
Mitochondrial Pathophysiology on Chronic Kidney Disease





Abstract
1. Introduction
2. Chronic Kidney Disease
3. Fibrosis in the Kidney
Possible Origin of Fibroblasts
4. Epithelial–Mesenchymal Transition (EMT)
Main Potentiators of EMT That Culminate in CKD
5. The Possible Mechanisms to Delay Fibrosis Progression—Pharmacological and Metabolic Approaches
5.1. Mechanisms Associated with Signaling Pathways
5.2. Metabolic Targeting Therapies
5.3. Natural Approaches
5.4. miRNA Approaches and Others
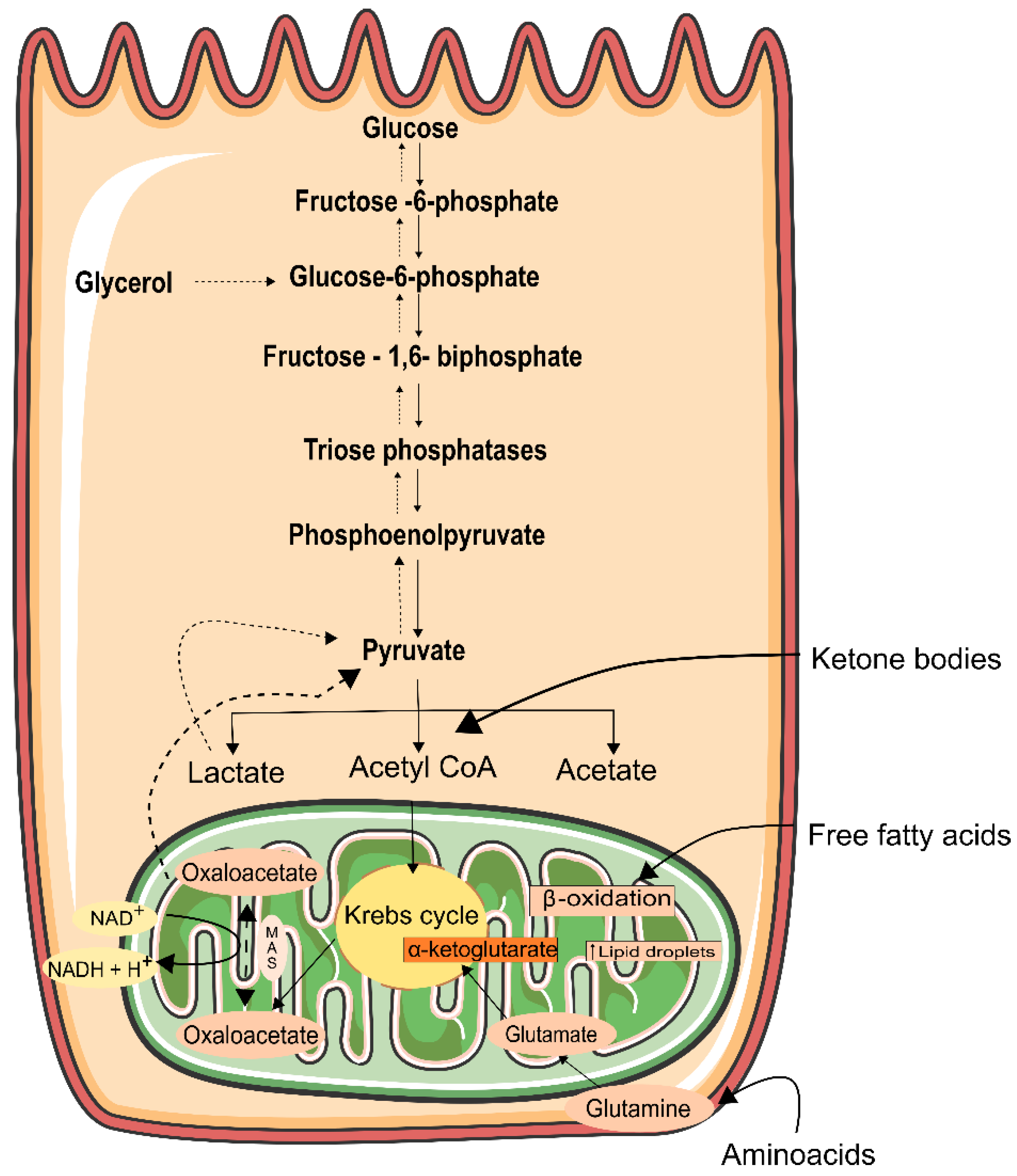
6. Energy Generation in the Kidney
7. Mitochondrial Function in the Kidney
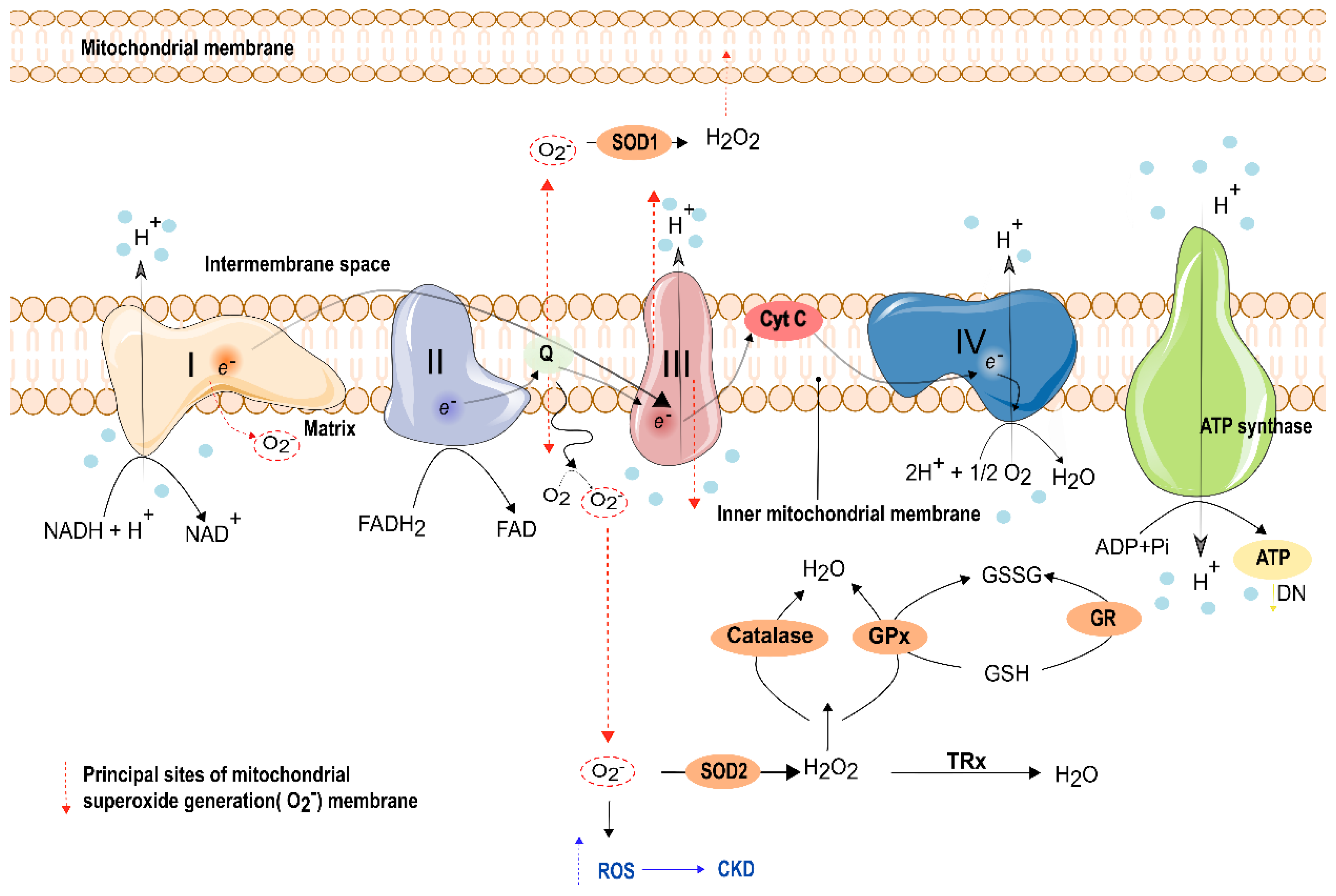
8. Oxidative Stress and Electron Transport Phosphorylation Impairment in Kidney Function
8.1. Oxidative Stress (OS) Percussions in the Kidney
8.2. Mitochondrial Oxidative Phosphorylation Impairment in the Kidney
9. Antioxidant Capacity in the Kidney
10. Targeting Kidney Mitochondria—Therapeutic Approaches
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bikbov, B.; Purcell, C.A.; Levey, A.S.; Smith, M.; Abdoli, A.; Abebe, M.; Adebayo, O.M.; Afarideh, M.; Agarwal, S.K.; Agudelo-Botero, M.; et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef]
- Leask, A. CCN3: A novel anti-fibrotic treatment in end-stage renal disease? J. Cell Commun. Signal. 2012, 6, 115–116. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, M.L.; McCulloch, T.A.; Harper, S.J.; Wheatley, T.J.; Edwards, C.M.; Feehally, J.; Furness, P.N. Early measurement of interstitial fibrosis predicts long-term renal function and graft survival in renal transplantation. Br. J. Surg. 1996, 83, 1082–1085. [Google Scholar] [CrossRef] [PubMed]
