
PCD Genes—From Patients to Model Organisms and Back to Humans

 , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction—Cilia Diversity
2. Single-Celled Models—The Power of Small and Simple
2.1. Green Alga, Chlamydomonas reinhardtii
2.2. Parasitic Protists, Trypanosoma spp.
2.3. Ciliate: Tetrahymena and Paramecium
3. Freshwater Planarian Schmidtea mediterranea—Matters Becoming a Little More Complicated
4. Aquatic Vertebrates–When a Plethora of Siblings and Fast Development Matter
5. Mice—Blood Is Thicker than Water—When Being a Mammal Matters
6. In Vitro Cell Culture
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 2019, 15, 199–219. [Google Scholar] [CrossRef] [PubMed]
- Ford, M.J.; Yeyati, P.L.; Mali, G.R.; Keighren, M.A.; Waddell, S.H.; Mjoseng, H.K.; Douglas, A.T.; Hall, E.A.; Sakaue-Sawano, A.; Miyawaki, A.; et al. A Cell/Cilia Cycle Biosensor for Single-Cell Kinetics Reveals Persistence of Cilia after G1/S Transition Is a General Property in Cells and Mice. Dev. Cell 2018, 47, 509–523.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wensel, T.G.; Zhang, Z.; Anastassov, I.A.; Gilliam, J.C.; He, F.; Schmid, M.F.; Robichaux, M.A. Structural and molecular bases of rod photoreceptor morphogenesis and disease. Prog. Retin. Eye Res. 2016, 55, 32–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wensel, T.G.; Potter, V.L.; Moye, A.; Zhang, Z.; Robichaux, M.A. Structure and dynamics of photoreceptor sensory cilia. Pflugers Arch. Eur. J. Physiol. 2021, 473, 1517–1537. [Google Scholar] [CrossRef] [PubMed]
- Falk, N.; Lösl, M.; Schröder, N.; Gießl, A. Specialized cilia in mammalian sensory systems. Cells 2015, 4, 500–519. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zhou, J. The Kinocilia of Cochlear Hair Cells: Structures, Functions, and Diseases. Front. Cell Dev. Biol. 2021, 9, 2134. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, P.M.; Mcewen, D.P.; Martens, J.R. Olfactory cilia: Linking sensory cilia function and human disease. Chem. Senses 2009, 34, 451–464. [Google Scholar] [CrossRef] [Green Version]
- Breeze, R.G.; Wheeldon, E.B. The cells of the pulmonary airways. Am. Rev. Respir. Dis. 1977, 116, 705–777. [Google Scholar] [CrossRef]
- Jafek, B.W. Ultrastructure of human nasal mucosa. Laryngoscope 1983, 93, 1576–1599. [Google Scholar] [CrossRef]
- Bustamante-Marin, X.M.; Ostrowski, L.E. Cilia and mucociliary clearance. Cold Spring Harb. Perspect. Biol. 2017, 9, 1–17. [Google Scholar] [CrossRef]
- Rodriguez, K.; Gaston, B.; Wasman, J.; Marozkina, N. Lessons From Unilateral Loss of Cilia: Early Nasal Nitric Oxide Gas Mixing and the Role of Sinus Patency in Determining Nasal Nitric Oxide. Clin. Med. Insights Ear Nose Throat 2017, 10, 117955061774636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzadeh, Z.; Kusne, Y.; Duran-Moreno, M.; Cabrales, E.; Gil-Perotin, S.; Ortiz, C.; Chen, B.; Garcia-Verdugo, J.M.; Sanai, N.; Alvarez-Buylla, A. Bi- and uniciliated ependymal cells define continuous floor-plate-derived tanycytic territories. Nat. Commun. 2017, 8, 13759. [Google Scholar] [CrossRef] [Green Version]
- Lorencova, M.; Mitro, A.; Jurikova, M.; Galfiova, P.; Mikusova, R.; Krivosikova, L.; Janegova, A.; Palkovic, M.; Polak, S. Ependymal cells surface of human third brain ventricle by scanning electron microscopy. Bratislava Med. J. 2020, 121, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Bruni, J.E.; Montemurro, D.G.; Clattenburg, R.E.; Singh, R.P. A scanning electron microscopic study of the ependymal surface of the third ventricle of the rabbit, rat, mouse and human brain. Anat. Rec. 1972, 174, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Worthington, W.C.; Cathcart, R.S. Ependymal cilia: Distribution and activity in the adult human brain. Science 1963, 139, 221–222. [Google Scholar] [CrossRef]
- Lyons, R.A.; Saridogan, E.; Djahanbakhch, O. The reproductive significance of human Fallopian tube cilia. Hum. Reprod. Update 2006, 12, 363–372. [Google Scholar] [CrossRef]
- Wang, S.; Burton, J.C.; Behringer, R.R.; Larina, I. V In vivo micro-scale tomography of ciliary behavior in the mammalian oviduct. Sci. Rep. 2015, 5, 13216. [Google Scholar] [CrossRef] [Green Version]
- Hoque, M.; Chen, D.; Hess, R.A.; Li, F.Q.; Takemaru, K.I. CEP164 is essential for efferent duct multiciliogenesis and male fertility. Reproduction 2021, 162, 129–139. [Google Scholar] [CrossRef]
- Yuan, S.; Liu, Y.; Peng, H.; Tang, C.; Hennig, G.W.; Wang, Z.; Wang, L.; Yu, T.; Klukovich, R.; Zhang, Y.; et al. Motile cilia of the male reproductive system require miR-34/miR-449 for development and function to generate luminal turbulence. Proc. Natl. Acad. Sci. USA. 2019, 116, 3584–3593. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Wu, H.; Zhu, D.; Wang, Y.; Shen, Q.; Cheng, H.; Zhang, J.; Geng, H.; Liu, Y.; He, X.; et al. Bi-allelic mutations in MCIDAS and CCNO cause human infertility associated with abnormal gamete transport. Clin. Genet. 2021, 100, 731–742. [Google Scholar] [CrossRef]
- Terré, B.; Lewis, M.; Gil-Gómez, G.; Han, Z.; Lu, H.; Aguilera, M.; Prats, N.; Roy, S.; Zhao, H.; Stracker, T.H. Defects in efferent duct multiciliogenesis underlie male infertility in GEMC1-, MCIDAS- or CCNO-deficient mice. Development 2019, 146, dev162628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dougherty, G.W.; Mizuno, K.; Nöthe-Menchen, T.; Ikawa, Y.; Boldt, K.; Ta-Shma, A.; Aprea, I.; Minegishi, K.; Pang, Y.P.; Pennekamp, P.; et al. CFAP45 deficiency causes situs abnormalities and asthenospermia by disrupting an axonemal adenine nucleotide homeostasis module. Nat. Commun. 2020, 11, 5520. [Google Scholar] [CrossRef] [PubMed]
- Aprea, I.; Nöthe-Menchen, T.; Dougherty, G.W.; Raidt, J.; Loges, N.T.; Kaiser, T.; Wallmeier, J.; Olbrich, H.; Strünker, T.; Kliesch, S.; et al. Motility of efferent duct cilia aids passage of sperm cells through the male reproductive system. Mol. Hum. Reprod. 2021, 27, 1–15. [Google Scholar] [CrossRef]
- Nonaka, S.; Tanaka, Y.; Okada, Y.; Takeda, S.; Harada, A.; Kanai, Y.; Kido, M.; Hirokawa, N. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell 1998, 95, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Wallmeier, J.; Nielsen, K.G.; Kuehni, C.E.; Lucas, J.S.; Leigh, M.W.; Zariwala, M.A.; Omran, H. Motile ciliopathies. Nat. Rev. Dis. Prim. 2020, 6, 77. [Google Scholar] [CrossRef]
- Serres, C.; Escalier, D.; David, G. Ultrastructural morphometry of the human sperm flagellum with a stereological analysis of the lengths of the dense fibres. Biol. Cell 1983, 49, 153–161. [Google Scholar] [CrossRef]
- Touré, A.; Martinez, G.; Kherraf, Z.E.; Cazin, C.; Beurois, J.; Arnoult, C.; Ray, P.F.; Coutton, C. The genetic architecture of morphological abnormalities of the sperm tail. Hum. Genet. 2021, 140, 21–42. [Google Scholar] [CrossRef]
- Shinohara, K.; Hamada, H. Cilia in left–right symmetry breaking. Cold Spring Harb. Perspect. Biol. 2017, 9, 1–9. [Google Scholar] [CrossRef]
- Gonçalves, J.; Pelletier, L. The ciliary transition zone: Finding the pieces and assembling the gate. Mol. Cells 2017, 40, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef]
- Kiesel, P.; Alvarez Viar, G.; Tsoy, N.; Maraspini, R.; Gorilak, P.; Varga, V.; Honigmann, A.; Pigino, G. The molecular structure of mammalian primary cilia revealed by cryo-electron tomography. Nat. Struct. Mol. Biol. 2020, 27, 1115–1124. [Google Scholar] [CrossRef]
- Osinka, A.; Poprzeczko, M.; Zielinska, M.M.; Fabczak, H.; Joachimiak, E.; Wloga, D. Ciliary Proteins: Filling the Gaps. Recent Advances in Deciphering the Protein Composition of Motile Ciliary Complexes. Cells 2019, 8, 730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuser, T.; Barber, C.F.; Lin, J.; Krell, J.; Rebesco, M.; Porter, M.E.; Nicastro, D. Cryoelectron tomography reveals doublet-specific structures and unique interactions in the I1 dynein. Proc. Natl. Acad. Sci. USA 2012, 109, E2067–E2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dymek, E.E.; Heuser, T.; Nicastro, D.; Smith, E.F. The CSC is required for complete radial spoke assembly and wild-type ciliary motility. Mol. Biol. Cell 2011, 22, 2520–2531. [Google Scholar] [CrossRef] [PubMed]
- Heuser, T.; Dymek, E.E.; Lin, J.; Smith, E.F.; Nicastro, D. The CSC connects three major axonemal complexes involved in dynein regulation. Mol. Biol. Cell 2012, 23, 3143–3155. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, R.; Song, K.; Yanagisawa, H.A.; Fox, L.; Yagi, T.; Wirschell, M.; Hirono, M.; Kamiya, R.; Nicastro, D.; Sale, W.S. The MIA complex is a conserved and novel dynein regulator essential for normal ciliary motility. J. Cell Biol. 2013, 201, 263–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazan, R.; Schröfel, A.; Joachimiak, E.; Poprzeczko, M.; Pigino, G.; Wloga, D. Ccdc113/Ccdc96 complex, a novel regulator of ciliary beating that connects radial spoke 3 to dynein g and the nexin link. PLoS Genet. 2021, 17, e1009388. [Google Scholar] [CrossRef]
- Samsel, Z.; Sekretarska, J.; Osinka, A.; Wloga, D.; Joachimiak, E. Central apparatus, the molecular kickstarter of ciliary and flagellar nanomachines. Int. J. Mol. Sci. 2021, 22, 3013. [Google Scholar] [CrossRef]
- Sun, S.; Fisher, R.L.; Bowser, S.S.; Pentecost, B.T.; Sui, H. Three-dimensional architecture of epithelial primary cilia. Proc. Natl. Acad. Sci. USA. 2019, 116, 9370–9379. [Google Scholar] [CrossRef] [Green Version]
- Flock, A.; Duvall, A.J. The ultrastructure of the kinocilium of the sensory cells in the inner ear and lateral line organs. J. Cell Biol. 1965, 25, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Sobkowicz, H.M.; Slapnick, S.M.; August, B.K. The kinocilium of auditory hair cells and evidence for its morphogenetic role during the regeneration of stereocilia and cuticular plates. J. Neurocytol. 1995, 24, 633–653. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Takasaka, T.; Tonosaki, A.; Watanabe, H. Fine structure of Guinea pig vestibular kinocilium. Acta Otolaryngol. 1989, 108, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.S.; Yehuda, B.S.; Moninger, T.O.; Kline, J.N.; Welsh, M.J. Motile cilia of human airway epithelia are chemosensory. Science 2009, 325, 1131–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, R.; Javidan-Nejad, C.; Alexander-Brett, J.; Horani, A.; Cabellon, M.C.; Walter, M.J.; Brody, S.L. Sensory functions of motile cilia and implication for bronchiectasis. Front. Biosci.-Sch. 2012, 4, 1088–1098. [Google Scholar] [CrossRef] [Green Version]
- Mitchison, H.M.; Valente, E.M. Motile and non-motile cilia in human pathology: From function to phenotypes. J. Pathol. 2017, 241, 294–309. [Google Scholar] [CrossRef]
- Ringers, C.; Olstad, E.W.; Jurisch-Yaksi, N. The role of motile cilia in the development and physiology of the nervous system. Philos. Trans. R. Soc. B Biol. Sci. 2019, 375, 20190156. [Google Scholar] [CrossRef] [Green Version]
