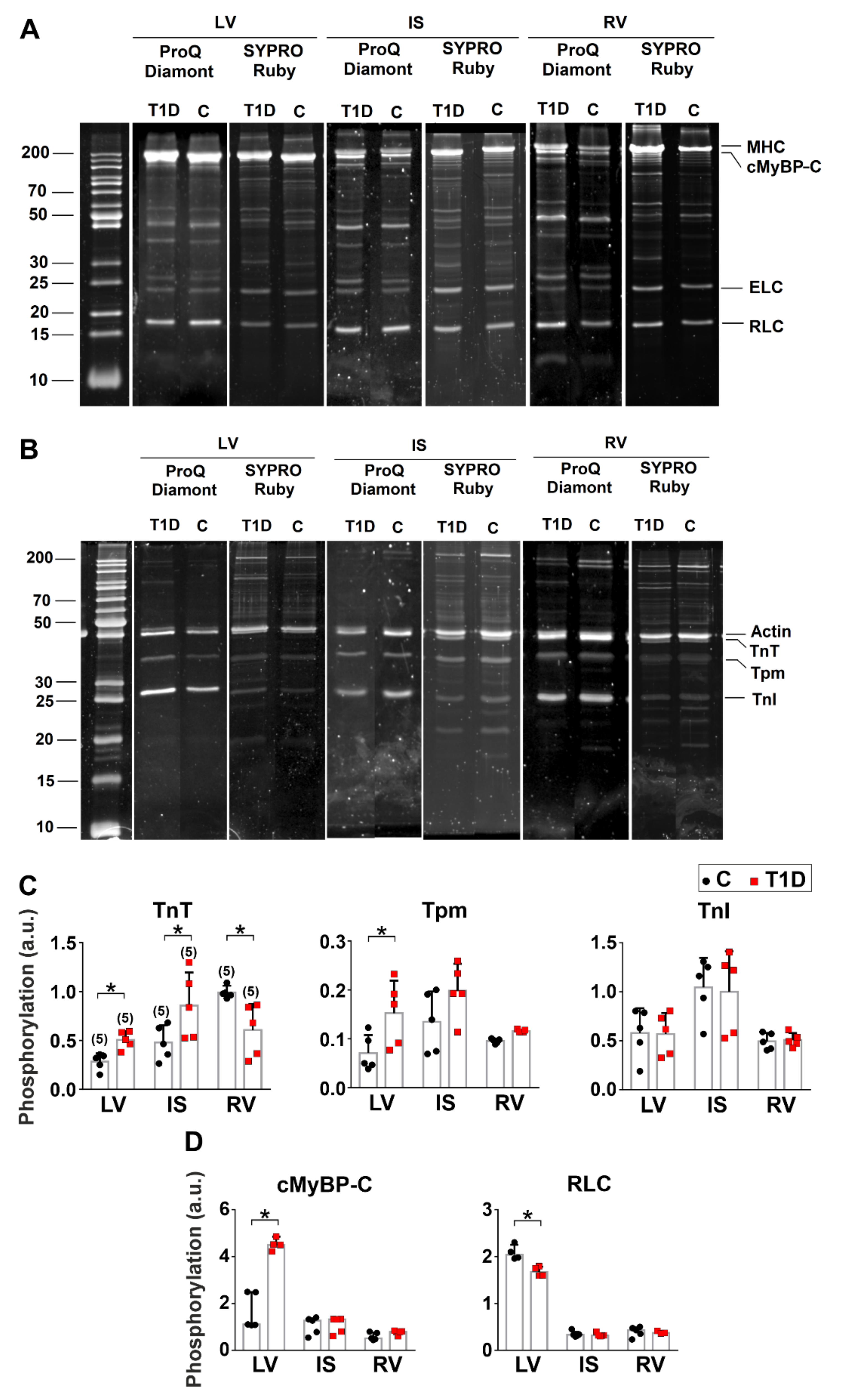
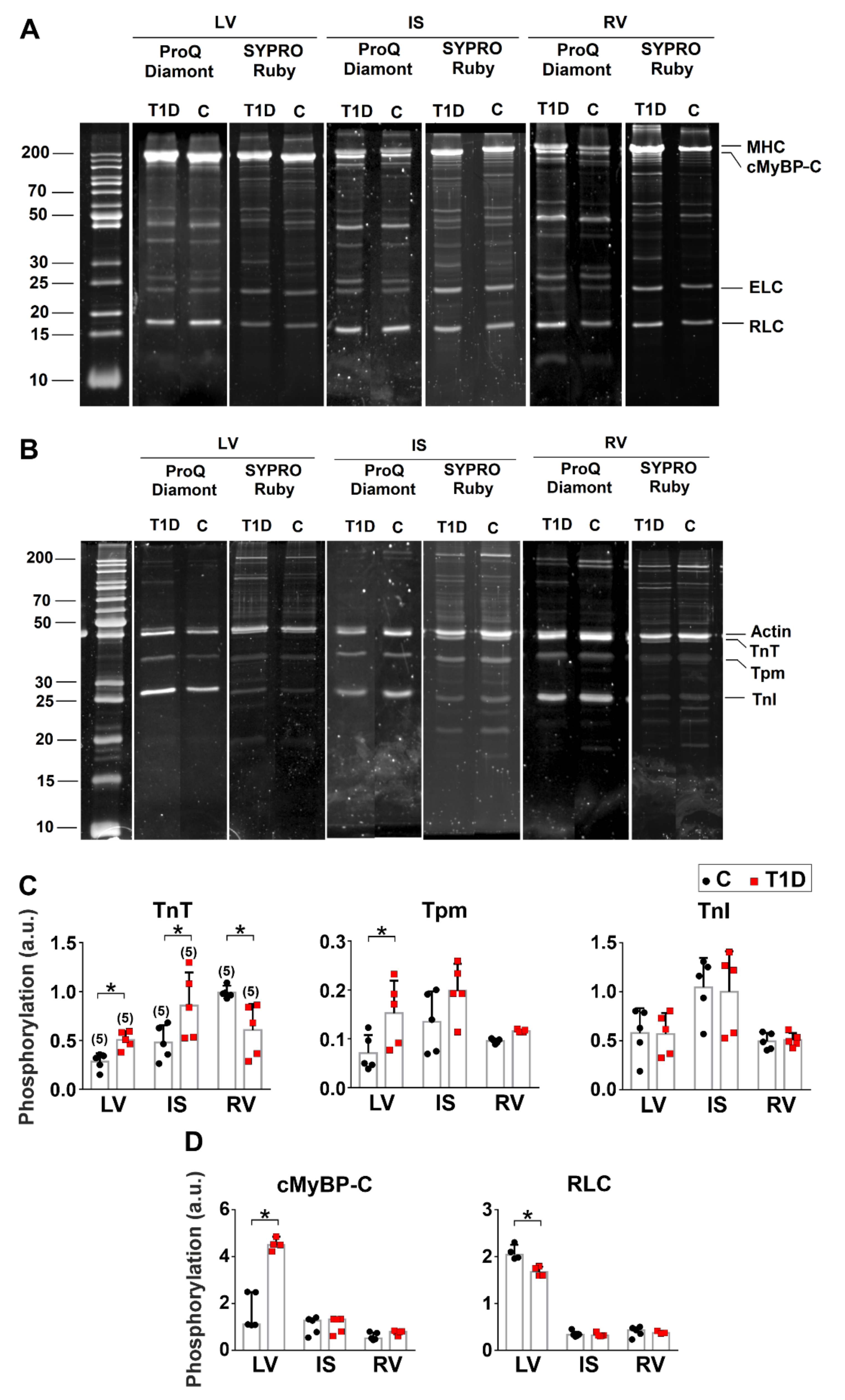
2.9. Protein Preparation and Phosphorylation Analysis
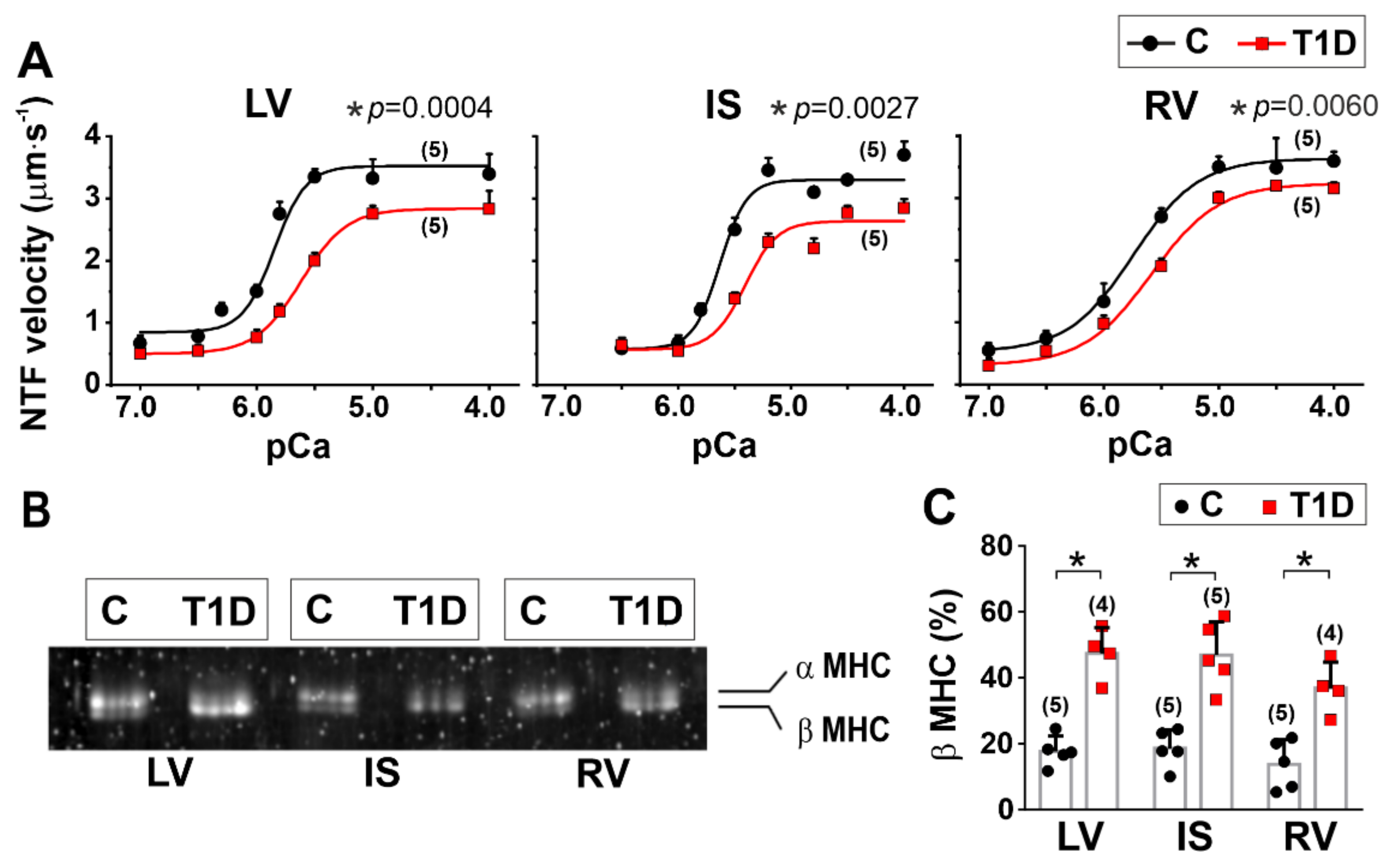
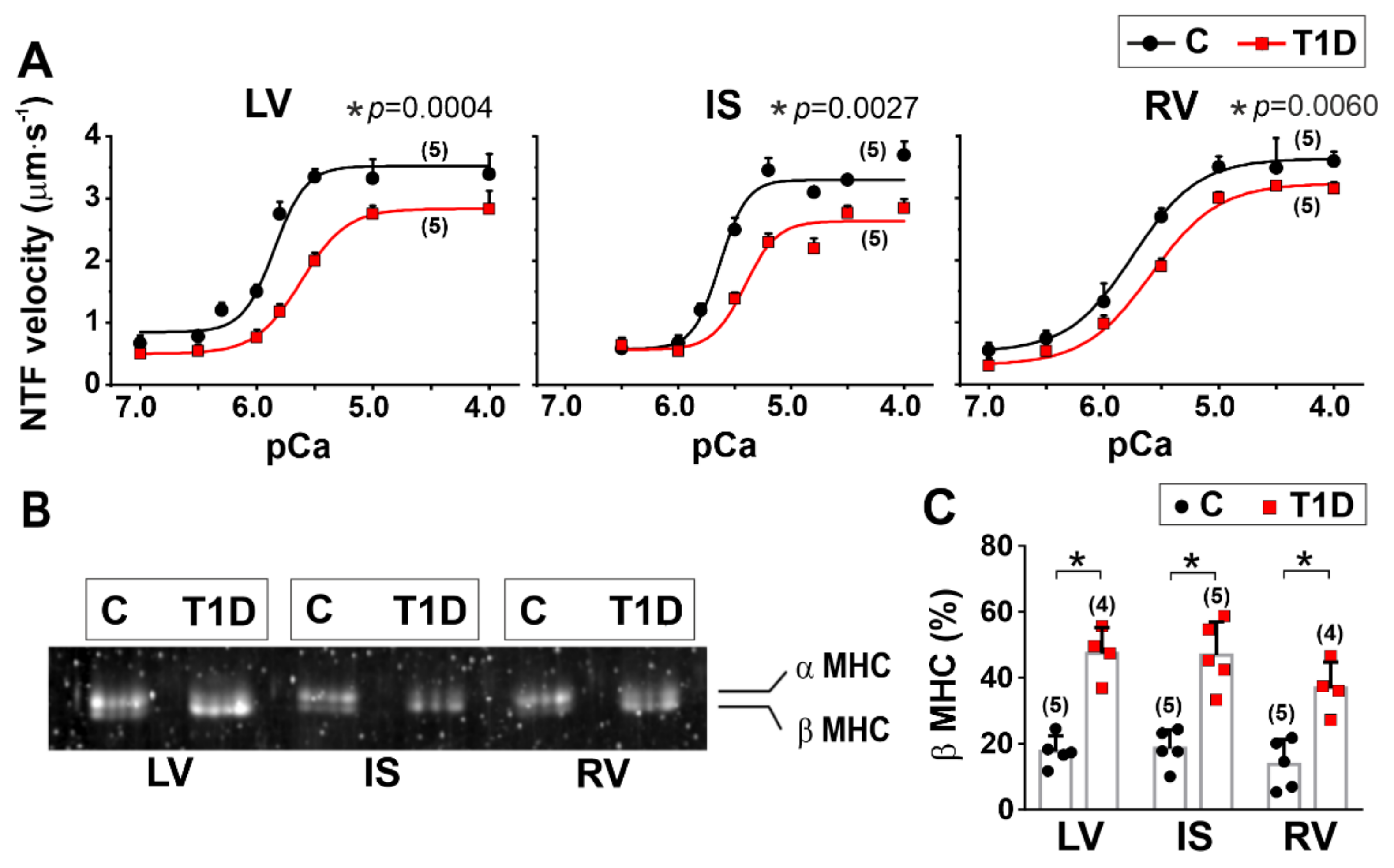
2.10. In Vitro Motility Assay
2.11. Statistics
3. Results
3.1. The Effects of Type 1 Diabetes on the Heart Function and Structure
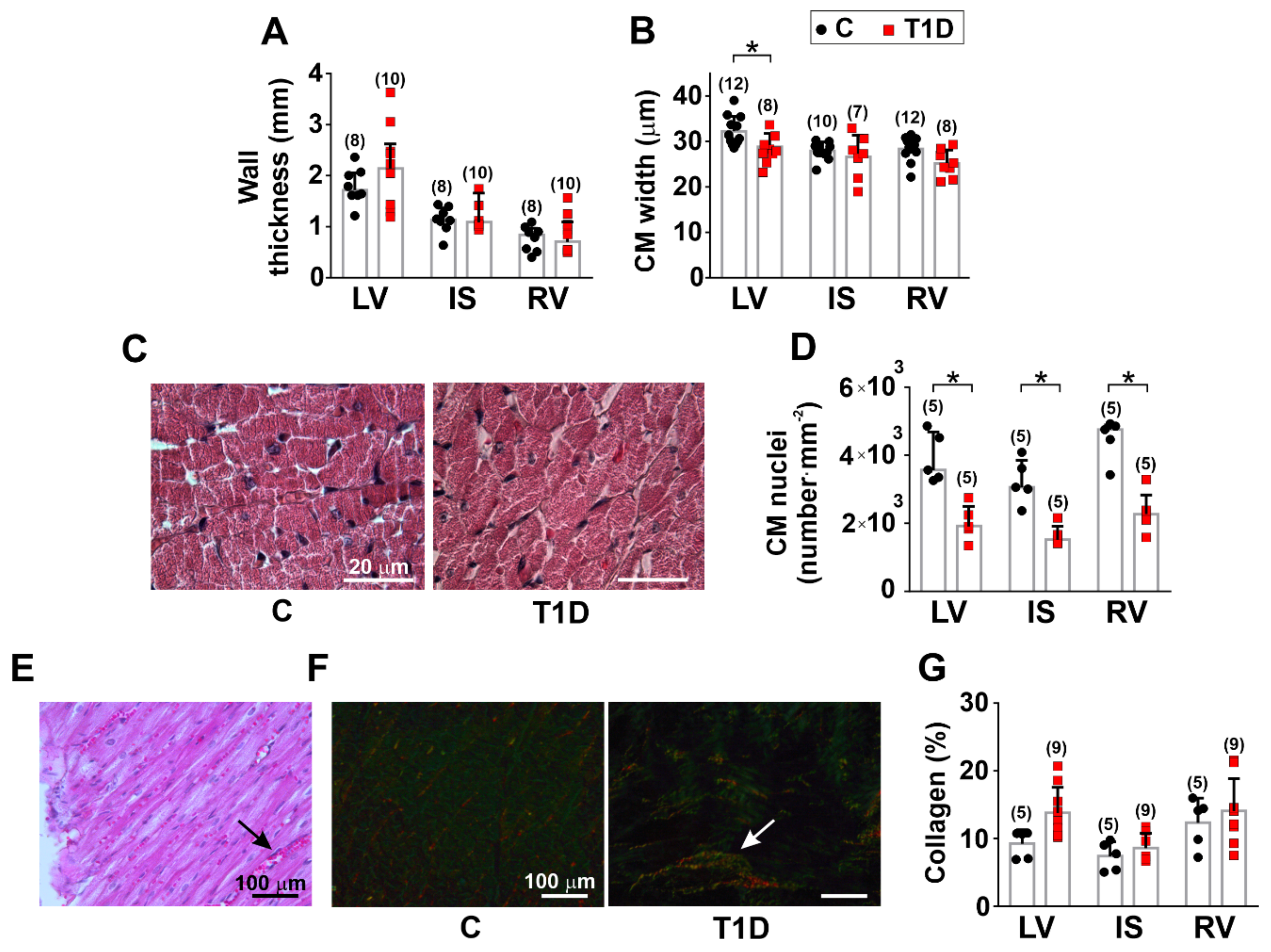
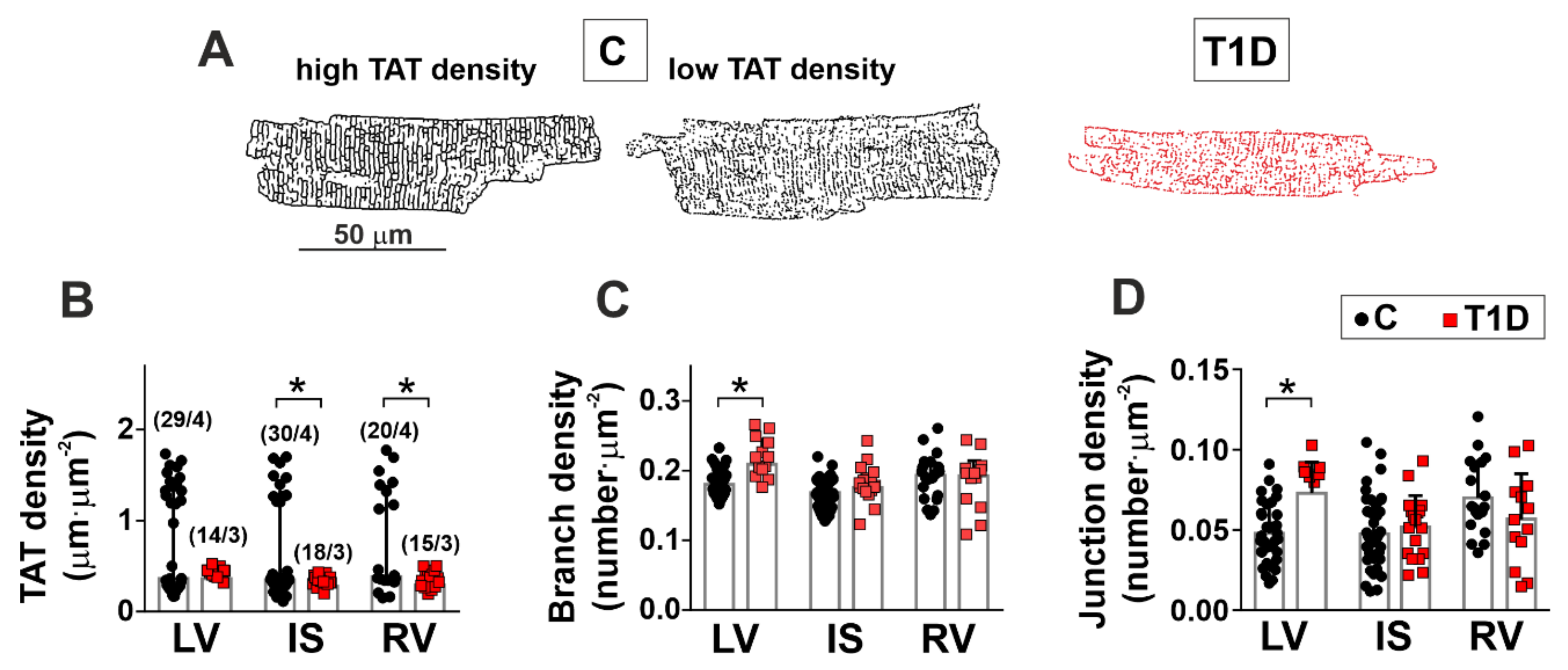
3.2. The Effects of Type 1 Diabetes on the Morphology of the Left and Right Ventricles
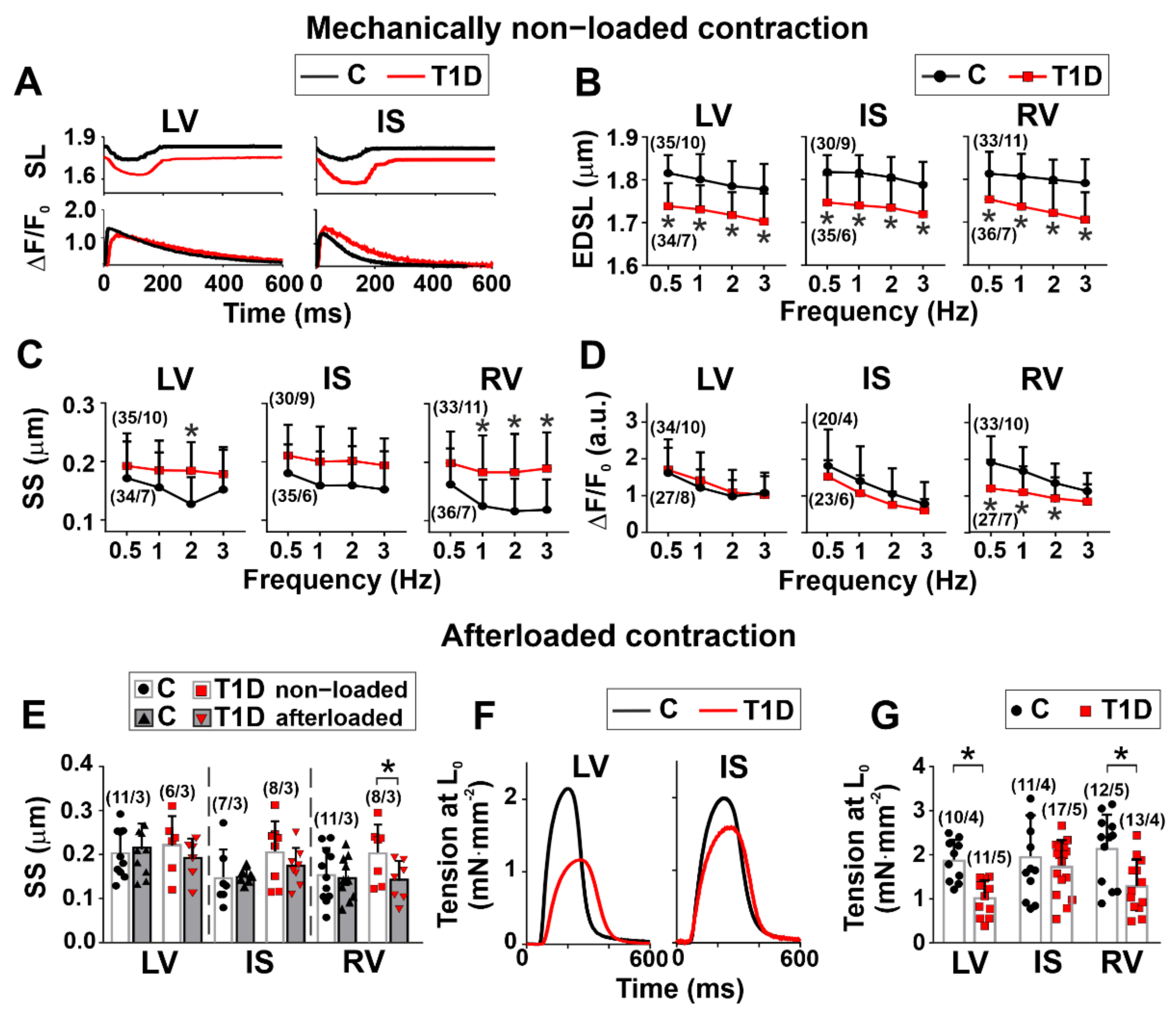
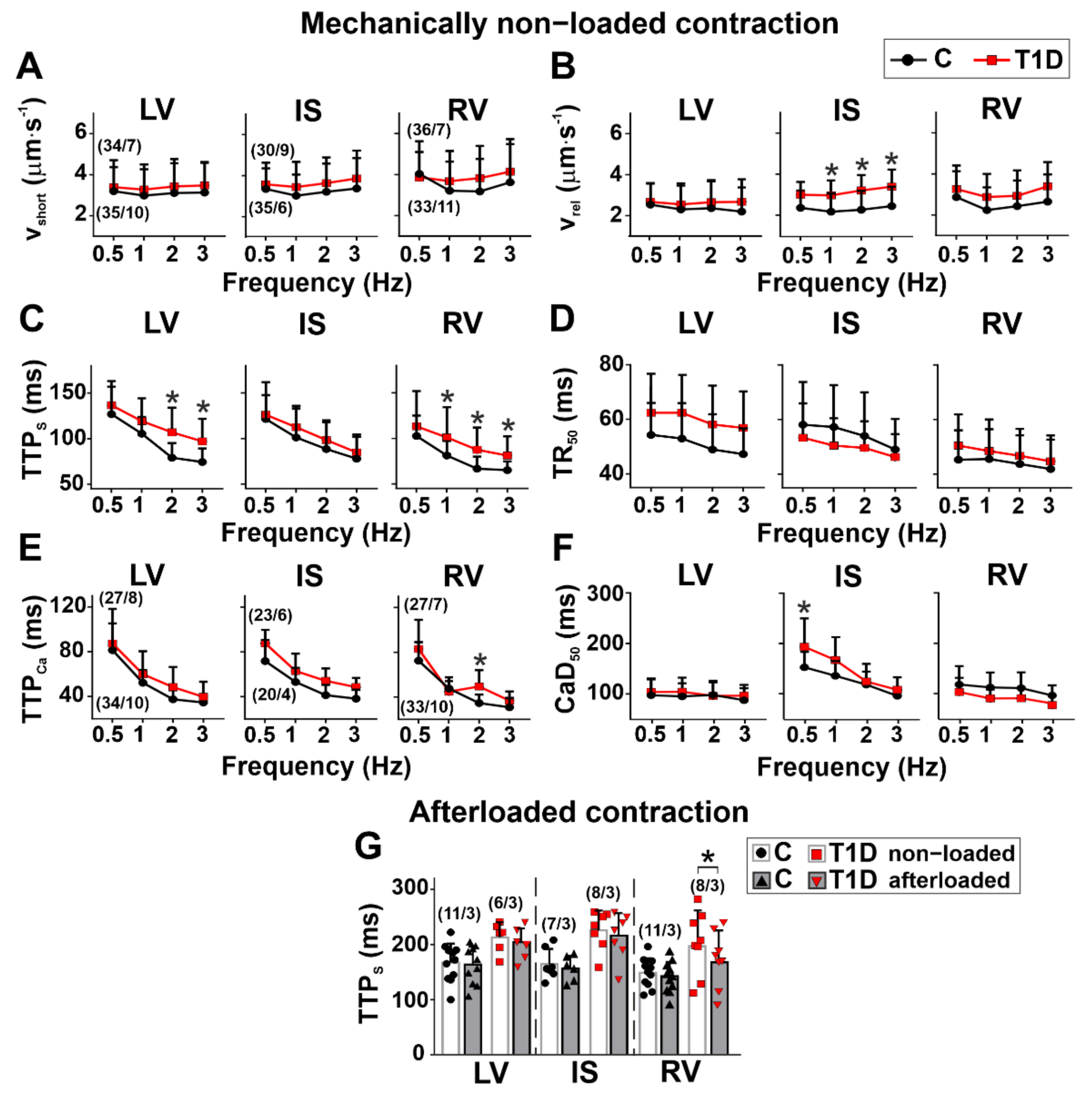
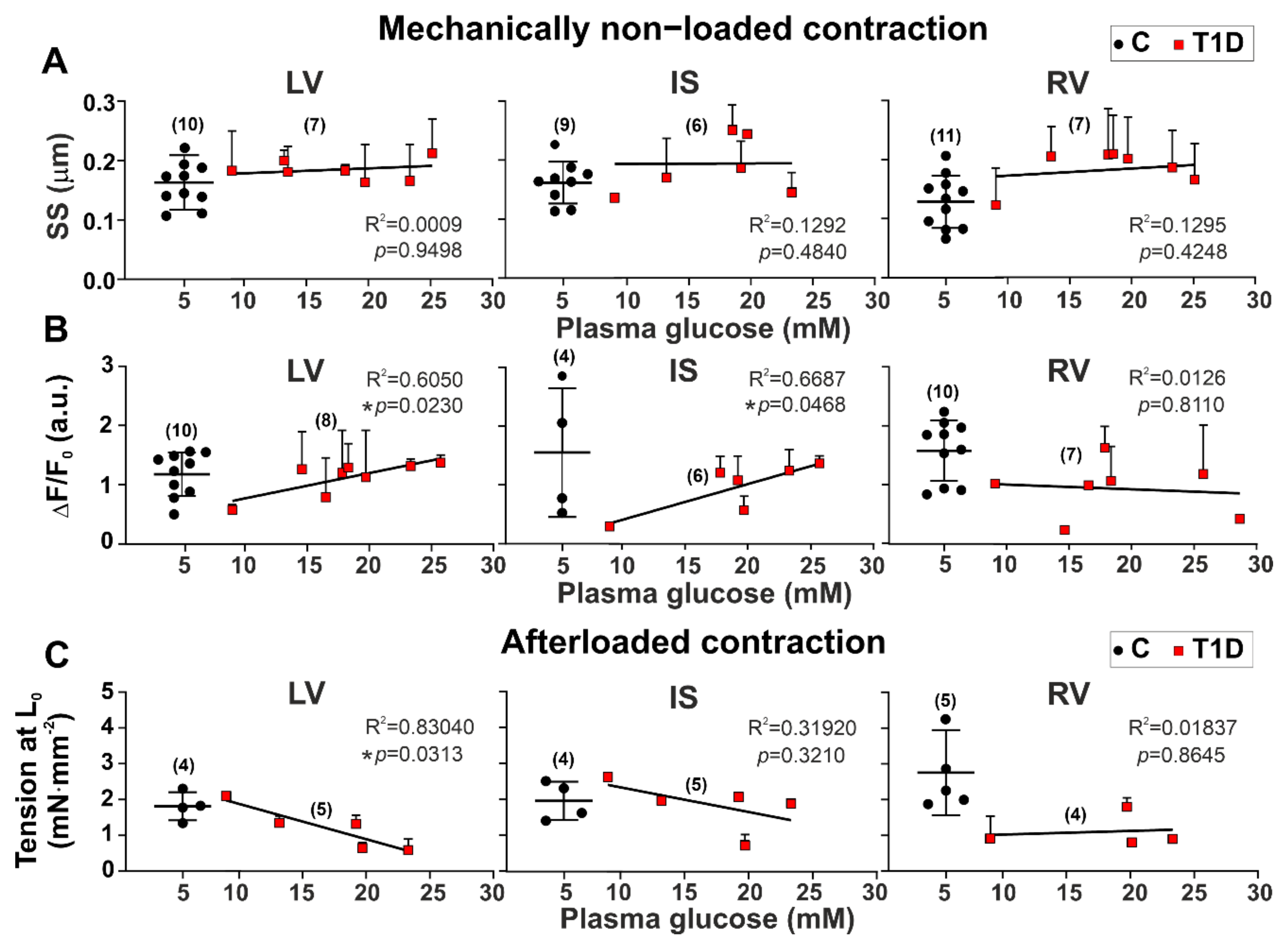
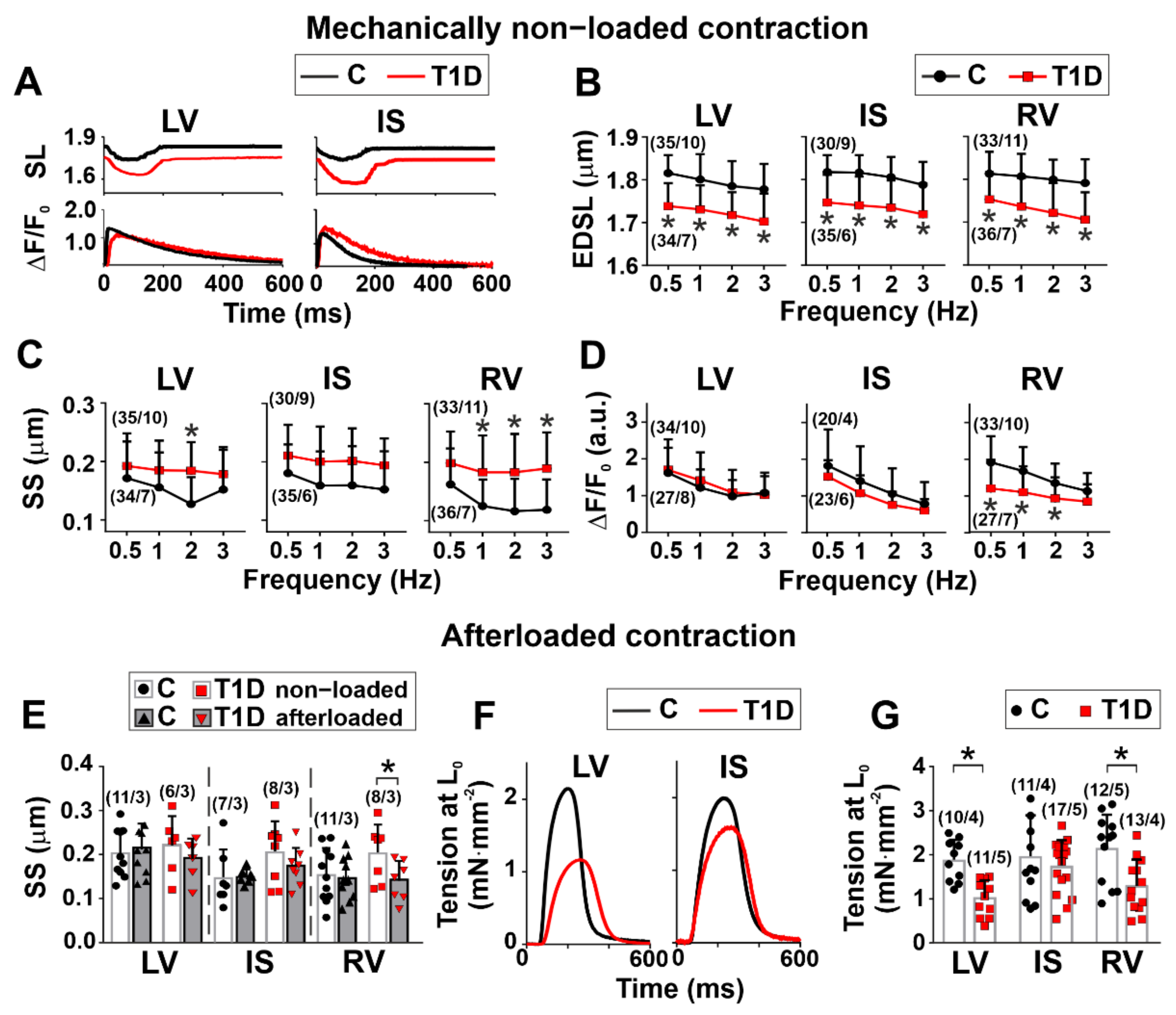
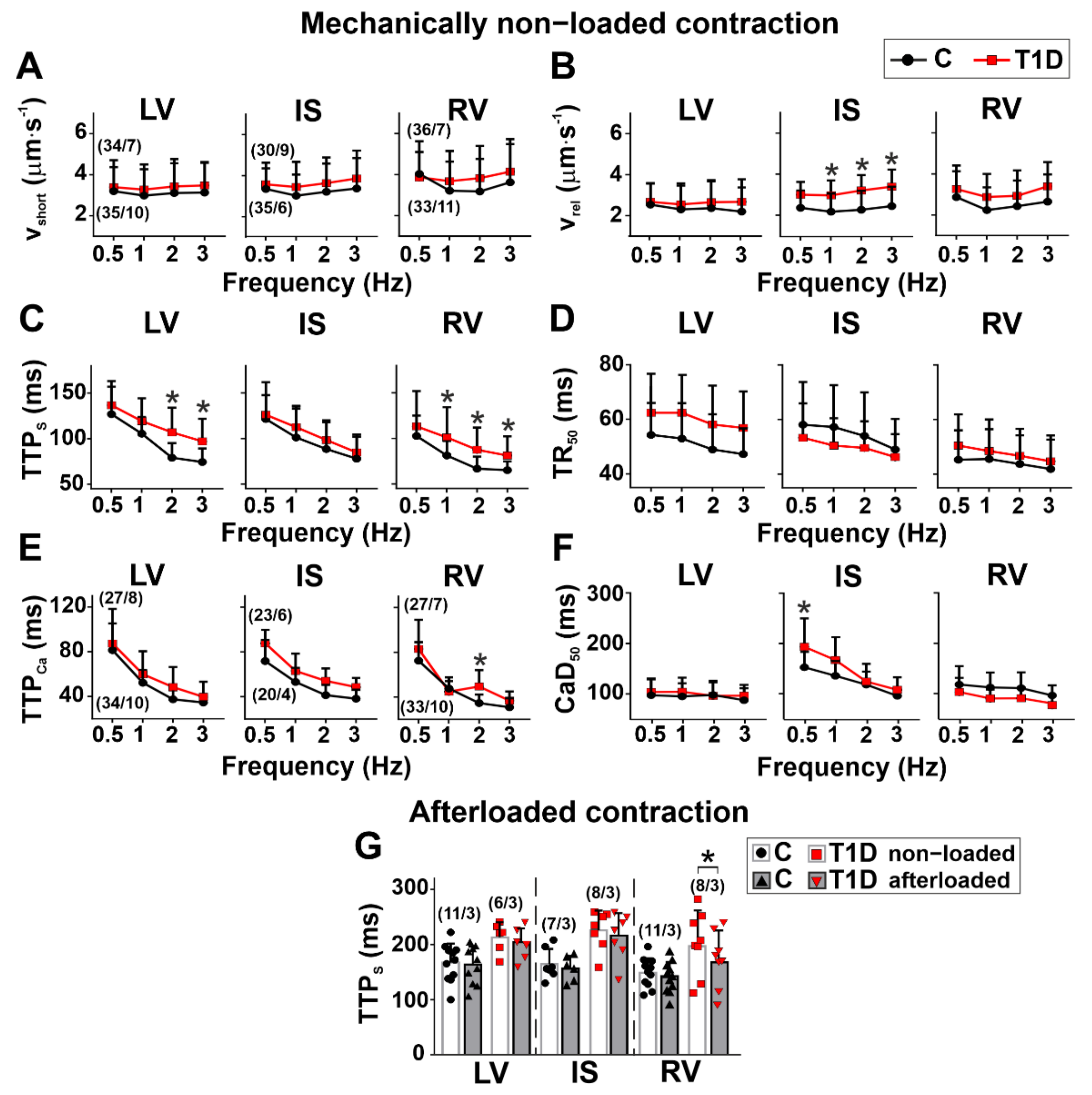
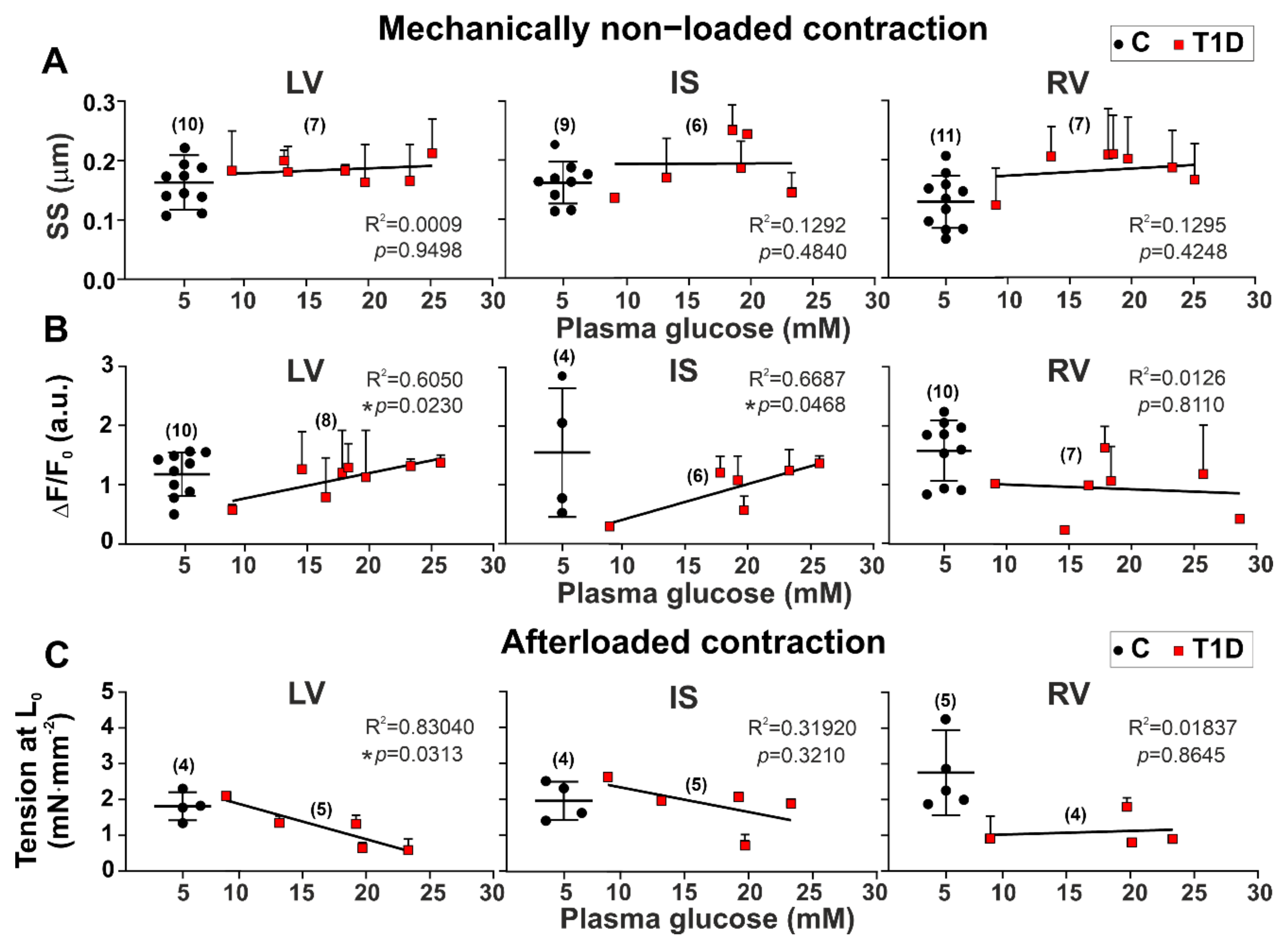
3.3. The Effects of Type 1 Diabetes on Contraction and [Ca2+]i Transients in Single Myocytes from the Left and Right Ventricles
3.4. The Effects of Type 1 Diabetes on Tension–Length and Velocity–Length Relationships in Single Myocytes from the Left and Right Ventricles
3.5. The Effects of Type 1 Diabetes on Actin-Myosin Interaction and Sarcomeric Protein Phosphorylation in the Left and Right Ventricles
4. Discussion
4.1. Interventricular Differences in the Healthy Heart