- Mutsaers, H.A.M.; Stribos, E.G.D.; Glorieux, G.; Vanholder, R.; Olinga, P. Chronic Kidney Disease and Fibrosis: The Role of Uremic Retention Solutes. Front. Med. 2015, 2, 60. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.Z.; Szeto, C.C. Mitochondrial dysfunction in diabetic kidney disease. Clin. Chim. Acta 2019, 496, 108–116. [Google Scholar] [CrossRef]
- Locatelli, F.; Canaud, B.; Eckardt, K.-U.; Stenvinkel, P.; Wanner, C.; Zoccali, C. Oxidative stress in end-stage renal disease: An emerging threat to patient outcome. Nephrol. Dial. Transplant. 2003, 18, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative Stress and Antioxidant Defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef]
- Sarko, D. Kidney-Specific Drug Delivery: Review of Opportunities, Achievements, and Challenges. J. Anal. Pharm. Res. 2016, 2. [Google Scholar] [CrossRef]
- Higgins, C.E.; Tang, J.; Mian, B.M.; Higgins, S.P.; Gifford, C.C.; Conti, D.J.; Meldrum, K.K.; Samarakoon, R.; Higgins, P.J. TGF-β1–p53 cooperativity regulates a profibrotic genomic program in the kidney: Molecular mechanisms and clinical implications. FASEB J. 2019, 33, 10596–10606. [Google Scholar] [CrossRef]
- Levey, A.S.; Coresh, J.; Balk, E.; Kausz, A.T.; Levin, A.; Steffes, M.W.; Hogg, R.J.; Perrone, R.D.; Lau, J.; Eknoyan, G. National Kidney Foundation Practice Guidelines for Chronic Kidney Disease: Evaluation, Classification, and Stratification. Ann. Intern. Med. 2003, 139, 137–147. [Google Scholar] [CrossRef]
- Weisbord, S.D.; Palevsky, P.M. Design of Clinical Trials in Acute Kidney Injury: Lessons from the Past and Future Directions. Semin. Nephrol. 2016, 36, 42–52. [Google Scholar] [CrossRef] [PubMed]
- El Nahas, A.M.; Bello, A.K. Chronic kidney disease: The global challenge. Lancet 2005, 365, 331–340. [Google Scholar] [CrossRef]