- Legendre, M.; Zaragosi, L.E.; Mitchison, H.M. Motile cilia and airway disease. Semin. Cell Dev. Biol. 2020, 110, 19–33. [Google Scholar] [CrossRef]
- Bustamante-Marin, X.M.; Yin, W.N.; Sears, P.R.; Werner, M.E.; Brotslaw, E.J.; Mitchell, B.J.; Jania, C.M.; Zeman, K.L.; Rogers, T.D.; Herring, L.E.; et al. Lack of GAS2L2 Causes PCD by Impairing Cilia Orientation and Mucociliary Clearance. Am. J. Hum. Genet. 2019, 104, 229–245. [Google Scholar] [CrossRef] [Green Version]
- Little, R.B.; Norris, D.P. Right, left and cilia: How asymmetry is established. Semin. Cell Dev. Biol. 2021, 110, 11–18. [Google Scholar] [CrossRef]
- Brennan, S.K.; Ferkol, T.W.; Davis, S.D. Emerging genotype-phenotype relationships in primary ciliary dyskinesia. Int. J. Mol. Sci. 2021, 22, 8272. [Google Scholar] [CrossRef]
- Focsa, I.O.; Budistenau, M.; Bǎlgrǎdean, M. Clinical and genetic heterogeneity of primary ciliopathies (Review). Int. J. Mol. Med. 2021, 48, 176. [Google Scholar] [CrossRef] [PubMed]
- Tobin, J.L.; Beales, P.L. The nonmotile ciliopathies. Genet. Med. 2009, 11, 386–402. [Google Scholar] [CrossRef] [Green Version]
- Horani, A.; Ferkol, T.W. Advances in the Genetics of Primary Ciliary Dyskinesia: Clinical Implications. Chest 2018, 154, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Antony, D.; Brunner, H.G.; Schmidts, M. Ciliary dyneins and dynein related ciliopathies. Cells 2021, 10, 1885. [Google Scholar] [CrossRef] [PubMed]
- Poprzeczko, M.; Bicka, M.; Farahat, H.; Bazan, R.; Osinka, A.; Fabczak, H.; Joachimiak, E.; Wloga, D. Rare Human Diseases: Model Organisms in Deciphering the Molecular Basis of Primary Ciliary Dyskinesia. Cells 2019, 8, 1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, M.; Stracker, T.H. Transcriptional regulation of multiciliated cell differentiation. Semin. Cell Dev. Biol. 2021, 110, 51–60. [Google Scholar] [CrossRef]
- Carvalho-Santos, Z.; Azimzadeh, J.; Pereira-Leal, J.B.; Bettencourt-Dias, M. Tracing the origins of centrioles, cilia, and flagella. J. Cell Biol. 2011, 194, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Scholey, J.M. Assembly, Functions and Evolution of Archaella, Flagella and Cilia. Curr. Biol. 2018, 28, R278–R292. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, D.R. Evolution of cilia. Cold Spring Harb. Perspect. Biol. 2017, 9, 1–12. [Google Scholar] [CrossRef]
- Horani, A.; Ferkol, T.W. Primary ciliary dyskinesia and associated sensory ciliopathies. Expert Rev. Respir. Med. 2016, 10, 569–576. [Google Scholar] [CrossRef] [Green Version]
- Brndiarova, M.; Kvassayova, J.; Vojtkova, J.; Igaz, M.; Buday, T.; Plevkova, J. Changes of Motile Ciliary Phenotype in Patients with Primary Ciliopathies. Adv. Exp. Med. Biol. 2021, 1335, 79–85. [Google Scholar]
- Kempeneers, C.; Chilvers, M.A. To beat, or not to beat, that is question! The spectrum of ciliopathies. Pediatr. Pulmonol. 2018, 53, 1122–1129. [Google Scholar] [CrossRef]
- Vertii, A.; Bright, A.; Delaval, B.; Hehnly, H.; Doxsey, S. New frontiers: Discovering cilia-independent functions of cilia proteins. EMBO Rep. 2015, 16, 1275–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, W.M.S.; Burch, R.L. The Principles of Humane Experimental Technique; Universities Federation for Animal Welfare: Hertfordshire, UK, 1959; ISBN 0-900767-78-2. [Google Scholar]
- Pazour, G.J.; Agrin, N.; Leszyk, J.; Witman, G.B. Proteomic analysis of a eukaryotic cilium. J. Cell Biol. 2005, 170, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.C.; Northey, J.G.B.; Garg, J.; Pearlman, R.E.; Siu, K.W.M. Robust method for proteome analysis by MS/MS using an entire translated genome: Demonstration on the ciliome of Tetrahymena thermophila. J. Proteome Res. 2005, 4, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Subota, I.; Julkowska, D.; Vincensini, L.; Reeg, N.; Buisson, J.; Blisnick, T.; Huet, D.; Perrot, S.; Santi-Rocca, J.; Duchateau, M.; et al. Proteomic analysis of intact flagella of procyclic Trypanosoma brucei cells identifies novel flagellar proteins with unique sub-localization and dynamics. Mol. Cell. Proteomics 2014, 13, 1769–1786. [Google Scholar] [CrossRef] [Green Version]
- Pigino, G.; Bui, K.H.; Maheshwari, A.; Lupetti, P.; Diener, D.; Ishikawa, T. Cryoelectron tomography of radial spokes in cilia and flagella. J. Cell Biol. 2011, 195, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Barber, C.F.; Heuser, T.; Carbajal-González, B.I.; Botchkarev, V.V.; Nicastro, D. Three-dimensional structure of the radial spokes reveals heterogeneity and interactions with dyneins in Chlamydomonas flagella. Mol. Biol. Cell 2012, 23, 111–120. [Google Scholar] [CrossRef]
- Tuxhorn, J.; Daise, T.; Dentler, W.L. Regulation of Flagellar Length in Chlamydomonas. Cytoskeleton 1998, 40, 133–146. [Google Scholar] [CrossRef]
- Salomé, P.A.; Merchant, S.S. A series of fortunate events: Introducing chlamydomonas as a reference organism. Plant Cell 2019, 31, 1682–1707. [Google Scholar] [CrossRef] [Green Version]
- Harris, E.H. Chlamydomonas as a model organism. Annu. Rev. Plant Biol. 2001, 52, 363–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinkerson, R.E.; Jonikas, M.C. Molecular techniques to interrogate and edit the Chlamydomonas nuclear genome. Plant J. 2015, 82, 393–412. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Dutcher, S.K. Genetic and genomic approaches to identify genes involved in flagellar assembly in Chlamydomonas reinhardtii. Methods Cell Biol. 2015, 127, 349–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, E.H. Chlamydomonas in the laboratory. In The Chlamydomonas Sourcebook: Introduction to Chlamydomonas and Its Laboratory Use; Elsevier Academic Press: Amsterdam, The Netherlands, 2008; pp. 241–302. ISBN 9780123708748. [Google Scholar]
- Li, X.; Zhang, R.; Patena, W.; Gang, S.S.; Blum, S.R.; Ivanova, N.; Yue, R.; Robertson, J.M.; Lefebvre, P.A.; Fitz-Gibbon, S.T.; et al. An Indexed, Mapped Mutant Library Enables Reverse Genetics Studies of Biological Processes in Chlamydomonas reinhardtii. Plant Cell 2016, 28, 367–387. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Patena, W.; Fauser, F.; Jinkerson, R.E.; Saroussi, S.; Meyer, M.T.; Ivanova, N.; Robertson, J.M.; Yue, R.; Zhang, R.; et al. A genome-wide algal mutant library and functional screen identifies genes required for eukaryotic photosynthesis. Nat. Genet. 2019, 51, 627–635. [Google Scholar] [CrossRef]
- Dhokane, D.; Bhadra, B.; Dasgupta, S. CRISPR based targeted genome editing of Chlamydomonas reinhardtii using programmed Cas9-gRNA ribonucleoprotein. Mol. Biol. Rep. 2020, 47, 8747–8755. [Google Scholar] [CrossRef]
- Shin, S.E.; Lim, J.M.; Koh, H.G.; Kim, E.K.; Kang, N.K.; Jeon, S.; Kwon, S.; Shin, W.S.; Lee, B.; Hwangbo, K.; et al. CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Sci. Rep. 2016, 6, 27810. [Google Scholar] [CrossRef]
- Guzmán-Zapata, D.; Sandoval-Vargas, J.M.; Macedo-Osorio, K.S.; Salgado-Manjarrez, E.; Castrejón-Flores, J.L.; Oliver-Salvador, M.D.C.; Durán-Figueroa, N.V.; Nogué, F.; Badillo-Corona, J.A. Efficient editing of the nuclear APT reporter gene in chlamydomonas reinhardtii via expression of a CRISPR-Cas9 module. Int. J. Mol. Sci. 2019, 20, 1247. [Google Scholar] [CrossRef] [Green Version]
- Picariello, T.; Hou, Y.; Kubo, T.; McNeill, N.A.; Yanagisawa, H.A.; Oda, T.; Witman, G.B. TIM, a targeted insertional mutagenesis method utilizing CRISPR/Cas9 in Chlamydomonas reinhardtii. PLoS ONE 2020, 15, e0232594. [Google Scholar] [CrossRef]
- Kindle, K.L. High-frequency nuclear transformation of Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA 1990, 87, 1228–1232. [Google Scholar] [CrossRef] [Green Version]
- Shimogawara, K.; Fujiwara, S.; Grossman, A.; Usuda, H. High-efficiency transformation of Chlamydomonas reinhardtii by electroporation. Genetics 1998, 148, 1821–1828. [Google Scholar] [CrossRef] [PubMed]
- Kindle, K.L.; Schnell, R.A.; Fernandez, E.; Lefebvre, P.A. Stable nuclear transformation of Chlamydomonas using the Chlamydomonas gene for nitrate reductase. J. Cell Biol. 1989, 109, 2589–2601. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.V.; Misquitta, R.W.; Reddy, V.S.; Rao, B.J.; Rajam, M.V. Genetic transformation of the green alga - Chlamydomonas reinhardtii by Agrobacterium tumefaciens. Plant Sci. 2004, 166, 731–738. [Google Scholar] [CrossRef]
- Park, R.V.; Asbury, H.; Miller, S.M. Modification of a Chlamydomonas reinhardtii CRISPR/Cas9 transformation protocol for use with widely available electroporation equipment. MethodsX 2020, 7, 100855. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Jeon, S.; Kim, S.; Chang, Y.K.; Kim, Y.C. Development of a pVEC peptide-based ribonucleoprotein (RNP) delivery system for genome editing using CRISPR/Cas9 in Chlamydomonas reinhardtii. Sci. Rep. 2020, 10, 22158. [Google Scholar] [CrossRef]
- Wakabayashi, K.I.; Kamiya, R. Axonemal motility in Chlamydomonas. Methods Cell Biol. 2015, 127, 387–402. [Google Scholar] [CrossRef]
- Oda, T.; Yanagisawa, H.; Kamiya, R.; Kikkawa, M. A molecular ruler determines the repeat length in eukaryotic cilia and flagella. Science 2014, 346, 857–860. [Google Scholar] [CrossRef]
- King, S.M.; Patel-King, R.S. The oligomeric outer dynein arm assembly factor CCDC103 is tightly integrated within the ciliary axoneme and exhibits periodic binding to microtubules. J. Biol. Chem. 2015, 290, 7388–7401. [Google Scholar] [CrossRef] [Green Version]
- Panizzi, J.R.; Becker-Heck, A.; Castleman, V.H.; Al-Mutairi, D.A.; Liu, Y.; Loges, N.T.; Pathak, N.; Austin-Tse, C.; Sheridan, E.; Schmidts, M.; et al. CCDC103 mutations cause primary ciliary dyskinesia by disrupting assembly of ciliary dynein arms. Nat. Genet. 2012, 44, 714–719. [Google Scholar] [CrossRef] [Green Version]
- Heuser, T.; Raytchev, M.; Krell, J.; Porter, M.E.; Nicastro, D. The dynein regulatory complex is the nexin link and a major regulatory node in cilia and flagella. J. Cell Biol. 2009, 187, 921–933. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Diener, D.R.; Yang, C.; Kohno, T.; Pazour, G.J.; Dienes, J.M.; Agrin, N.S.; King, S.M.; Sale, W.S.; Kamiya, R.; et al. Radial spoke proteins of Chlamydomonas flagella. J. Cell Sci. 2006, 119, 1165–1174. [Google Scholar] [CrossRef] [Green Version]
- Castleman, V.H.; Romio, L.; Chodhari, R.; Hirst, R.A.; de Castro, S.C.P.; Parker, K.A.; Ybot-Gonzalez, P.; Emes, R.D.; Wilson, S.W.; Wallis, C.; et al. Mutations in radial spoke head protein genes RSPH9 and RSPH4A cause primary ciliary dyskinesia with central-microtubular-pair abnormalities. Am. J. Hum. Genet. 2008, 84, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, B.D.; Velleca, M.A.; Curry, A.M.; Rosenbaum, J.L. Molecular cloning and sequence analysis of the Chlamydomonas gene coding for radial spoke protein 3: Flagellar mutation pf-14 is an ochre allele. J. Cell Biol. 1989, 109, 235–245. [Google Scholar] [CrossRef]