4.2. Interventricular Alterations Promoted by Type 1 Diabetes
5. Conclusions
6. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rawshani, A.; Sattar, N.; Franzén, S.; Rawshani, A.; Hattersley, A.T.; Svensson, A.-M.; Eliasson, B.; Gudbjörnsdottir, S. Excess mortality and cardiovascular disease in young adults with type 1 diabetes in relation to age at onset: A nationwide, register-based cohort study. Lancet 2018, 392, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Petrie, J.R.; Sattar, N. Excess Cardiovascular Risk in Type 1 Diabetes Mellitus: Role for a Dysfunctional Immune Response? Circulation 2019, 139, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Janeczko, D.; Czyzyk, A.; Kopczynski, J.; Krzyzanowski, M. Risk factors of cardiovascular death in diabetic patients. Diabet. Med. 1991, 8, S100–S103. [Google Scholar] [CrossRef]
- Mizamtsidi, M.; Paschou, S.A.; Grapsa, J.; Vryonidou, A. Diabetic cardiomyopathy: A clinical entity or a cluster of molecular heart changes? Eur. J. Clin. Investig. 2016, 46, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Tate, M.; Grieve, D.J.; Ritchie, R.H. Are targeted therapies for diabetic cardiomyopathy on the horizon? Clin. Sci. 2017, 131, 897–915. [Google Scholar] [CrossRef] [PubMed]