- Cooper, M.E.; Jandeleit-Dahm, K.; Thomas, M.C. Targets to retard the progression of diabetic nephropathy. Kidney Int. 2005, 68, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Uchino, S.; Kellum, J.A.; Bellomo, R.; Doig, G.S.; Morimatsu, H.; Morgera, S.; Schetz, M.; Tan, I.; Bouman, C.; Macedo, E.; et al. Acute Renal Failure in Critically Ill PatientsA Multinational, Multicenter Study. JAMA 2005, 294, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Bagshaw, S.M.; George, C.; Gibney, N.; Bellomo, R. A Multi-Center Evaluation of Early Acute Kidney Injury in Critically Ill Trauma Patients. Ren. Fail. 2008, 30, 581–589. [Google Scholar] [CrossRef]
- Emlet, D.R.; Shaw, A.D.; Kellum, J.A. Sepsis-Associated AKI: Epithelial Cell Dysfunction. Semin. Nephrol. 2015, 35, 85–95. [Google Scholar] [CrossRef]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A Common Pathway to Organ Injury and Failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef]
- Flores, J.C.; Alvo, M.; Münzenmayer, J.; Borja, H.; Morales, J.; Vega, J.; Zuñiga, C.; Müller, H. Enfermedad renal crónica: Clasificación, identificación, manejo y complicaciones. Rev. Méd. Chile 2009, 137, 137–177. [Google Scholar] [CrossRef][Green Version]
- Zoccali, C.; Vanholder, R.; Massy, Z.A.; Ortiz, A.; Sarafidis, P.; Dekker, F.W.; Fliser, D.; Fouque, D.; Heine, G.H.; Jager, K.J.; et al. The systemic nature of CKD. Nat. Rev. Nephrol. 2017, 13, 344–358. [Google Scholar] [CrossRef]
- Forbes, J.M.; Cooper, M.E. Mechanisms of Diabetic Complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef]
- Reidy, K.; Kang, H.M.; Hostetter, T.; Susztak, K. Molecular mechanisms of diabetic kidney disease. J. Clin. Investig. 2014, 124, 2333–2340. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R.; Bakris, G.L.; Bilous, R.W.; Chiang, J.L.; de Boer, I.H.; Goldstein-Fuchs, J.; Hirsch, I.B.; Kalantar-Zadeh, K.; Narva, A.S.; Navaneethan, S.D.; et al. Diabetic Kidney Disease: A Report From an ADA Consensus Conference. Diabetes Care 2014, 37, 2864–2883. [Google Scholar] [CrossRef] [PubMed]