- Bustamante-Marin, X.M.; Horani, A.; Stoyanova, M.; Charng, W.L.; Bottier, M.; Sears, P.R.; Yin, W.N.; Daniels, L.A.; Bowen, H.; Conrad, D.F.; et al. Mutation of CFAP57, a protein required for the asymmetric targeting of a subset of inner dynein arms in Chlamydomonas, causes primary ciliary dyskinesia. PLoS Genet. 2020, 16, e1008691. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Le, T.V.; Augspurger, K.; Tritschler, D.; Bower, R.; Fu, G.; Perrone, C.; O’Toole, E.T.; Mills, K.V.; Dymek, E.; et al. FAP57/WDR65 targets assembly of a subset of inner arm dyneins and connects to regulatory hubs in cilia. Mol. Biol. Cell 2019, 30, 2659–2680. [Google Scholar] [CrossRef]
- Zhang, H.; Mitchell, D.R. Cpc1, a Chlamydomonas central pair protein with an adenylate kinase domain. J. Cell Sci. 2004, 117, 4179–4188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiPetrillo, C.G.; Smith, E.F. Pcdp1 is a central apparatus protein that binds Ca2+-calmodulin and regulates ciliary motility. J. Cell Biol. 2010, 189, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Lechtreck, K.F.; Witman, G.B. Chlamydomonas reinhardtii hydin is a central pair protein required for flagellar motility. J. Cell Biol. 2007, 176, 473–482. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.F.; Lefebvre, P.A. PF16 encodes a protein with armadillo repeats and localizes to a single microtubule of the central apparatus in Chlamydomonas flagella. J. Cell Biol. 1996, 132, 359–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, G.; Zhao, L.; Dymek, E.; Hou, Y.; Song, K.; Phan, N.; Shang, Z.; Smith, E.F.; Witman, G.B.; Nicastro, D. Structural organization of the C1a-e-c supercomplex within the ciliary central apparatus. J. Cell Biol. 2019, 218, 4236–4251. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, J.; Cheng, H.; Gao, Y.; Liu, W.; Zhang, Z.; Jiang, H.; Li, W.; Zhu, F.; Lv, M.; et al. Patients with severe asthenoteratospermia carrying SPAG6 or RSPH3 mutations have a positive pregnancy outcome following intracytoplasmic sperm injection. J. Assist. Reprod. Genet. 2020, 37, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Rupp, G.; O’Toole, E.; Gardner, L.C.; Mitchell, B.F.; Porter, M.E. The sup-pf-2 mutations of Chlamydomonas alter the activity of the outer dynein arms by modification of the gamma-dynein heavy chain. J. Cell Biol. 1996, 135, 1853–1865. [Google Scholar] [CrossRef] [Green Version]
- Wilkerson, C.G.; King, S.M.; Koutoulis, A.; Pazour, G.J.; Witman, G.B. The 78,000 M(r) intermediate chain of Chlamydomonas outer arm dynein isa WD-repeat protein required for arm assembly. J. Cell Biol. 1995, 129, 169–178. [Google Scholar] [CrossRef]
- Mitchell, D.R.; Kang, Y. Reversion analysis of dynein intermediate chain function. J. Cell Sci. 1993, 105, 1069–1078. [Google Scholar] [CrossRef]
- Pennarun, G.; Escudier, E.; Chapelin, C.; Bridoux, A.M.; Cacheux, V.; Roger, G.; Clément, A.; Goossens, M.; Amselem, S.; Duriez, B. Loss-of-function mutations in a human gene related to Chlamydomonas reinhardtii dynein IC78 result in primary ciliary dyskinesia. Am. J. Hum. Genet. 1999, 65, 1508–1519. [Google Scholar] [CrossRef] [Green Version]
- Olbrich, H.; Häffner, K.; Kispert, A.; Völkel, A.; Volz, A.; Sasmaz, G.; Reinhardt, R.; Hennig, S.; Lehrach, H.; Konietzko, N.; et al. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat. Genet. 2002, 30, 143–144. [Google Scholar] [CrossRef]
- Loges, N.T.; Olbrich, H.; Fenske, L.; Mussaffi, H.; Horvath, J.; Fliegauf, M.; Kuhl, H.; Baktai, G.; Peterffy, E.; Chodhari, R.; et al. DNAI2 mutations cause primary ciliary dyskinesia with defects in the outer dynein arm. Am. J. Hum. Genet. 2008, 83, 547–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leigh, M.W.; Pittman, J.E.; Carson, J.L.; Ferkol, T.W.; Dell, S.D.; Davis, S.D.; Knowles, M.R.; Zariwala, M.A. Clinical and genetic aspects of primary ciliary dyskinesia/kartagener syndrome. Genet. Med. 2009, 11, 473–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin-Tse, C.; Halbritter, J.; Zariwala, M.A.; Gilberti, R.M.; Gee, H.Y.; Hellman, N.; Pathak, N.; Liu, Y.; Panizzi, J.R.; Patel-King, R.S.; et al. Zebrafish ciliopathy screen plus human mutational analysis identifies C21orf59 and CCDC65 defects as causing primary ciliary dyskinesia. Am. J. Hum. Genet. 2013, 93, 672–686. [Google Scholar] [CrossRef] [Green Version]
- Fassad, M.R.; Shoemark, A.; le Borgne, P.; Koll, F.; Patel, M.; Dixon, M.; Hayward, J.; Richardson, C.; Frost, E.; Jenkins, L.; et al. C11orf70 Mutations Disrupting the Intraflagellar Transport-Dependent Assembly of Multiple Axonemal Dyneins Cause Primary Ciliary Dyskinesia. Am. J. Hum. Genet. 2018, 102, 956–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duquesnoy, P.; Escudier, E.; Vincensini, L.; Freshour, J.; Bridoux, A.M.; Coste, A.; Deschildre, A.; de Blic, J.; Legendre, M.; Montantin, G.; et al. Loss-of-Function Mutations in the Human Ortholog of Chlamydomonas reinhardtii ODA7 Disrupt Dynein Arm Assembly and Cause Primary Ciliary Dyskinesia. Am. J. Hum. Genet. 2009, 85, 890–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamiya, R. Mutations at twelve independent loci result in absence of outer dynein arms in Chylamydomonas reinhardtii. J. Cell Biol. 1988, 107, 2253–2258. [Google Scholar] [CrossRef] [PubMed]
- Freshour, J.; Yokoyama, R.; Mitchell, D.R. Chlamydomonas flagellar outer row dynein assembly protein ODA7 interacts with both outer row and I1 inner row dyneins. J. Biol. Chem. 2007, 282, 5404–5412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Piperno, G.; Luck, D.J.L. Paralyzed flagella mutants of Chlamydomonas reinhardtii. Defective for axonemal doublet microtubule arms. J. Biol. Chem. 1979, 254, 3091–3099. [Google Scholar] [CrossRef]
- Omran, H.; Kobayashi, D.; Olbrich, H.; Tsukahara, T.; Loges, N.T.; Hagiwara, H.; Zhang, Q.; Leblond, G.; O’Toole, E.; Hara, C.; et al. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature 2008, 456, 611–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, R.; Yanagi, S.; Nagao, M.; Yamasaki, Y.; Tanaka, Y.; Sale, W.S.; Yagi, T.; Kon, T. Mutations in PIH proteins MOT48, TWI1 and PF13 define common and unique steps for preassembly of each, different ciliary dynein. PLoS Genet. 2020, 16, e1009126. [Google Scholar] [CrossRef]
- Mitchison, H.M.; Schmidts, M.; Loges, N.T.; Freshour, J.; Dritsoula, A.; Hirst, R.A.; O’Callaghan, C.; Blau, H.; Al Dabbagh, M.; Olbrich, H.; et al. Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nat. Genet. 2012, 44, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, R.; Obbineni, J.M.; Alford, L.M.; Ide, T.; Owa, M.; Hwang, J.; Kon, T.; Inaba, K.; James, N.; King, S.M.; et al. Chlamydomonas DYX1C1/PF23 is essential for axonemal assembly and proper morphology of inner dynein arms. PLoS Genet. 2017, 13, e1006996. [Google Scholar] [CrossRef] [Green Version]
- Horani, A.; Druley, T.E.; Zariwala, M.A.; Patel, A.C.; Levinson, B.T.; Van Arendonk, L.G.; Thornton, K.C.; Giacalone, J.C.; Albee, A.J.; Wilson, K.S.; et al. Whole-exome capture and sequencing identifies HEATR2 mutation as a cause of primary ciliary dyskinesia. Am. J. Hum. Genet. 2012, 91, 685–693. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, R.; Hirono, M.; Kamiya, R. Discrete PIH proteins function in the cytoplasmic preassembly of different subsets of axonemal dyneins. J. Cell Biol. 2010, 190, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Desai, P.B.; Freshour, J.R.; Mitchell, D.R. Chlamydomonas axonemal dynein assembly locus ODA8 encodes a conserved flagellar protein needed for cytoplasmic maturation of outer dynein arm complexes. Cytoskeleton 2015, 72, 16–28. [Google Scholar] [CrossRef] [Green Version]
- Fabczak, H.; Osinka, A. Role of the novel Hsp90 co-chaperones in dynein arms’ preassembly. Int. J. Mol. Sci. 2019, 20, 6174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owa, M.; Uchihashi, T.; aki Yanagisawa, H.; Yamano, T.; Iguchi, H.; Fukuzawa, H.; ichi Wakabayashi, K.; Ando, T.; Kikkawa, M. Inner lumen proteins stabilize doublet microtubules in cilia and flagella. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasudevan, K.K.; Song, K.; Alford, L.M.; Sale, W.S.; Dymek, E.E.; Smith, E.F.; Hennessey, T.; Joachimiak, E.; Urbanska, P.; Wloga, D.; et al. FAP206 is a microtubule-docking adapter for ciliary radial spoke 2 and dynein c. Mol. Biol. Cell 2015, 26, 696–710. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.; Wang, Q.; Phan, N.; Urbanska, P.; Joachimiak, E.; Lin, J.; Wloga, D.; Nicastro, D. The I1 dynein-associated tether and tether head complex is a conserved regulator of ciliary motility. Mol. Biol. Cell 2018, 29, 1048–1059. [Google Scholar] [CrossRef] [PubMed]
- Rotureau, B.; Subota, I.; Bastin, P. Molecular bases of cytoskeleton plasticity during the Trypanosoma brucei parasite cycle. Cell. Microbiol. 2011, 13, 705–716. [Google Scholar] [CrossRef] [Green Version]
- Bertiaux, E.; Mallet, A.; Rotureau, B.; Bastin, P. Intraflagellar transport during assembly of flagella of different length in Trypanosoma brucei isolated from tsetse flies. J. Cell Sci. 2020, 133, jcs248989. [Google Scholar] [CrossRef]
- An, T.; Zhou, Q.; Hu, H.; Cormaty, H.; Li, Z. FAZ27 cooperates with FLAM3 and ClpGM6 to maintain cell morphology in Trypanosoma brucei. J. Cell Sci. 2020, 133, jcs245258. [Google Scholar] [CrossRef]
- Langousis, G.; Hill, K.L. Motility and more: The flagellum of Trypanosoma brucei. Nat. Rev. Microbiol. 2014, 12, 505–518. [Google Scholar] [CrossRef] [Green Version]
- Oberholzer, M.; Lopez, M.A.; Ralston, K.S.; Hill, K.L. Approaches for functional analysis of flagellar proteins in African trypanosomes. Methods Cell Biol. 2009, 93, 21–57. [Google Scholar] [CrossRef] [Green Version]
- Wirtz, E.; Leal, S.; Ochatt, C.; Cross, G.M. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 1999, 99, 89–101. [Google Scholar] [CrossRef]
- Santi-Rocca, J.; Chenouard, N.; Fort, C.; Lagache, T.; Olivo-Marin, J.C.; Bastin, P. Imaging intraflagellar transport in trypanosomes. Methods Cell Biol. 2015, 127, 487–508. [Google Scholar] [CrossRef] [PubMed]
- Burkard, G.; Fragoso, C.M.; Roditi, I. Highly efficient stable transformation of bloodstream forms of Trypanosoma brucei. Mol. Biochem. Parasitol. 2007, 153, 220–223. [Google Scholar] [CrossRef] [PubMed]
- MacGregor, P.; Rojas, F.; Dean, S.; Matthews, K.R. Stable transformation of pleomorphic bloodstream form Trypanosoma brucei. Mol. Biochem. Parasitol. 2013, 190, 60–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachmaier, S.; Thanner, T.; Boshart, M. Culturing and Transfection of Pleomorphic Trypanosoma brucei. Methods Mol. Biol. 2020, 2116, 23–38. [Google Scholar] [PubMed]