- De Blasio, M.J.; Huynh, N.; Deo, M.; Dubrana, L.E.; Walsh, J.; Willis, A.; Prakoso, D.; Kiriazis, H.; Donner, D.G.; Chatham, J.C. Defining the progression of diabetic cardiomyopathy in a mouse model of type 1 diabetes. Front. Physiol. 2020, 11, 124. [Google Scholar] [CrossRef] [Green Version]
- Basu, R.; Oudit, G.Y.; Wang, X.; Zhang, L.; Ussher, J.R.; Lopaschuk, G.D.; Kassiri, Z. Type 1 diabetic cardiomyopathy in the Akita (Ins2 WT/C96Y) mouse model is characterized by lipotoxicity and diastolic dysfunction with preserved systolic function. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H2096–H2108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [Green Version]
- Marwick, T.H.; Ritchie, R.; Shaw, J.E.; Kaye, D. Implications of underlying mechanisms for the recognition and management of diabetic cardiomyopathy. J. Am. Coll. Cardiol. 2018, 71, 339–351. [Google Scholar] [CrossRef]
- Damiano, R., Jr.; La Follette, P., Jr.; Cox, J.L.; Lowe, J.; Santamore, W. Significant left ventricular contribution to right ventricular systolic function. Am. J. Physiol. Heart Circ. Physiol. 1991, 261, H1514–H1524. [Google Scholar] [CrossRef]