- Yagoubat, A.; Corrales, R.M.; Bastien, P.; Lévêque, M.F.; Sterkers, Y. Gene Editing in Trypanosomatids: Tips and Tricks in the CRISPR-Cas9 Era. Trends Parasitol. 2020, 36, 745–760. [Google Scholar] [CrossRef]
- Medeiros, L.C.S.; South, L.; Peng, D.; Bustamante, J.M.; Wang, W.; Bunkofske, M.; Perumal, N.; Sanchez-Valdez, F.; Tarleton, R.L. Rapid, selection-free, high-efficiency genome editing in protozoan parasites using CRISPR-cas9 ribonucleoproteins. MBio 2017, 8, e01788-17. [Google Scholar] [CrossRef] [Green Version]
- Lander, N.; Chiurillo, M.A. State-of-the-art CRISPR/Cas9 Technology for Genome Editing in Trypanosomatids. J. Eukaryot. Microbiol. 2019, 66, 981–991. [Google Scholar] [CrossRef]
- Baron, D.M.; Kabututu, Z.P.; Hill, K.L. Stuck in reverse: Loss of LC1 in Trypanosoma brusei disrupts outer dynein arms and leads to reverse flagellar beat and backward movement. J. Cell Sci. 2007, 120, 1513–1520. [Google Scholar] [CrossRef] [Green Version]
- Ralston, K.S.; Lerner, A.G.; Diener, D.R.; Hill, K.L. Flagellar motility contributes to cytokinesis in Trypanosoma brucei and is modulated by an evolutionarily conserved dynein regulatory system. Eukaryot. Cell 2006, 5, 696–711. [Google Scholar] [CrossRef] [Green Version]
- Branche, C.; Kohl, L.; Toutirais, G.; Buisson, J.; Cosson, J.; Bastin, P. Conserved and specific functions of axoneme components in trypanosome motility. J. Cell Sci. 2006, 119, 3443–3455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krüger, T.; Engstler, M. Motility Analysis of Trypanosomatids. Methods Mol. Biol. 2020, 2116, 409–423. [Google Scholar] [PubMed]
- Höög, J.L.; Bouchet-Marquis, C.; McIntosh, J.R.; Hoenger, A.; Gull, K. Cryo-electron tomography and 3-D analysis of the intact flagellum in Trypanosoma brucei. J. Struct. Biol. 2012, 178, 189–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imhof, S.; Zhang, J.; Wang, H.; Bui, K.H.; Nguyen, H.; Atanasov, I.; Hui, W.H.; Yang, S.K.; Zhou, Z.H.; Hill, K.L. Cryo electron tomography with volta phase plate reveals novel structural foundations of the 96-nm axonemal repeat in the pathogen trypanosoma brucei. Elife 2019, 8, e52058. [Google Scholar] [CrossRef]
- Motta, M.C.M.; Catta-Preta, C.M.C. Electron Microscopy Techniques Applied to Symbiont-Harboring Trypanosomatids: The Association of the Bacterium with Host Organelles. Methods Mol. Biol. 2020, 2116, 425–447. [Google Scholar]
- Kabututu, Z.P.; Thayer, M.; Melehani, J.H.; Hill, K.L. CMF70 is a subunit of the dynein regulatory complex. J. Cell Sci. 2010, 123, 3587–3595. [Google Scholar] [CrossRef] [Green Version]
- Hutchings, N.R.; Donelson, J.E.; Hill, K.L. Trypanin is a cytoskeletal linker protein and is required for cell motility in African trypanosomes. J. Cell Biol. 2002, 156, 867–877. [Google Scholar] [CrossRef] [Green Version]
- Dawe, H.R.; Shaw, M.K.; Farr, H.; Gull, K. The hydrocephalus inducing gene product, Hydin, positions axonemal central pair microtubules. BMC Biol. 2007, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Bonnefoy, S.; Watson, C.M.; Kernohan, K.D.; Lemos, M.; Hutchinson, S.; Poulter, J.A.; Crinnion, L.A.; Berry, I.; Simmonds, J.; Vasudevan, P.; et al. Biallelic Mutations in LRRC56, Encoding a Protein Associated with Intraflagellar Transport, Cause Mucociliary Clearance and Laterality Defects. Am. J. Hum. Genet. 2018, 103, 727–739. [Google Scholar] [CrossRef] [Green Version]
- Aprea, I.; Raidt, J.; Höben, I.M.; Loges, N.T.; Nöthe-Menchen, T.; Pennekamp, P.; Olbrich, H.; Kaiser, T.; Biebach, L.; Tüttelmann, F.; et al. Defects in the cytoplasmic assembly of axonemal dynein arms cause morphological abnormalities and dysmotility in sperm cells leading to male infertility. PLoS Genet. 2021, 17, e1009306. [Google Scholar] [CrossRef]
- Kott, E.; Duquesnoy, P.; Copin, B.; Legendre, M.; Dastot-Le Moal, F.; Montantin, G.; Jeanson, L.; Tamalet, A.; Papon, J.F.; Siffroi, J.P.; et al. Loss-of-function mutations in LRRC6, a gene essential for proper axonemal assembly of inner and outer dynein arms, cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2012, 91, 958–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, G.W.; Denny, P.W.; Vaughan, S.; Goulding, D.; Jeffries, T.R.; Smith, D.F.; Gull, K.; Field, M.C. An Evolutionarily Conserved Coiled-Coil Protein Implicated in Polycystic Kidney Disease Is Involved in Basal Body Duplication and Flagellar Biogenesis in Trypanosoma brucei. Mol. Cell. Biol. 2005, 25, 3774–3783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coutton, C.; Vargas, A.S.; Amiri-Yekta, A.; Kherraf, Z.E.; Mustapha, S.F.B.; Le Tanno, P.; Wambergue-Legrand, C.; Karaouzène, T.; Martinez, G.; Crouzy, S.; et al. Mutations in CFAP43 and CFAP44 cause male infertility and flagellum defects in Trypanosoma and human. Nat. Commun. 2018, 9, 686. [Google Scholar] [CrossRef] [PubMed]
- Martinez, G.; Beurois, J.; Dacheux, D.; Cazin, C.; Bidart, M.; Kherraf, Z.E.; Robinson, D.R.; Satre, V.; Le Gac, G.; Ka, C.; et al. Biallelic variants in MAATS1 encoding CFAP91, a calmodulin-associated and spoke-associated complex protein, cause severe astheno-teratozoospermia and male infertility. J. Med. Genet. 2020, 57, 708–716. [Google Scholar] [CrossRef]
- Kherraf, Z.E.; Amiri-Yekta, A.; Dacheux, D.; Karaouzène, T.; Coutton, C.; Christou-Kent, M.; Martinez, G.; Landrein, N.; Le Tanno, P.; Mustapha, S.F.B.; et al. A Homozygous Ancestral SVA-Insertion-Mediated Deletion in WDR66 Induces Multiple Morphological Abnormalities of the Sperm Flagellum and Male Infertility. Am. J. Hum. Genet. 2018, 103, 400–412. [Google Scholar] [CrossRef] [Green Version]
- Lorès, P.; Dacheux, D.; Kherraf, Z.E.; Mbango, J.F.N.; Coutton, C.; Stouvenel, L.; Ialy-Radio, C.; Amiri-Yekta, A.; Whitfield, M.; Schmitt, A.; et al. Mutations in TTC29, Encoding an Evolutionarily Conserved Axonemal Protein, Result in Asthenozoospermia and Male Infertility. Am. J. Hum. Genet. 2019, 105, 1148–1167. [Google Scholar] [CrossRef]
- Lynn, D. Ciliates. Encycl. Microbiol. 2009, 578–592. [Google Scholar]
- Wloga, D.; Frankel, J. From Molecules to Morphology: Cellular Organization of Tetrahymena thermophila. Methods Cell Biol. 2012, 109, 83–140. [Google Scholar]
- Aubusson-Fleury, A.; Cohen, J.; Lemullois, M. Ciliary heterogeneity within a single cell: The Paramecium model. Methods Cell Biol. 2015, 127, 457–485. [Google Scholar]
- Tassin, A.M.; Lemullois, M.; Aubusson-Fleury, A. Paramecium tetraurelia basal body structure. Cilia 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Valentine, M.; Houten, J. Van Using paramecium as a model for ciliopathies. Genes (Basel) 2021, 12, 1493. [Google Scholar] [CrossRef] [PubMed]
- Wloga, D.; Camba, A.; Rogowski, K.; Manning, G.; Jerka-Dziadosz, M.; Gaertig, J. Members of the NIMA-related kinase family promote disassembly of cilia by multiple mechanisms. Mol. Biol. Cell 2006, 17, 2799–2810. [Google Scholar] [CrossRef] [PubMed]
- Cezar Alves, H.; da Consolação Diniz Javaroti, D.; Roberto Ferreira, J.; Helena Regali Seleghim, M. Optimized culture and growth curves of two ciliated protozoan strains of Paramecium caudatum Ehrenberg, 1833 to use in ecotoxicologycal assays. Rev. Bras. Zoociências 2016, 17, 77–90. [Google Scholar]
- Ishida, M.; Hori, M. Improved isolation method to establish axenic strains of Paramecium. Japanese J. Protozool. 2017, 50, 1–14. [Google Scholar] [CrossRef]
- Gaertig, J.; Wloga, D.; Vasudevan, K.K.; Guha, M.; Dentler, W. Discovery and functional evaluation of ciliary proteins in tetrahymena thermophila. Methods Enzymol. 2013, 525, 265–284. [Google Scholar]
- Jiang, Y.Y.; Lechtreck, K.; Gaertig, J. Total internal reflection fluorescence microscopy of intraflagellar transport in Tetrahymena thermophila. Methods Cell Biol. 2015, 127, 445–456. [Google Scholar] [CrossRef] [Green Version]
- Gogendeau, D.; Lemullois, M.; Le Borgne, P.; Castelli, M.; Aubusson-Fleury, A.; Arnaiz, O.; Cohen, J.; Vesque, C.; Schneider-Maunoury, S.; Bouhouche, K.; et al. MKS-NPHP module proteins control ciliary shedding at the transition zone. PLoS Biol. 2020, 18, e3000640. [Google Scholar] [CrossRef] [Green Version]
- Hazime, K.S.; Zhou, Z.; Joachimiak, E.; Bulgakova, N.A.; Wloga, D.; Malicki, J.J. STORM imaging reveals the spatial arrangement of transition zone components and IFT particles at the ciliary base in Tetrahymena. Sci. Rep. 2021, 11, 7899. [Google Scholar] [CrossRef]
- Winey, M.; Stemm-Wolf, A.J.; Giddings, T.H.; Pearson, C.G. Cytological Analysis of Tetrahymena thermophila. Methods Cell Biol. 2012, 109, 357–378. [Google Scholar]
- Joachimiak, E.; Osinka, A.; Farahat, H.; Świderska, B.; Sitkiewicz, E.; Poprzeczko, M.; Fabczak, H.; Wloga, D. Composition and function of the C1b/C1f region in the ciliary central apparatus. Sci. Rep. 2021, 11, 11760. [Google Scholar] [CrossRef]
- Shang, Y.; Song, X.; Bowen, J.; Corstanje, R.; Gao, Y.; Gaertig, J.; Gorovsky, M.A. A robust inducible-repressible promoter greatly facilitates gene knockouts, conditional expression, and overexpression of homologous and heterologous genes in Tetrahymena thermophila. Proc. Natl. Acad. Sci. USA 2002, 99, 3734–3739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dave, D.; Wloga, D.; Gaertig, J. Manipulating ciliary protein-encoding genes in Tetrahymena thermophila. Methods Cell Biol. 2009, 93, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Gotesman, M.; Hosein, R.E.; Williams, S.A. Using a Hand-Held Gene Gun for Genetic Transformation of Tetrahymena thermophila. Methods Mol. Biol. 2022, 2364, 349–361. [Google Scholar] [PubMed]
- Rajagopalan, V.; Corpuz, E.O.; Hubenschmidt, M.J.; Townsend, C.R.; Asai, D.J.; Wilkes, D.E. Analysis of properties of cilia using Tetrahymena thermophila. Methods Mol. Biol. 2009, 586, 283–299. [Google Scholar] [CrossRef] [PubMed]
- Chalker, D.L. Transformation and Strain Engineering of Tetrahymena. Methods Cell Biol. 2012, 109, 327–345. [Google Scholar]
- Galvani, A.; Sperling, L. RNA interference by feeding in Paramecium. Trends Genet. 2002, 18, 11–12. [Google Scholar] [CrossRef]
- Carradec, Q.; Götz, U.; Arnaiz, O.; Pouch, J.; Simon, M.; Meyer, E.; Marker, S. Primary and secondary siRNA synthesis triggered by RNAs from food bacteria in the ciliate Paramecium tetraurelia. Nucleic Acids Res. 2015, 43, 1818–1833. [Google Scholar] [CrossRef] [Green Version]
- Dougherty, G.W.; Loges, N.T.; Klinkenbusch, J.A.; Olbrich, H.; Pennekamp, P.; Menchen, T.; Raidt, J.; Wallmeier, J.; Werner, C.; Westermann, C.; et al. DNAH11 localization in the proximal region of respiratory cilia defines distinct outer dynein arm complexes. Am. J. Respir. Cell Mol. Biol. 2016, 55, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Fliegauf, M.; Olbrich, H.; Horvath, J.; Wildhaber, J.H.; Zariwala, M.A.; Kennedy, M.; Knowles, M.R.; Omran, H. Mislocalization of DNAH5 and DNAH9 in respiratory cells from patients with primary ciliary dyskinesia. Am. J. Respir. Crit. Care Med. 2005, 171, 1343–1349. [Google Scholar] [CrossRef] [Green Version]
- Fassad, M.R.; Shoemark, A.; Legendre, M.; Hirst, R.A.; Koll, F.; le Borgne, P.; Louis, B.; Daudvohra, F.; Patel, M.P.; Thomas, L.; et al. Mutations in Outer Dynein Arm Heavy Chain DNAH9 Cause Motile Cilia Defects and Situs Inversus. Am. J. Hum. Genet. 2018, 103, 984–994. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Shen, X.; Chi, Y.; Shen, Y. Primary ciliary dyskinesia relative protein ZMYND10 is involved in regulating ciliary function and intraflagellar transport in Paramecium tetraurelia. Eur. J. Protistol. 2021, 77, 125756. [Google Scholar] [CrossRef]
- Thomas, L.; Bouhouche, K.; Whitfield, M.; Thouvenin, G.; Coste, A.; Louis, B.; Szymanski, C.; Bequignon, E.; Papon, J.F.; Castelli, M.; et al. TTC12 Loss-of-Function Mutations Cause Primary Ciliary Dyskinesia and Unveil Distinct Dynein Assembly Mechanisms in Motile Cilia Versus Flagella. Am. J. Hum. Genet. 2020, 106, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Zariwala, M.A.; Gee, H.Y.; Kurkowiak, M.; Al-Mutairi, D.A.; Leigh, M.W.; Hurd, T.W.; Hjeij, R.; Dell, S.D.; Chaki, M.; Dougherty, G.W.; et al. ZMYND10 is mutated in primary ciliary dyskinesia and interacts with LRRC6. Am. J. Hum. Genet. 2013, 93, 336–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, D.J.; Onoufriadis, A.; Shoemark, A.; Simpson, M.A.; Zur Lage, P.I.; De Castro, S.C.; Bartoloni, L.; Gallone, G.; Petridi, S.; Woollard, W.J.; et al. Mutations in ZMYND10, a gene essential for proper axonemal assembly of inner and outer dynein arms in humans and flies, cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2013, 93, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Kurkowiak, M.; Ziętkiewicz, E.; Greber, A.; Voelkel, K.; Wojda, A.; Pogorzelski, A.; Witt, M. ZMYND10 - Mutation analysis in Slavic patients with primary ciliary dyskinesia. PLoS ONE 2016, 11, e0148067. [Google Scholar] [CrossRef]
- Zietkiewicz, E.; Bukowy-Bieryllo, Z.; Rabiasz, A.; Daca-Roszak, P.; Wojda, A.; Voelkel, K.; Rutkiewicz, E.; Pogorzelski, A.; Rasteiro, M.; Witt, M. CFAP300: Mutations in slavic patients with primary ciliary dyskinesia and a role in ciliary dynein arms trafficking. Am. J. Respir. Cell Mol. Biol. 2019, 61, 400–449. [Google Scholar] [CrossRef]
- Mali, G.R.; Yeyati, P.L.; Mizuno, S.; Dodd, D.O.; Tennant, P.A.; Keighren, M.A.; Lage, P.Z.; Shoemark, A.; Garcia-Munoz, A.; Shimada, A.; et al. ZMYND10 functions in a chaperone relay during axonemal dynein assembly. Elife 2018, 7, e34389. [Google Scholar] [CrossRef]
- Liu, W.; Sha, Y.; Li, Y.; Mei, L.; Lin, S.; Huang, X.; Lu, J.; Ding, L.; Kong, S.; Lu, Z. Loss-of-function mutations in SPEF2 cause multiple morphological abnormalities of the sperm flagella (MMAF). J. Med. Genet. 2019, 56, 678–684. [Google Scholar] [CrossRef]
- Tu, C.; Nie, H.; Meng, L.; Wang, W.; Li, H.; Yuan, S.; Cheng, D.; He, W.; Liu, G.; Du, J.; et al. Novel mutations in SPEF2 causing different defects between flagella and cilia bridge: The phenotypic link between MMAF and PCD. Hum. Genet. 2020, 139, 257–271. [Google Scholar] [CrossRef]
- Liu, C.; Lv, M.; He, X.; Zhu, Y.; Amiri-Yekta, A.; Li, W.; Wu, H.; Kherraf, Z.E.; Liu, W.; Zhang, J.; et al. Homozygous mutations in SPEF2 induce multiple morphological abnormalities of the sperm flagella and male infertility. J. Med. Genet. 2020, 57, 31–37. [Google Scholar] [CrossRef]
- Sha, Y.; Liu, W.; Wei, X.; Zhu, X.; Luo, X.; Liang, L.; Guo, T. Biallelic mutations in Sperm flagellum 2 cause human multiple morphological abnormalities of the sperm flagella (MMAF) phenotype. Clin. Genet. 2019, 96, 385–393. [Google Scholar] [CrossRef]
- Dong, F.N.; Amiri-Yekta, A.; Martinez, G.; Saut, A.; Tek, J.; Stouvenel, L.; Lorès, P.; Karaouzène, T.; Thierry-Mieg, N.; Satre, V.; et al. Absence of CFAP69 Causes Male Infertility due to Multiple Morphological Abnormalities of the Flagella in Human and Mouse. Am. J. Hum. Genet. 2018, 102, 636–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Li, W.; Wu, H.; Lv, M.; Liu, W.; Liu, C.; Zhu, F.; Li, C.; Fang, Y.; Yang, C.; et al. Novel homozygous CFAP69 mutations in humans and mice cause severe asthenoteratospermia with multiple morphological abnormalities of the sperm flagella. J. Med. Genet. 2019, 56, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Martinez, G.; Liu, H.; Beurois, J.; Wu, H.; Amiri-Yekta, A.; Liang, D.; Kherraf, Z.E.; Bidart, M.; Cazin, C.; et al. Bi-allelic truncating variants in CFAP206 cause male infertility in human and mouse. Hum. Genet. 2021, 140, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.Y.; Yang, Y.H.; Chen, S.R. Molecular genetics of infertility: Loss-of-function mutations in humans and corresponding knockout/mutated mice. Hum. Reprod. Update 2021, 27, 154–189. [Google Scholar] [CrossRef] [PubMed]
- Urbanska, P.; Joachimiak, E.; Bazan, R.; Fu, G.; Poprzeczko, M.; Fabczak, H.; Nicastro, D.; Wloga, D. Ciliary proteins Fap43 and Fap44 interact with each other and are essential for proper cilia and flagella beating. Cell. Mol. Life Sci. 2018, 75, 4479–4493. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Wang, X.; Li, W.; Yang, X.; Li, Z.; Liu, W.; Li, C.; Zhu, Z.; Wang, L.; Wang, J.; et al. Biallelic Mutations in CFAP43 and CFAP44 Cause Male Infertility with Multiple Morphological Abnormalities of the Sperm Flagella. Am. J. Hum. Genet. 2017, 100, 854–864. [Google Scholar] [CrossRef] [Green Version]
- Sha, Y.W.; Wang, X.; Xu, X.; Su, Z.Y.; Cui, Y.; Bin Mei, L.; Huang, X.J.; Chen, J.; He, X.M.; Ji, Z.Y.; et al. Novel Mutations in CFAP44 and CFAP43 Cause Multiple Morphological Abnormalities of the Sperm Flagella (MMAF). Reprod. Sci. 2019, 26, 26–34. [Google Scholar] [CrossRef]
- Khan, I.; Shah, B.; Dil, S.; Ullah, N.; Zhou, J.T.; Zhao, D.R.; Zhang, Y.W.; Jiang, X.H.; Khan, R.; Khan, A.; et al. Novel biallelic loss-of-function mutations in CFAP43 cause multiple morphological abnormalities of the sperm flagellum in Pakistani families. Asian J. Androl. 2021, 23, 627–632. [Google Scholar] [CrossRef]
- Wu, H.; Li, W.; He, X.; Liu, C.; Fang, Y.; Zhu, F.; Jiang, H.; Liu, W.; Song, B.; Wang, X.; et al. NovelCFAP43 andCFAP44 mutations cause male infertility with multiple morphological abnormalities of the sperm flagella (MMAF). Reprod. Biomed. Online 2019, 38, 769–778. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, J.; Kherraf, Z.E.; Sun, S.; Zhang, X.; Cazin, C.; Coutton, C.; Zouari, R.; Zhao, S.; Hu, F.; et al. CFAP61 is required for sperm flagellum formation and male fertility in human and mouse. Development 2021, 148, dev199805. [Google Scholar] [CrossRef]
- Urbanska, P.; Song, K.; Joachimiak, E.; Krzemien-Ojak, L.; Koprowski, P.; Hennessey, T.; Jerka-Dziadosz, M.; Fabczak, H.; Gaertig, J.; Nicastro, D.; et al. The CSC proteins FAP61 and FAP251 build the basal substructures of radial spoke 3 in cilia. Mol. Biol. Cell 2015, 26, 1463–1475. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; He, X.; Yang, S.; Liu, C.; Wu, H.; Liu, W.; Lv, M.; Tang, D.; Tan, J.; Tang, S.; et al. Biallelic mutations of CFAP251 cause sperm flagellar defects and human male infertility. J. Hum. Genet. 2019, 64, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Lobo, D.; Beane, W.S.; Levin, M. Modeling planarian regeneration: A primer for reverse-engineering the worm. PLoS Comput. Biol. 2012, 8, e1002481. [Google Scholar] [CrossRef]
- Azimzadeh, J.; Basquin, C. Basal bodies across eukaryotes series: Basal bodies in the freshwater planarian Schmidtea mediterranea. Cilia 2016, 5, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Guo, F.; Jing, Q.; Zhu, X.; Yan, X. Characterisation of centriole biogenesis during multiciliation in planarians. Biol. Cell 2020, 112, 398–408. [Google Scholar] [CrossRef]
- Lesko, S.L.; Rouhana, L. Dynein assembly factor with WD repeat domains 1 (DAW1) is required for the function of motile cilia in the planarian Schmidtea mediterranea. Dev. Growth Differ. 2020, 62, 423–437. [Google Scholar] [CrossRef]
- Kyuji, A.; Patel-King, R.S.; Hisabori, T.; King, S.M.; Wakabayashi, K.I. Cilia Loss and Dynein Assembly Defects in Planaria Lacking an Outer Dynein Arm-Docking Complex Subunit. Zoolog. Sci. 2020, 37, 7–13. [Google Scholar] [CrossRef]
- Basquin, C.; Orfila, A.M.; Azimzadeh, J. The planarian Schmidtea mediterranea as a model for studying motile cilia and multiciliated cells. Methods Cell Biol. 2015, 127, 243–262. [Google Scholar] [CrossRef]
- Rink, J.C.; Vu, H.T.K.; Alvarado, A.S. The maintenance and regeneration of the planarian excretory system are regulated by EGFR signaling. Development 2011, 138, 3769–3780. [Google Scholar] [CrossRef] [Green Version]
- McKanna, J.A. Fine structure of the protonephridial system in planaria - I. Flame cells. Zeitschrift für Zellforsch. Mikroskopische Anat. 1968, 92, 509–523. [Google Scholar] [CrossRef]
- MacRae, E.K. The fine structure of sensory receptor processes in the auricular epithelium of the planarian, Dugesia tigrina. Zeitschrift für Zellforsch. Mikroskopische Anat. 1967, 82, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Brooks, E.R.; Wallingford, J.B. Multiciliated Cells. Curr. Biol. 2014, 24, R973–R982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merryman, M.S.; Alvarado, A.S.; Jenkin, J.C. Culturing planarians in the laboratory. Methods Mol. Biol. 2018, 1774, 241–258. [Google Scholar] [PubMed]
- Sousa, N.; Adell, T. Maintenance of Schmidtea mediterranea in the Laboratory. BIO-PROTOCOL 2018, 8, e3040. [Google Scholar] [CrossRef]
- Rompolas, P.; Azimzadeh, J.; Marshall, W.F.; King, S.M. Analysis of ciliary assembly and function in planaria. Methods Enzymol. 2013, 525, 245–264. [Google Scholar]
- King, S.M.; Patel-King, R.S. Planaria as a model system for the analysis of ciliary assembly and motility. Methods Mol. Biol. 2016, 1454, 245–254. [Google Scholar]
- Vij, S.; Rink, J.C.; Ho, H.K.; Babu, D.; Eitel, M.; Narasimhan, V.; Tiku, V.; Westbrook, J.; Schierwater, B.; Roy, S. Evolutionarily Ancient Association of the FoxJ1 Transcription Factor with the Motile Ciliogenic Program. PLoS Genet. 2012, 8, e1003019. [Google Scholar] [CrossRef]