- Kondo, R.P.; Dederko, D.A.; Teutsch, C.; Chrast, J.; Catalucci, D.; Chien, K.R.; Giles, W.R. Comparison of contraction and calcium handling between right and left ventricular myocytes from adult mouse heart: A role for repolarization waveform. J. Physiol. 2006, 571, 131–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, C.E.; Heijman, J.; Dobrev, D. Differences in left versus right ventricular electrophysiological properties in cardiac dysfunction and arrhythmogenesis. Arrhythmia Electrophysiol. Rev. 2016, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Meurs, K.M.; Lacombe, V.A.; Dryburgh, K.; Fox, P.R.; Reiser, P.R.; Kittleson, M.D. Differential expression of the cardiac ryanodine receptor in normal and arrhythmogenic right ventricular cardiomyopathy canine hearts. Hum. Genet. 2006, 120, 111–118. [Google Scholar] [CrossRef]
- Cadete, V.J.; Lin, H.b.; Sawicka, J.; Wozniak, M.; Sawicki, G. Proteomic analysis of right and left cardiac ventricles under aerobic conditions and after ischemia/reperfusion. Proteomics 2012, 12, 2366–2377. [Google Scholar] [CrossRef]
- Belin, R.J.; Sumandea, M.P.; Sievert, G.A.; Harvey, L.A.; Geenen, D.L.; Solaro, R.J.; de Tombe, P.P. Interventricular differences in myofilament function in experimental congestive heart failure. Pflügers Arch. Eur. J. Physiol. 2011, 462, 795–809. [Google Scholar] [CrossRef] [Green Version]