- Höben, I.M.; Hjeij, R.; Olbrich, H.; Dougherty, G.W.; Nöthe-Menchen, T.; Aprea, I.; Frank, D.; Pennekamp, P.; Dworniczak, B.; Wallmeier, J.; et al. Mutations in C11orf70 Cause Primary Ciliary Dyskinesia with Randomization of Left/Right Body Asymmetry Due to Defects of Outer and Inner Dynein Arms. Am. J. Hum. Genet. 2018, 102, 973–984. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.T.; Gao, C.; Lucker, B.F.; Cole, D.G.; Mitchell, D.R. ODA16 aids axonemal outer row dynein assembly through an interaction with the intraflagellar transport machinery. J. Cell Biol. 2008, 183, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Taschner, M.; Mourão, A.; Awasthi, M.; Basquin, J.; Lorentzen, E. Structural basis of outer dynein arm intraflagellar transport by the transport adaptor protein ODA16 and the intraflagellar transport protein IFT46. J. Biol. Chem. 2017, 292, 7462–7473. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Witman, G.B. The N-terminus of IFT46 mediates intraflagellar transport of outer arm dynein and its cargo-adaptor ODA16. Mol. Biol. Cell 2017, 28, 2420–2433. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.T.; Mitchell, D.R. ODA16p, a chlamydomonas flagellar protein needed for dynein assembly. Mol. Biol. Cell 2005, 16, 5004–5012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C.; Wang, G.; Amack, J.D.; Mitchell, D.R. Oda16/Wdr69 is essential for axonemal dynein assembly and ciliary motility during zebrafish embryogenesis. Dev. Dyn. 2010, 239, 2190–2197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, G.M.; Francis, R.; Chu, K.K.; Birket, S.E.; Gabriel, G.; Trombley, J.E.; Lemke, K.L.; Klena, N.; Turner, B.; Tearney, G.J.; et al. Assessment of ciliary phenotype in primary ciliary dyskinesia by micro-optical coherence tomography. JCI insight 2017, 2, e91702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995, 203, 253–310. [Google Scholar] [CrossRef]
- Kalueff, A.V.; Stewart, A.M.; Gerlai, R. Zebrafish as an emerging model for studying complex brain disorders. Trends Pharmacol. Sci. 2014, 35, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Olmstead, A.W.; Korte, J.J.; Woodis, K.K.; Bennett, B.A.; Ostazeski, S.; Degitz, S.J. Reproductive maturation of the tropical clawed frog: Xenopus tropicalis. Gen. Comp. Endocrinol. 2009, 160, 117–123. [Google Scholar] [CrossRef]
- Wlizla, M.; McNamara, S.; Horb, M.E. Generation and care of Xenopus laevis and Xenopus tropicalis embryos. Methods Mol. Biol. 2018, 1865, 19–32. [Google Scholar]
- Blitz, I.L.; Cho, K.W.Y. Control of zygotic genome activation in Xenopus. Curr. Top. Dev. Biol. 2021, 145, 167–204. [Google Scholar] [CrossRef]
- Blum, M.; Ott, T. Xenopus: An undervalued model organism to study and model human genetic disease. Cells Tissues Organs 2019, 205, 303–312. [Google Scholar] [CrossRef]
- Session, A.M.; Uno, Y.; Kwon, T.; Chapman, J.A.; Toyoda, A.; Takahashi, S.; Fukui, A.; Hikosaka, A.; Suzuki, A.; Kondo, M.; et al. Genome evolution in the allotetraploid frog Xenopus laevis. Nature 2016, 538, 336–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostiuk, V.; Khokha, M.K. Xenopus as a platform for discovery of genes relevant to human disease. Curr. Top. Dev. Biol. 2021, 145, 277–312. [Google Scholar] [CrossRef] [PubMed]
- Kramer-Zucker, A.G.; Olale, F.; Haycraft, C.J.; Yoder, B.K.; Schier, A.F.; Drummond, I.A. Cilia-driven fluid flow in the zebrafish pronephros, brain and Kupffer’s vesicle is required for normal organogenesis. Development 2005, 132, 1907–1921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavares, B.; Jacinto, R.; Sampaio, P.; Pestana, S.; Pinto, A.; Vaz, A.; Roxo-Rosa, M.; Gardner, R.; Lopes, T.; Schilling, B.; et al. Notch/Her12 signalling modulates, motile/immotile cilia ratio downstream of Foxj1a in zebrafish left-right organizer. Elife 2017, 6, e25165. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.; Manning, M.L.; Amack, J.D. Quantitative description of fluid flows produced by left-right cilia in zebrafish. Methods Cell Biol. 2015, 127, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Ruzicka, L.; Howe, D.G.; Ramachandran, S.; Toro, S.; Van Slyke, C.E.; Bradford, Y.M.; Eagle, A.; Fashena, D.; Frazer, K.; Kalita, P.; et al. The Zebrafish Information Network: New support for non-coding genes, richer Gene Ontology annotations and the Alliance of Genome Resources. Nucleic Acids Res. 2019, 47, D867–D873. [Google Scholar] [CrossRef]
- Bowes, J.B.; Snyder, K.A.; Segerdell, E.; Gibb, R.; Jarabek, C.; Noumen, E.; Pollet, N.; Vize, P.D. Xenbase: A xenopus biology and genomics resource. Nucleic Acids Res. 2008, 36, D761–D767. [Google Scholar] [CrossRef] [Green Version]
- Olstad, E.W.; Ringers, C.; Hansen, J.N.; Wens, A.; Brandt, C.; Wachten, D.; Yaksi, E.; Jurisch-Yaksi, N. Ciliary Beating Compartmentalizes Cerebrospinal Fluid Flow in the Brain and Regulates Ventricular Development. Curr. Biol. 2019, 29, 229–241. [Google Scholar] [CrossRef] [Green Version]
- Pinto, A.L.; Rasteiro, M.; Bota, C.; Pestana, S.; Sampaio, P.; Hogg, C.; Burgoyne, T.; Lopes, S.S. Zebrafish motile cilia as a model for primary ciliary dyskinesia. Int. J. Mol. Sci. 2021, 22, 8361. [Google Scholar] [CrossRef]
- Liu, Y.; Pathak, N.; Kramer-Zucker, A.; Drummond, I.A. Notch signaling controls the differentiation of transporting epithelia and multiciliated cells in the zebrafish pronephros. Development 2007, 134, 1111–1122. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Park, A.; Sun, Z. Intraflagellar transport proteins are essential for cilia formation and for planar cell polarity. J. Am. Soc. Nephrol. 2010, 21, 1326–1333. [Google Scholar] [CrossRef] [Green Version]
- Malicki, J.; Avanesov, A.; Li, J.; Yuan, S.; Sun, Z. Analysis of Cilia Structure and Function in Zebrafish. Methods Cell Biol. 2011, 101, 39–74. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Zhang, X.; Jia, S.; Yelick, P.C.; Zhao, C. Zebrafish as a Model for Human Ciliopathies. J. Genet. Genomics 2016, 43, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Boskovski, M.T.; Yuan, S.; Pedersen, N.B.; Goth, C.K.; Makova, S.; Clausen, H.; Brueckner, M.; Khokha, M.K. The heterotaxy gene GALNT11 glycosylates Notch to orchestrate cilia type and laterality. Nature 2013, 504, 456–459. [Google Scholar] [CrossRef] [Green Version]
- Blum, M.; Beyer, T.; Weber, T.; Vick, P.; Andre, P.; Bitzer, E.; Schweickert, A. Xenopus, an ideal model system to study vertebrate left-right asymmetry. Dev. Dyn. 2009, 238, 1215–1225. [Google Scholar] [CrossRef]
- Schweickert, A.; Weber, T.; Beyer, T.; Vick, P.; Bogusch, S.; Feistel, K.; Blum, M. Cilia-Driven Leftward Flow Determines Laterality in Xenopus. Curr. Biol. 2007, 17, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Chung, M.I.; Peyrot, S.M.; LeBoeuf, S.; Park, T.J.; McGary, K.L.; Marcotte, E.M.; Wallingford, J.B. RFX2 is broadly required for ciliogenesis during vertebrate development. Dev. Biol. 2012, 363, 155–165. [Google Scholar] [CrossRef]
- Rao, V.G.; Kulkarni, S.S. Xenopus to the rescue: A model to validate and characterize candidate ciliopathy genes. Genesis 2021, 59, e23414. [Google Scholar] [CrossRef]
- Werner, M.E.; Mitchell, B.J. Understanding ciliated epithelia: The power of Xenopus. Genesis 2012, 50, 176–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Mitchell, B.J. Basal bodies in Xenopus. Cilia 2016, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Wallmeier, J.; Shiratori, H.; Dougherty, G.W.; Edelbusch, C.; Hjeij, R.; Loges, N.T.; Menchen, T.; Olbrich, H.; Pennekamp, P.; Raidt, J.; et al. TTC25 Deficiency Results in Defects of the Outer Dynein Arm Docking Machinery and Primary Ciliary Dyskinesia with Left-Right Body Asymmetry Randomization. Am. J. Hum. Genet. 2016, 99, 460–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweickert, A.; Feistel, K. The Xenopus Embryo: An Ideal Model System to Study Human Ciliopathies. Curr. Pathobiol. Rep. 2015 32 2015, 3, 115–127. [Google Scholar] [CrossRef]
- Werner, M.E.; Mitchell, B.J. Using xenopus skin to study cilia development and function. Methods Enzymol. 2013, 525, 191–217. [Google Scholar]
- Walentek, P.; Quigley, I.K. What we can learn from a tadpole about ciliopathies and airway diseases: Using systems biology in Xenopus to study cilia and mucociliary epithelia. Genesis 2017, 55, e23001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagenlocher, C.; Walentek, P.; Müller, C.; Thumberger, T.; Feistel, K. Ciliogenesis and cerebrospinal fluid flow in the developing Xenopus brain are regulated by foxj1. Cilia 2013, 2, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leventea, E.; Hazime, K.; Zhao, C.; Malicki, J. Analysis of cilia structure and function in zebrafish. Methods Cell Biol. 2016, 133, 179–227. [Google Scholar] [CrossRef]
- Brooks, E.R.; Wallingford, J.B. In vivo investigation of cilia structure and function using Xenopus. Methods Cell Biol. 2015, 127, 131–159. [Google Scholar] [CrossRef] [Green Version]
- Heasman, J. Morpholino oligos: Making sense of antisense? Dev. Biol. 2002, 243, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Auer, T.O.; Bene, F. Del CRISPR/Cas9 and TALEN-mediated knock-in approaches in zebrafish. Methods 2014, 69, 142–150. [Google Scholar] [CrossRef]
- Tandon, P.; Conlon, F.; Furlow, J.D.; Horb, M.E. Expanding the genetic toolkit in Xenopus: Approaches and opportunities for human disease modeling. Dev. Biol. 2017, 426, 325–335. [Google Scholar] [CrossRef] [Green Version]
- Salanga, C.M.; Salanga, M.C. Genotype to phenotype: Crispr gene editing reveals genetic compensation as a mechanism for phenotypic disjunction of morphants and mutants. Int. J. Mol. Sci. 2021, 22, 3472. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Ng, C.P.; Habacher, H.; Roy, S. Foxj1 transcription factors are master regulators of the motile ciliogenic program. Nat. Genet. 2008, 40, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, J.L.; Oishi, I.; Belmonte, J.C.I.; Kintner, C. The forkhead protein Foxj1 specifies node-like cilia in Xenopus and zebrafish embryos. Nat. Genet. 2008, 40, 1454–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Rayamajhi, D.; Ravi, V.; Narasimhan, V.; Chong, Y.L.; Lu, H.; Venkatesh, B.; Roy, S. Conservation as well as divergence in Mcidas function underlies the differentiation of multiciliated cells in vertebrates. Dev. Biol. 2020, 465, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Hjeij, R.; Onoufriadis, A.; Watson, C.M.; Slagle, C.E.; Klena, N.T.; Dougherty, G.W.; Kurkowiak, M.; Loges, N.T.; Diggle, C.P.; Morante, N.F.C.; et al. CCDC151 mutations cause primary ciliary dyskinesia by disruption of the outer dynein arm docking complex formation. Am. J. Hum. Genet. 2014, 95, 257–274. [Google Scholar] [CrossRef] [Green Version]
- Hjeij, R.; Lindstrand, A.; Francis, R.; Zariwala, M.A.; Liu, X.; Li, Y.; Damerla, R.; Dougherty, G.W.; Abouhamed, M.; Olbrich, H.; et al. ARMC4 mutations cause primary ciliary dyskinesia with randomization of left/right body asymmetry. Am. J. Hum. Genet. 2013, 93, 357–367. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Cao, J.; Huang, S.; Feng, D.; Zhang, W.; Zhu, X.; Yan, X. Characterization of tetratricopeptide repeat-containing proteins critical for cilia formation and function. PLoS ONE 2015, 10, e0124378. [Google Scholar] [CrossRef] [Green Version]