- Bódi, B.; Kovács, Á.; Gulyás, H.; Mártha, L.; Tóth, A.; Mátyás, C.; Barta, B.A.; Oláh, A.; Merkely, B.; Radovits, T. Long-Term PDE-5A Inhibition Improves Myofilament Function in Left and Right Ventricular Cardiomyocytes through Partially Different Mechanisms in Diabetic Rat Hearts. Antioxidants 2021, 10, 1776. [Google Scholar] [CrossRef]
- Schreckenberg, R.; Rebelo, M.; Deten, A.; Weber, M.; Rohrbach, S.; Pipicz, M.; Csonka, C.; Ferdinandy, P.; Schulz, R.; Schlüter, K.-D. Specific mechanisms underlying right heart failure: The missing upregulation of superoxide dismutase-2 and its decisive role in antioxidative defense. Antioxid. Redox Signal. 2015, 23, 1220–1232. [Google Scholar] [CrossRef]
- Hegyi, B.; Shimkunas, R.; Jian, Z.; Izu, L.T.; Bers, D.M.; Chen-Izu, Y. Mechanoelectric coupling and arrhythmogenesis in cardiomyocytes contracting under mechanical afterload in a 3D viscoelastic hydrogel. Proc. Natl. Acad. Sci. USA 2021, 118, e2108484118. [Google Scholar] [CrossRef]
- Kranstuber, A.; del Rio, C.; Biesiadecki, B.; Hamlin, R.; Ottobre, J.; Gyorke, S.; Lacombe, V.A. Advanced glycation end product cross-link breaker attenuates diabetes-induced cardiac dysfunction by improving sarcoplasmic reticulum calcium handling. Front. Physiol. 2012, 3, 292. [Google Scholar] [CrossRef] [Green Version]
- Szkudelski, T. The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol. Res. 2001, 50, 537–546. [Google Scholar]
- Danilova, I.; Sarapultsev, P.; Medvedeva, S.; Gette, I.; Bulavintceva, T.; Sarapultsev, A. Morphological restructuring of myocardium during the early phase of experimental diabetes mellitus. Anat. Rec. 2015, 298, 396–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danilova, I.; Medvedeva, S.; Shmakova, S.; Chereshneva, M.; Sarapultsev, A.; Sarapultsev, P. Pathological changes in the cellular structures of retina and choroidea in the early stages of alloxan-induced diabetes. World J. Diabetes 2018, 9, 239. [Google Scholar] [CrossRef] [PubMed]
- Konopelski, P.; Ufnal, M. Electrocardiography in rats: A comparison to human. Physiol. Res. 2016, 65, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Íñigo, L.; Navarro-González, D.; Fernández-Montero, A.; Pastrana-Delgado, J.; Martínez, J.A. The TyG index may predict the development of cardiovascular events. Eur. J. Clin. Investig. 2016, 46, 189–197. [Google Scholar] [CrossRef]
- Kiernan, J. Sirius Red Staining Protocol for Collagen. 2014. Available online: http://www.ihcworld.com/_protocols/special_stains/sirius_red.htm (accessed on 5 December 2021).