- Becker-Heck, A.; Zohn, I.E.; Okabe, N.; Pollock, A.; Lenhart, K.B.; Sullivan-Brown, J.; McSheene, J.; Loges, N.T.; Olbrich, H.; Haeffner, K.; et al. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat. Genet. 2011, 43, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Colantonio, J.R.; Vermot, J.; Wu, D.; Langenbacher, A.D.; Fraser, S.; Chen, J.N.; Hill, K.L. The dynein regulatory complex is required for ciliary motility and otolith biogenesis in the inner ear. Nature 2009, 457, 205–209. [Google Scholar] [CrossRef]
- Evron, T.; Philipp, M.; Lu, J.; Meloni, A.R.; Burkhalter, M.; Chen, W.; Caron, M.G. Growth arrest specific 8 (Gas8) and G protein-coupled receptor kinase 2 (GRK2) cooperate in the control of smoothened signaling. J. Biol. Chem. 2011, 286, 27676–27686. [Google Scholar] [CrossRef] [Green Version]
- Sedykh, I.; Teslaa, J.J.; Tatarsky, R.L.; Keller, A.N.; Toops, K.A.; Lakkaraju, A.; Nyholm, M.K.; Wolman, M.A.; Grinblat, Y. Novel roles for the radial spoke head protein 9 in neural and neurosensory cilia. Sci. Rep. 2016, 6, 34437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, E.H.; Huh, H.J.; Jeong, I.; Lee, N.Y.; Koh, W.J.; Park, H.C.; Ki, C.S. A nonsense variant in NME5 causes human primary ciliary dyskinesia with radial spoke defects. Clin. Genet. 2020, 98, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.W.; Nguyen, C.T.; Chen, M.H.; Yang, J.H.; Gacayan, R.; Huang, J.; Chen, J.N.; Chuang, P.T. Fused has evolved divergent roles in vertebrate Hedgehog signalling and motile ciliogenesis. Nature 2009, 459, 98–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falkenberg, L.G.; Beckman, S.A.; Ravisankar, P.; Dohn, T.E.; Waxman, J.S. Ccdc103 promotes myeloid cell proliferation and migration independent of motile cilia. DMM Dis. Model. Mech. 2021, 14, dmm048439. [Google Scholar] [CrossRef]
- Pazour, G.J.; Witman, G.B. The vertebrate primary cilium is a sensory organelle. Curr. Opin. Cell Biol. 2003, 15, 105–110. [Google Scholar] [CrossRef]
- Afzelius, B.A.; Ewetz, L.; Palmblad, J.; Udén, A.-M.; Venizelos, N. Structure and Function of Neutrophil Leukocytes from Patients with the Immotile-Cilia Syndrome. Acta Med. Scand. 1980, 208, 145–154. [Google Scholar] [CrossRef]
- Cockx, M.; Gouwy, M.; Ruytinx, P.; Lodewijckx, I.; Van Hout, A.; Knoops, S.; Pörtner, N.; Ronsse, I.; Vanbrabant, L.; Godding, V.; et al. Monocytes from patients with Primary Ciliary Dyskinesia show enhanced inflammatory properties and produce higher levels of pro-inflammatory cytokines. Sci. Rep. 2017, 7, 14657. [Google Scholar] [CrossRef] [Green Version]
- Cockx, M.; Blanter, M.; Gouwy, M.; Ruytinx, P.; Salama, S.A.; Knoops, S.; Pörtner, N.; Vanbrabant, L.; Lorent, N.; Boon, M.; et al. The antimicrobial activity of peripheral blood neutrophils is altered in patients with primary ciliary dyskinesia. Int. J. Mol. Sci. 2021, 22, 6172. [Google Scholar] [CrossRef]
- Stubbs, J.L.; Vladar, E.K.; Axelrod, J.D.; Kintner, C. Multicilin promotes centriole assembly and ciliogenesis during multiciliate cell differentiation. Nat. Cell Biol. 2012, 14, 140–147. [Google Scholar] [CrossRef] [Green Version]
- Wallmeier, J.; Al-Mutairi, D.A.; Chen, C.T.; Loges, N.T.; Pennekamp, P.; Menchen, T.; Ma, L.; Shamseldin, H.E.; Olbrich, H.; Dougherty, G.W.; et al. Mutations in CCNO result in congenital mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat. Genet. 2014, 46, 646–651. [Google Scholar] [CrossRef]
- Amirav, I.; Wallmeier, J.; Loges, N.T.; Menchen, T.; Pennekamp, P.; Mussaffi, H.; Abitbul, R.; Avital, A.; Bentur, L.; Dougherty, G.W.; et al. Systematic Analysis of CCNO Variants in a Defined Population: Implications for Clinical Phenotype and Differential Diagnosis. Hum. Mutat. 2016, 37, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, K.M.; Grimes, D.T.; Schottenfeld-Roames, J.; Werner, M.E.; Ku, T.S.J.; Kim, S.K.; Pelliccia, J.L.; Morante, N.F.C.; Mitchell, B.J.; Burdine, R.D. C21orf59/kurly Controls Both Cilia Motility and Polarization. Cell Rep. 2016, 14, 1841–1849. [Google Scholar] [CrossRef] [Green Version]
- Rachev, E.; Schuster-Gossler, K.; Fuhl, F.; Ott, T.; Tveriakhina, L.; Beckers, A.; Hegermann, J.; Boldt, K.; Mai, M.; Kremmer, E.; et al. CFAP43 modulates ciliary beating in mouse and Xenopus. Dev. Biol. 2020, 459, 109–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, L.; Ostrowski, L.E. Motile cilia genetics and cell biology: Big results from little mice. Cell. Mol. Life Sci. 2021, 78, 769–797. [Google Scholar] [CrossRef] [PubMed]
- Sosa, M.A.G.; De Gasperi, R.; Elder, G.A. Modeling human neurodegenerative diseases in transgenic systems. Hum. Genet. 2012, 131, 535–563. [Google Scholar] [CrossRef]
- Vincensini, L.; Blisnick, T.; Bastin, P. 1001 Model Organisms To Study Cilia and Flagella. Biol. Cell 2011, 103, 109–130. [Google Scholar] [CrossRef]
- Peters, L.L.; Robledo, R.F.; Bult, C.J.; Churchill, G.A.; Paigen, B.J.; Svenson, K.L. The mouse as a model for human biology: A resource guide for complex trait analysis. Nat. Rev. Genet. 2007, 8, 58–69. [Google Scholar] [CrossRef]
- Rosenthal, N.; Brown, S. The mouse ascending: Perspectives for human-disease models. Nat. Cell Biol. 2007, 9, 993–999. [Google Scholar] [CrossRef]
- Vladar, E.K.; Brody, S.L. Analysis of Ciliogenesis in Primary Culture Mouse Tracheal Epithelial Cells. Methods Enzymol. 2013, 525, 285–309. [Google Scholar]
- Eenjes, E.; Mertens, T.C.J.; Buscop-Van Kempen, M.J.; Van Wijck, Y.; Taube, C.; Rottier, R.J.; Hiemstra, P.S. A novel method for expansion and differentiation of mouse tracheal epithelial cells in culture. Sci. Rep. 2018, 8, 7349. [Google Scholar] [CrossRef] [Green Version]
- Gabrion, J.B.; Herbuté, S.; Bouillé, C.; Maurel, D.; Kuchler-Bopp, S.; Laabich, A.; Delaunoy, J.P. Ependymal and choroidal cells in culture: Characterization and functional differentiation. Microsc. Res. Tech. 1998, 41, 124–157. [Google Scholar] [CrossRef]
- Delgehyr, N.; Meunier, A.; Faucourt, M.; Grau, M.B.; Strehl, L.; Janke, C.; Spassky, N. Ependymal cell differentiation, from monociliated to multiciliated cells. Methods Cell Biol. 2015, 127, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Grondona, J.M.; Granados-Durán, P.; Fernández-Llebrez, P.; López-Ávalos, M.D. A simple method to obtain pure cultures of multiciliated ependymal cells from adult rodents. Histochem. Cell Biol. 2013, 139, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.Y.; Rosenthal, J.; Zhao, X.-Q.; Francis, R.J.; Chatterjee, B.; Sabol, S.L.; Linask, K.L.; Bracero, L.; Connelly, P.S.; Daniels, M.P.; et al. Heterotaxy and complex structural heart defects in a mutant mouse model of primary ciliary dyskinesia. J. Clin. Invest. 2007, 117, 3742–3752. [Google Scholar] [CrossRef] [Green Version]
- Ermakov, A.; Stevens, J.L.; Whitehill, E.; Robson, J.E.; Pieles, G.; Brooker, D.; Goggolidou, P.; Powles-Glover, N.; Hacker, T.; Young, S.R.; et al. Mouse mutagenesis identifies novel roles for left-right patterning genes in pulmonary, craniofacial, ocular, and limb development. Dev. Dyn. 2009, 238, 581–594. [Google Scholar] [CrossRef]
- Burnicka-Turek, O.; Steimle, J.D.; Huang, W.; Felker, L.; Kamp, A.; Kweon, J.; Peterson, M.; Reeves, R.H.; Maslen, C.L.; Gruber, P.J.; et al. Cilia gene mutations cause atrioventricular septal defects by multiple mechanisms. Hum. Mol. Genet. 2016, 25, 3011–3028. [Google Scholar] [CrossRef] [PubMed]
- Abdelhamed, Z.; Vuong, S.M.; Hill, L.; Shula, C.; Timms, A.; Beier, D.; Campbell, K.; Mangano, F.T.; Stottmann, R.W.; Goto, J. A mutation in Ccdc39 causes neonatal hydrocephalus with abnormal motile cilia development in mice. Development 2018, 145, dev154500. [Google Scholar] [CrossRef] [Green Version]
- Ha, S.; Lindsay, A.M.; Timms, A.E.; Beier, D.R. Mutations in Dnaaf1 and Lrrc48 cause hydrocephalus, laterality defects, and sinusitis in mice. G3 Genes, Genomes, Genet. 2016, 6, 2479–2487. [Google Scholar] [CrossRef] [Green Version]
- Sironen, A.; Kotaja, N.; Mulhern, H.; Wyatt, T.A.; Sisson, J.H.; Pavlik, J.A.; Miiluniemi, M.; Fleming, M.D.; Lee, L. Loss of SPEF2 function in mice results in spermatogenesis defects and primary ciliary dyskinesia. Biol. Reprod. 2011, 85, 690–701. [Google Scholar] [CrossRef] [Green Version]
- Cheong, A.; Degani, R.; Tremblay, K.D.; Mager, J. A null allele of Dnaaf2 displays embryonic lethality and mimics human ciliary dyskinesia. Hum. Mol. Genet. 2019, 28, 2775–2784. [Google Scholar] [CrossRef]
- Matsuo, M.; Shimada, A.; Koshida, S.; Saga, Y.; Takeda, H. The establishment of rotational polarity in the airway and ependymal cilia: Analysis with a novel cilium motility mutant mouse. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2013, 304, L736–L745. [Google Scholar] [CrossRef] [Green Version]
- Yin, W.; Livraghi-Butrico, A.; Sears, P.R.; Rogers, T.D.; Burns, K.A.; Grubb, B.R.; Ostrowski, L.E. Mice with a deletion of Rsph1 exhibit a low level of mucociliary clearance and develop a primary ciliary dyskinesia phenotype. Am. J. Respir. Cell Mol. Biol. 2019, 61, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Tarkar, A.; Loges, N.T.; Slagle, C.E.; Francis, R.; Dougherty, G.W.; Tamayo, J.V.; Shook, B.; Cantino, M.; Schwartz, D.; Jahnke, C.; et al. DYX1C1 is required for axonemal dynein assembly and ciliary motility. Nat. Genet. 2013, 45, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Lehti, M.S.; Henriksson, H.; Rummukainen, P.; Wang, F.; Uusitalo-Kylmälä, L.; Kiviranta, R.; Heino, T.J.; Kotaja, N.; Sironen, A. Cilia-related protein SPEF2 regulates osteoblast differentiation. Sci. Rep. 2018, 8, 859. [Google Scholar] [CrossRef] [PubMed]
- Lehti, M.S.; Zhang, F.P.; Kotaja, N.; Sironen, A. SPEF2 functions in microtubule-mediated transport in elongating spermatids to ensure proper male germ cell differentiation. Development 2017, 144, 2683–2693. [Google Scholar] [CrossRef]
- Oji, A.; Noda, T.; Fujihara, Y.; Miyata, H.; Kim, Y.J.; Muto, M.; Nozawa, K.; Matsumura, T.; Isotani, A.; Ikawa, M. CRISPR/Cas9 mediated genome editing in ES cells and its application for chimeric analysis in mice. Sci. Rep. 2016, 6, 31666. [Google Scholar] [CrossRef]
- Lu, H.; Anujan, P.; Zhou, F.; Zhang, Y.; Chong, Y.L.; Bingle, C.D.; Roy, S. Mcidas mutant mice reveal a two-step process for the specification and differentiation of multiciliated cells in mammals. Development 2019, 146, dev172643. [Google Scholar] [CrossRef] [Green Version]
- Muthusamy, N.; Vijayakumar, A.; Cheng, G.; Ghashghaei, H.T. A Knock-in Foxj1CreERT2:: GFP mouse for recombination in epithelial cells with motile cilia. Genesis 2014, 52, 350–358. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, F.; Burtscher, I.; Stauber, M.; Gossler, A.; Lickert, H. A novel Cre-inducible knock-in ARL13B-tRFP fusion cilium reporter. Genesis 2017, 55, e23073. [Google Scholar] [CrossRef]