- Butova, X.; Myachina, T.; Khokhlova, A. A combined Langendorff-injection technique for simultaneous isolation of single cardiomyocytes from atria and ventricles of the rat heart. MethodsX 2021, 8, 101189. [Google Scholar] [CrossRef]
- Myachina, T.; Butova, K.; Lookin, O. Development and Program Implementation of an Algorithm to Estimate the Mean Sarcomere Length of a Cardiomyocyte. Biophysics 2019, 64, 732–737. [Google Scholar] [CrossRef]
- Khokhlova, A.; Konovalov, P.; Iribe, G.; Solovyova, O.; Katsnelson, L. The effects of mechanical preload on transmural differences in mechano-calcium-electric feedback in single cardiomyocytes: Experiments and mathematical models. Front. Physiol. 2020, 11, 171. [Google Scholar] [CrossRef]
- Volzhaninov, D.; Khokhlova, A. Parallel Control of Two Digital Micromanipulators for Biomechanical Experiments Using LabVIEW. In Proceedings of the 2019 Ural Symposium on Biomedical Engineering, Radioelectronics and Information Technology (USBEREIT), Yekaterinburg, Russia, 25−26 April 2019; pp. 167–170. [Google Scholar]
- Nishimura, S.; Yasuda, S.-i.; Katoh, M.; Yamada, K.P.; Yamashita, H.; Saeki, Y.; Sunagawa, K.; Nagai, R.; Hisada, T.; Sugiura, S. Single cell mechanics of rat cardiomyocytes under isometric, unloaded, and physiologically loaded conditions. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H196–H202. [Google Scholar] [CrossRef] [Green Version]
- Wagner, E.; Brandenburg, S.; Kohl, T.; Lehnart, S.E. Analysis of tubular membrane networks in cardiac myocytes from atria and ventricles. J. Vis. Exp. JoVE 2014, 92, e51823. [Google Scholar] [CrossRef]
- Margossian, S.S.; Lowey, S. [7] Preparation of myosin and its subfragments from rabbit skeletal muscle. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1982; Volume 85, pp. 55–71. [Google Scholar]
- Reiser, P.J.; Kline, W.O. Electrophoretic separation and quantitation of cardiac myosin heavy chain isoforms in eight mammalian species. Am. J. Physiol. Heart Circ. Physiol. 1998, 274, H1048–H1053. [Google Scholar] [CrossRef]
- Pardee, J.D.; Aspudich, J. [18] Purification of muscle actin. Methods Enzymol. 1982, 85, 164–181. [Google Scholar]
- Matyushenko, A.M.; Shchepkin, D.V.; Kopylova, G.V.; Popruga, K.E.; Artemova, N.V.; Pivovarova, A.V.; Bershitsky, S.Y.; Levitsky, D.I. Structural and functional effects of cardiomyopathy-causing mutations in the troponin T-binding region of cardiac tropomyosin. Biochemistry 2017, 56, 250–259. [Google Scholar] [CrossRef]
- Mashanov, G.; Molloy, J. Automatic detection of single fluorophores in live cells. Biophys. J. 2007, 92, 2199–2211. [Google Scholar] [CrossRef] [Green Version]
- Nollet, E.E.; Manders, E.M.; Goebel, M.; Jansen, V.; Brockmann, C.; Osinga, J.; van der Velden, J.; Helmes, M.; Kuster, D.W. Large-Scale Contractility Measurements Reveal Large Atrioventricular and Subtle Interventricular Differences in Cultured Unloaded Rat Cardiomyocytes. Front. Physiol. 2020, 11, 815. [Google Scholar] [CrossRef]
- Sathish, V.; Xu, A.; Karmazyn, M.; Sims, S.M.; Narayanan, N. Mechanistic basis of differences in Ca2+-handling properties of sarcoplasmic reticulum in right and left ventricles of normal rat myocardium. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H88–H96. [Google Scholar] [CrossRef] [Green Version]
- Schlüter, K.-D.; Kutsche, H.S.; Hirschhäuser, C.; Schreckenberg, R.; Schulz, R. Review on chamber-specific differences in right and left heart reactive oxygen species handling. Front. Physiol. 2018, 9, 1799. [Google Scholar] [CrossRef]
- Moss, R.L.; Fitzsimons, D.P.; Ralphe, J.C. Cardiac MyBP-C regulates the rate and force of contraction in mammalian myocardium. Circ. Res. 2015, 116, 183–192. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Liu, Y.; Sharma, V.; Dirksen, R.T.; Waugh, R.; Sheu, S.-S.; Min, W. ASK1 associates with troponin T and induces troponin T phosphorylation and contractile dysfunction in cardiomyocytes. Am. J. Pathol. 2003, 163, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Bollensdorff, C.; Lookin, O.; Kohl, P. Assessment of contractility in intact ventricular cardiomyocytes using the dimensionless ‘Frank–Starling Gain’index. Pflügers Arch. Eur. J. Physiol. 2011, 462, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.; Thai, K.; Park, K.W.; Wang, P.; Makwana, O.; Lovett, D.H.; Simpson, P.C.; Baker, A.J. Intraventricular and interventricular cellular heterogeneity of inotropic responses to α1-adrenergic stimulation. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H946–H953. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Qin, M.; Jiang, W.; Zhang, Y.; Liu, X. Electrophysiological characteristics of pressure overload-induced cardiac hypertrophy and its influence on ventricular arrhythmias. PLoS ONE 2017, 12, e0183671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlüter, K.-D. Ways to study the biology of cardiomyocytes. In Cardiomyocytes–Active Players in Cardiac Disease; Springer: Berlin/Heidelberg, Germany, 2016; pp. 3–23. [Google Scholar]
- Rouleau, J.-L.; Paradis, P.; Shenasa, H.; Juneau, C. Faster time to peak tension and velocity of shortening in right versus left ventricular trabeculae and papillary muscles of dogs. Circ. Res. 1986, 59, 556–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matyushenko, A.M.; Shchepkin, D.V.; Kopylova, G.V.; Bershitsky, S.Y.; Levitsky, D.I. Unique functional properties of slow skeletal muscle tropomyosin. Biochimie 2020, 174, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, B.; Li, L.; Eason, J.C.; Efimov, I.R.; Trayanova, N.A. Differences between left and right ventricular chamber geometry affect cardiac vulnerability to electric shocks. Circ. Res. 2005, 97, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.-Y.; McCloskey, D.T.; Turcato, S.; Swigart, P.M.; Simpson, P.C.; Baker, A.J. Contrasting inotropic responses to α1-adrenergic receptor stimulation in left versus right ventricular myocardium. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2013–H2017. [Google Scholar] [CrossRef] [Green Version]
- Cagalinec, M.; Waczulikova, I.; Ulicna, O.; Chorvat, D., Jr. Morphology and contractility of cardiac myocytes in early stages of streptozotocin-induced diabetes mellitus in rats. Physiol. Res. 2013, 62, 489. [Google Scholar] [CrossRef]
- Toepfer, C.; Caorsi, V.; Kampourakis, T.; Sikkel, M.B.; West, T.G.; Leung, M.-C.; Al-Saud, S.A.; MacLeod, K.T.; Lyon, A.R.; Marston, S.B. Myosin regulatory light chain (RLC) phosphorylation change as a modulator of cardiac muscle contraction in disease. J. Biol. Chem. 2013, 288, 13446–13454. [Google Scholar] [CrossRef] [Green Version]