- Hua, X.; Zeman, K.L.; Zhou, B.; Hua, Q.; Senior, B.A.; Tilley, S.L.; Bennett, W.D. Noninvasive real-time measurement of nasal mucociliary clearance in mice by pinhole gamma scintigraphy. J. Appl. Physiol. 2010, 108, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Grubb, B.R.; Livraghi-Butrico, A.; Rogers, T.D.; Yin, W.; Button, B.; Ostrowski, L.E. Reduced mucociliary clearance in old mice is associated with a decrease in muc5b mucin. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2016, 310, L860–L867. [Google Scholar] [CrossRef] [Green Version]
- Veres, T.Z.; Kopcsányi, T.; Tirri, M.; Braun, A.; Miyasaka, M.; Germain, R.N.; Jalkanen, S.; Salmi, M. Intubation-free in vivo imaging of the tracheal mucosa using two-photon microscopy. Sci. Rep. 2017, 7, 694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnelley, M.; Morgan, K.S.; Siu, K.K.W.; Fouras, A.; Farrow, N.R.; Carnibella, R.P.; Parsons, D.W. Tracking extended mucociliary transport activity of individual deposited particles: Longitudinal synchrotron X-ray imaging in live mice. J. Synchrotron Radiat. 2014, 21, 768–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Jin, X.; Prasad, R.M.; Sari, Y.; Nauli, S.M. Three types of ependymal cells with intracellular calcium oscillation are characterized by distinct cilia beating properties. J. Neurosci. Res. 2014, 92, 1199–1204. [Google Scholar] [CrossRef]
- Ansari, R.; Buj, C.; Pieper, M.; König, P.; Schweikard, A.; Hüttmann, G. Micro-anatomical and functional assessment of ciliated epithelium in mouse trachea using optical coherence phase microscopy. Opt. Express 2015, 23, 23217. [Google Scholar] [CrossRef]
- Lee, L. Riding the wave of ependymal cilia: Genetic susceptibility to hydrocephalus in primary ciliary dyskinesia. J. Neurosci. Res. 2013, 91, 1117–1132. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.; Evans, C.C.; Lee, L. Strain-dependent brain defects in mouse models of primary ciliary dyskinesia with mutations in Pcdp1 and Spef2. Neuroscience 2014, 277, 552–567. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, C.W.; Preston, C.C.; Finn, R.; Eyster, K.M.; Faustino, R.S.; Lee, L. Strain-specific differences in brain gene expression in a hydrocephalic mouse model with motile cilia dysfunction. Sci. Rep. 2018, 8, 13370. [Google Scholar] [CrossRef]
- Dixon, M.; Shoemark, A. Secondary defects detected by transmission electron microscopy in primary ciliary dyskinesia diagnostics. Ultrastruct. Pathol. 2017, 41, 390–398. [Google Scholar] [CrossRef] [Green Version]
- Hirst, R.A.; Jackson, C.L.; Coles, J.L.; Williams, G.; Rutman, A.; Goggin, P.M.; Adam, E.C.; Page, A.; Evans, H.J.; Lackie, P.M.; et al. Culture of Primary Ciliary Dyskinesia Epithelial Cells at Air-Liquid Interface Can Alter Ciliary Phenotype but Remains a Robust and Informative Diagnostic Aid. PLoS ONE 2014, 9, e89675. [Google Scholar] [CrossRef] [Green Version]
- Coles, J.L.; Thompson, J.; Horton, K.L.; Hirst, R.A.; Griffin, P.; Williams, G.M.; Goggin, P.; Doherty, R.; Lackie, P.M.; Harris, A.; et al. A Revised Protocol for Culture of Airway Epithelial Cells as a Diagnostic Tool for Primary Ciliary Dyskinesia. J. Clin. Med. 2020, 9, 3753. [Google Scholar] [CrossRef]
- Hirst, R.A.; Rutman, A.; Williams, G.; O’Callaghan, C. Ciliated Air-Liquid Cultures as an Aid to Diagnostic Testing of Primary Ciliary Dyskinesia. Chest 2010, 138, 1441–1447. [Google Scholar] [CrossRef] [PubMed]
- Schamberger, A.C.; Staab-Weijnitz, C.A.; Mise-Racek, N.; Eickelberg, O. Cigarette smoke alters primary human bronchial epithelial cell differentiation at the air-liquid interface. Sci. Rep. 2015, 5, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pifferi, M.; Montemurro, F.; Cangiotti, A.M.; Ragazzo, V.; Di Cicco, M.; Vinci, B.; Vozzi, G.; Macchia, P.; Boner, A.L. Simplified cell culture method for the diagnosis of atypical primary ciliary dyskinesia. Thorax 2009, 64, 1077–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neugebauer, P.; Endepols, H.; Mickenhagen, A.; Walger, M. Ciliogenesis in submersion and suspension cultures of human nasal epithelial cells. Eur. Arch. Oto-Rhino-Laryngology 2003, 260, 325–330. [Google Scholar] [CrossRef]
- Marthin, J.K.; Stevens, E.M.; Larsen, L.A.; Christensen, S.T.; Nielsen, K.G. Patient-specific three-dimensional explant spheroids derived from human nasal airway epithelium: A simple methodological approach for ex vivo studies of primary ciliary dyskinesia. Cilia 2017, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- de Jong, P.M.; van Sterkenburg, M.A.; Hesseling, S.C.; Kempenaar, J.A.; Mulder, A.A.; Mommaas, A.M.; Dijkman, J.H.; Ponec, M. Ciliogenesis in human bronchial epithelial cells cultured at the air-liquid interface. Am. J. Respir. Cell Mol. Biol. 1994, 10, 271–277. [Google Scholar] [CrossRef]
- Cao, X.; Coyle, J.P.; Xiong, R.; Wang, Y.; Heflich, R.H.; Ren, B.; Gwinn, W.M.; Hayden, P.; Rojanasakul, L. Invited review: Human air-liquid-interface organotypic airway tissue models derived from primary tracheobronchial epithelial cells—Overview and perspectives. Vitr. Cell. Dev. Biol.-Anim. 2021, 57, 104–132. [Google Scholar] [CrossRef]
- Horani, A.; Brody, S.L.; Ferkol, T.W.; Shoseyov, D.; Wasserman, M.G.; Ta-shma, A.; Wilson, K.S.; Bayly, P.V.; Amirav, I.; Cohen-Cymberknoh, M.; et al. CCDC65 Mutation Causes Primary Ciliary Dyskinesia with Normal Ultrastructure and Hyperkinetic Cilia. PLoS ONE 2013, 8, e72299. [Google Scholar] [CrossRef]
- Loges, N.T.; Antony, D.; Maver, A.; Deardorff, M.A.; Güleç, E.Y.; Gezdirici, A.; Nöthe-Menchen, T.; Höben, I.M.; Jelten, L.; Frank, D.; et al. Recessive DNAH9 Loss-of-Function Mutations Cause Laterality Defects and Subtle Respiratory Ciliary-Beating Defects. Am. J. Hum. Genet. 2018, 103, 995–1008. [Google Scholar] [CrossRef] [Green Version]
- Munye, M.M.; Shoemark, A.; Hirst, R.A.; Delhove, J.M.; Sharp, T.V.; McKay, T.R.; O’Callaghan, C.; Baines, D.L.; Howe, S.J.; Hart, S.L. BMI-1 extends proliferative potential of human bronchial epithelial cells while retaining their mucociliary differentiation capacity. Am. J. Physiol. Cell. Mol. Physiol. 2017, 312, L258–L267. [Google Scholar] [CrossRef]
- Cindrić, S.; Dougherty, G.W.; Olbrich, H.; Hjeij, R.; Loges, N.T.; Amirav, I.; Philipsen, M.C.; Marthin, J.K.; Nielsen, K.G.; Sutharsan, S.; et al. SPEF2- and HYDIN -Mutant Cilia Lack the Central Pair–associated Protein SPEF2, Aiding Primary Ciliary Dyskinesia Diagnostics. Am. J. Respir. Cell Mol. Biol. 2020, 62, 382–396. [Google Scholar] [CrossRef] [PubMed]
- Wallmeier, J.; Frank, D.; Shoemark, A.; Nöthe-Menchen, T.; Cindric, S.; Olbrich, H.; Loges, N.T.; Aprea, I.; Dougherty, G.W.; Pennekamp, P.; et al. De Novo Mutations in FOXJ1 Result in a Motile Ciliopathy with Hydrocephalus and Randomization of Left/Right Body Asymmetry. Am. J. Hum. Genet. 2019, 105, 1030–1039. [Google Scholar] [CrossRef]
- Hockemeyer, D.; Jaenisch, R. Induced Pluripotent Stem Cells Meet Genome Editing. Cell Stem Cell 2016, 18, 573–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firth, A.L.; Dargitz, C.T.; Qualls, S.J.; Menon, T.; Wright, R.; Singer, O.; Gage, F.H.; Khanna, A.; Verma, I.M. Generation of multiciliated cells in functional airway epithelia from human induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2014, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, E.; Sansac, C.; Fieldes, M.; Bergougnoux, A.; Bourguignon, C.; Mianné, J.; Arnould, C.; Vachier, I.; Assou, S.; Bourdin, A.; et al. Generation of the induced pluripotent stem cell line UHOMi001-A from a patient with mutations in CCDC40 gene causing Primary Ciliary Dyskinesia (PCD). Stem Cell Res. 2018, 33, 15–19. [Google Scholar] [CrossRef]
- Drick, N.; Dahlmann, J.; Sahabian, A.; Haase, A.; Göhring, G.; Lachmann, N.; Ringshausen, F.C.; Welte, T.; Martin, U.; Olmer, R. Generation of two human induced pluripotent stem cell lines (MHHi017-A, MHHi017-B) from a patient with primary ciliary dyskinesia carrying a homozygous mutation (c.7915C > T [p.Arg2639*]) in the DNAH5 gene. Stem Cell Res. 2020, 46, 101848. [Google Scholar] [CrossRef] [PubMed]
- Sahabian, A.; von Schlehdorn, L.; Drick, N.; Pink, I.; Dahlmann, J.; Haase, A.; Göhring, G.; Welte, T.; Martin, U.; Ringshausen, F.C.; et al. Generation of two hiPSC clones (MHHi019-A, MHHi019-B) from a primary ciliary dyskinesia patient carrying a homozygous deletion in the NME5 gene (c.415delA (p.Ile139Tyrfs*8)). Stem Cell Res. 2020, 48, 101988. [Google Scholar] [CrossRef]
- Xu, X.; Dong, L.; Ma, C.; Xu, X.; Wu, H.; Wang, J.; Zhou, P.; Cao, Y.; Wei, Z. Establishment of an induced pluripotent stem cell line from a patient with primary ciliary dyskinesia carrying biallelic mutations in CCNO. Stem Cell Res. 2021, 53, 102372. [Google Scholar] [CrossRef]
- Dahlmann, J.; Sahabian, A.; Drick, N.; Haase, A.; Göhring, G.; Lachmann, N.; Ringshausen, F.C.; Welte, T.; Martin, U.; Olmer, R. Generation of two hiPSC lines (MHHi016-A, MHHi016-B) from a primary ciliary dyskinesia patient carrying a homozygous 5 bp duplication (c.248_252dup (p.Gly85Cysfs*11)) in exon 1 of the CCNO gene. Stem Cell Res. 2020, 46, 101850. [Google Scholar] [CrossRef]
- Suzuki, S.; Hawkins, F.J.; Barillà, C.; Beermann, M.L.; Kotton, D.N.; Davis, B.R. Differentiation of human pluripotent stem cells into functional airway basal stem cells. STAR Protoc. 2021, 2, 100683. [Google Scholar] [CrossRef]
- Hawkins, F.J.; Suzuki, S.; Beermann, M.L.; Barillà, C.; Wang, R.; Villacorta-Martin, C.; Berical, A.; Jean, J.C.; Le Suer, J.; Matte, T.; et al. Derivation of Airway Basal Stem Cells from Human Pluripotent Stem Cells. Cell Stem Cell 2021, 28, 79–95.e8. [Google Scholar] [CrossRef] [PubMed]


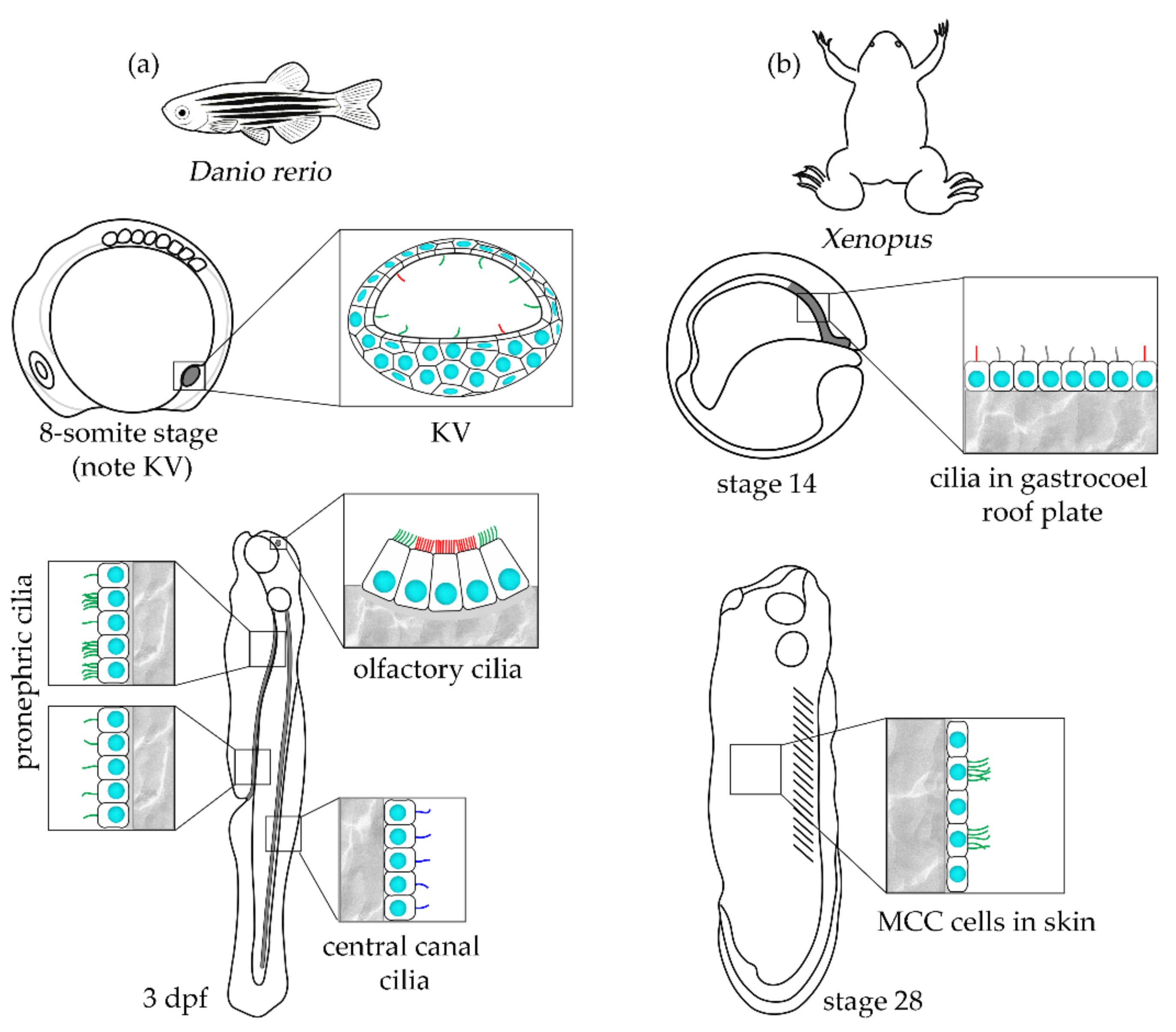
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niziolek, M.; Bicka, M.; Osinka, A.; Samsel, Z.; Sekretarska, J.; Poprzeczko, M.; Bazan, R.; Fabczak, H.; Joachimiak, E.; Wloga, D. PCD Genes—From Patients to Model Organisms and Back to Humans. Int. J. Mol. Sci. 2022, 23, 1749. https://doi.org/10.3390/ijms23031749
Niziolek M, Bicka M, Osinka A, Samsel Z, Sekretarska J, Poprzeczko M, Bazan R, Fabczak H, Joachimiak E, Wloga D. PCD Genes—From Patients to Model Organisms and Back to Humans. International Journal of Molecular Sciences. 2022; 23(3):1749. https://doi.org/10.3390/ijms23031749
Chicago/Turabian StyleNiziolek, Michal, Marta Bicka, Anna Osinka, Zuzanna Samsel, Justyna Sekretarska, Martyna Poprzeczko, Rafal Bazan, Hanna Fabczak, Ewa Joachimiak, and Dorota Wloga. 2022. "PCD Genes—From Patients to Model Organisms and Back to Humans" International Journal of Molecular Sciences 23, no. 3: 1749. https://doi.org/10.3390/ijms23031749
APA StyleNiziolek, M., Bicka, M., Osinka, A., Samsel, Z., Sekretarska, J., Poprzeczko, M., Bazan, R., Fabczak, H., Joachimiak, E., & Wloga, D. (2022). PCD Genes—From Patients to Model Organisms and Back to Humans. International Journal of Molecular Sciences, 23(3), 1749. https://doi.org/10.3390/ijms23031749