- Sadayappan, S.; Osinska, H.; Klevitsky, R.; Lorenz, J.N.; Sargent, M.; Molkentin, J.D.; Seidman, C.E.; Seidman, J.G.; Robbins, J. Cardiac myosin binding protein C phosphorylation is cardioprotective. Proc. Natl. Acad. Sci. USA 2006, 103, 16918–16923. [Google Scholar] [CrossRef] [Green Version]
- Kopylova, G.V.; Matyushenko, A.M.; Berg, V.Y.; Levitsky, D.I.; Bershitsky, S.Y.; Shchepkin, D.V. Acidosis modifies effects of phosphorylated tropomyosin on the actin-myosin interaction in the myocardium. J. Muscle Res. Cell Motil. 2021, 42, 343–353. [Google Scholar] [CrossRef]
- Modesti, P.A.; Vanni, S.; Bertolozzi, I.; Cecioni, I.; Lumachi, C.; Perna, A.M.; Boddi, M.; Gensini, G.F. Different growth factor activation in the right and left ventricles in experimental volume overload. Hypertension 2004, 43, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Havlenova, T.; Skaroupkova, P.; Miklovic, M.; Behounek, M.; Chmel, M.; Jarkovska, D.; Sviglerova, J.; Stengl, M.; Kolar, M.; Novotny, J. Right versus left ventricular remodeling in heart failure due to chronic volume overload: Pressure-volume and proteome analysis. Sci. Rep. 2021, 11, 17136. [Google Scholar] [CrossRef]
- Sequeira, V.; Nijenkamp, L.L.; Regan, J.A.; van der Velden, J. The physiological role of cardiac cytoskeleton and its alterations in heart failure. Biochim. Biophys. Acta Biomembr. 2014, 1838, 700–722. [Google Scholar] [CrossRef] [Green Version]
- Louch, W.E.; Bito, V.; Heinzel, F.R.; Macianskiene, R.; Vanhaecke, J.; Flameng, W.; Mubagwa, K.; Sipido, K.R. Reduced synchrony of Ca2+ release with loss of T-tubules—a comparison to Ca2+ release in human failing cardiomyocytes. Cardiovasc. Res. 2004, 62, 63–73. [Google Scholar] [CrossRef]
- Gadeberg, H.C.; Bond, R.C.; Kong, C.H.; Chanoit, G.P.; Ascione, R.; Cannell, M.B.; James, A.F. Heterogeneity of T-tubules in pig hearts. PLoS ONE 2016, 11, e0156862. [Google Scholar] [CrossRef] [Green Version]
- Canton, M.; Menazza, S.; Sheeran, F.L.; Polverino de Laureto, P.; Di Lisa, F.; Pepe, S. Oxidation of myofibrillar proteins in human heart failure. J. Am. Coll. Cardiol. 2011, 57, 300–309. [Google Scholar] [CrossRef]
- Toya, T.; Ito, K.; Kagami, K.; Osaki, A.; Sato, A.; Kimura, T.; Horii, S.; Yasuda, R.; Namba, T.; Ido, Y. Impact of oxidative posttranslational modifications of SERCA2 on heart failure exacerbation in young patients with non-ischemic cardiomyopathy: A pilot study. IJC Heart Vasc. 2020, 26, 100437. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Origin of Myosin | vF-actin (µm·s−1) | vmax (µm·s−1) | v0 (µm·s−1) | pCa50 | h | |
|---|---|---|---|---|---|---|
| LV | C | 3.36 (0.21) | 3.52 (0.10) | 0.84 (0.08) | 5.85 (0.05) | 1.4 (0.5) |
| T1D | 2.10 (0.08) * (p = 0.006) | 2.83 (0.05) * (p = 0.0004) | 0.50 (0.02) * (p = 0.0020) | 5.62 (0.01) * (p = 0.0014) | 1.1 (0.2) (p = 0.3892) | |
| IS | C | 3.09 (0.11) | 3.30 (0.03) | 0.58 (0.04) | 5.63 (0.01) | 1.7 (0.4) |
| T1D | 2.35 (0.14) * (p = 0.0020) | 2.68 (0.16) * (p = 0.0027) | 0.56 (0.13) (p = 0.8115) | 5.42 (0.02) (p < 0.0001) | 1.2 (0.2) (p = 0.1249) | |
| RV | C | 3.03 (0.22) | 3.63 (0.05) | 0.53 (0.04) | 5.75(0.03) | 1.1 (0.2) |
| T1D | 2.23 (0.15) * (p = 0.0065) | 3.23 (0.12) * (p = 0.0060) | 0.31 (0.09) * (p = 0.0180) | 5.59 (0.01) * (p = 0.0009) | 1.1 (0.1) (p > 0.9999) | |
| Characteristics | LV | IS | RV |
|---|---|---|---|
| structural features | |||
| wall thickness | – | – | – |
| collagen content | small foci of interstitial fibrosis | ||
| CM width | ↓ | – | – |
| CM length | – | – | – |
| CM nuclei density | ↓ | ↓ | ↓ |
| TAT network density | – | ↓ | ↓ |
| TAT network branch density | ↑ | – | – |
| TAT network junction density | ↑ | – | – |
| contractile function of single CM | |||
| mechanically non-loaded CM | |||
| EDSL | ↓ | ↓ | ↓ |
| Sarcomere-shortening amplitude | ↑ | – | ↑ |
| TTPS | ↑ | – | ↑ |
| TR50 | – | – | – |
| [Ca2+]i transient amplitude | – | – | ↓ |
| TTPCa | – | – | ↑ |
| CaD50 | – | ↑ (0.5 Hz) | – |
| afterloaded/preloaded CM at 1 Hz | |||
| afterload affects sarcomere-shortening amplitude and TTPS | – | – | yes |
| auxotonic cell tension at L0 | ↓ | – | ↓ |
| EDTLR slope | ↓ | ↓ | ↓ |
| ESTLR slope | ↓ | ↓ | ↓ |
| ATLR slope | ↓ | – | ↓ |
| vshortLR slope | ↓ | – | ↓ |
| vrelLR slope | ↓ | – | ↓ |
| myofilament function | |||
| maximum NTF sliding velocity over myosin | ↓ | ↓ | ↓ |
| Ca2+-sensitivity of pCa-NTF velocity dependence. | ↓ | ↓ | ↓ |
| maximum NTF fraction | ↓ | – | – |
| TnT phosphorylation | ↑ | ↑ | ↓ |
| TnI phosphorylation | – | – | – |
| Tpm phosphorylation | ↑ | – | – |
| cMyBP-C phosphorylation | ↑ | – | – |
| RLC phosphorylation | ↓ | – | – |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khokhlova, A.; Myachina, T.; Volzhaninov, D.; Butova, X.; Kochurova, A.; Berg, V.; Gette, I.; Moroz, G.; Klinova, S.; Minigalieva, I.; et al. Type 1 Diabetes Impairs Cardiomyocyte Contractility in the Left and Right Ventricular Free Walls but Preserves It in the Interventricular Septum. Int. J. Mol. Sci. 2022, 23, 1719. https://doi.org/10.3390/ijms23031719
Khokhlova A, Myachina T, Volzhaninov D, Butova X, Kochurova A, Berg V, Gette I, Moroz G, Klinova S, Minigalieva I, et al. Type 1 Diabetes Impairs Cardiomyocyte Contractility in the Left and Right Ventricular Free Walls but Preserves It in the Interventricular Septum. International Journal of Molecular Sciences. 2022; 23(3):1719. https://doi.org/10.3390/ijms23031719
Chicago/Turabian StyleKhokhlova, Anastasia, Tatiana Myachina, Denis Volzhaninov, Xenia Butova, Anastasia Kochurova, Valentina Berg, Irina Gette, Gleb Moroz, Svetlana Klinova, Ilzira Minigalieva, and et al. 2022. "Type 1 Diabetes Impairs Cardiomyocyte Contractility in the Left and Right Ventricular Free Walls but Preserves It in the Interventricular Septum" International Journal of Molecular Sciences 23, no. 3: 1719. https://doi.org/10.3390/ijms23031719
APA StyleKhokhlova, A., Myachina, T., Volzhaninov, D., Butova, X., Kochurova, A., Berg, V., Gette, I., Moroz, G., Klinova, S., Minigalieva, I., Solovyova, O., Danilova, I., Sokolova, K., Kopylova, G., & Shchepkin, D. (2022). Type 1 Diabetes Impairs Cardiomyocyte Contractility in the Left and Right Ventricular Free Walls but Preserves It in the Interventricular Septum. International Journal of Molecular Sciences, 23(3), 1719. https://doi.org/10.3390/ijms23031719