- Grupp, C.; Müller, G.A. Renal Fibroblast Culture. Nephron Exp. Nephrol. 1999, 7, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Bonner, G.; Maeshima, Y.; Colorado, P.; Müller, G.A.; Strutz, F.; Kalluri, R. Renal Fibrosis. Am. J. Pathol. 2001, 159, 1313–1321. [Google Scholar] [CrossRef]
- Oldfield, M.D.; Bach, L.A.; Forbes, J.M.; Nikolic-Paterson, D.; McRobert, A.; Thallas, V.; Atkins, R.C.; Osicka, T.; Jerums, G.; Cooper, M.E. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). J. Clin. Investig. 2001, 108, 1853–1863. [Google Scholar] [CrossRef]
- Rastaldi, M.P.; Ferrario, F.; Giardino, L.; Dell’Antonio, G.; Grillo, C.; Grillo, P.; Strutz, F.; Müller, G.A.; Colasanti, G.; D’Amico, G. Epithelial-mesenchymal transition of tubular epithelial cells in human renal biopsies. Kidney Int. 2002, 62, 137–146. [Google Scholar] [CrossRef]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef]
- Van Blijderveen, J.C.; Straus, S.M.; Zietse, R.; Stricker, B.H.; Sturkenboom, M.C.; Verhamme, K.M. A population-based study on the prevalence and incidence of chronic kidney disease in the Netherlands. Int. Urol. Nephrol. 2013, 46, 583–592. [Google Scholar] [CrossRef]
- Kuncio, G.S.; Neilson, E.G.; Haverty, T. Mechanisms of tubulointerstitial fibrosis. Kidney Int. 1991, 39, 550–556. [Google Scholar] [CrossRef]
- Eddy, A.A. Molecular insights into renal interstitial fibrosis. J. Am. Soc. Nephrol. 1996, 7, 2495–2508. [Google Scholar] [CrossRef]
- Humphreys, B.D. Mechanisms of Renal Fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Potenta, S.E.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Fibroblasts in Kidney Fibrosis EmergeviaEndothelial-to-Mesenchymal Transition. J. Am. Soc. Nephrol. 2008, 19, 2282–2287. [Google Scholar] [CrossRef]
- Österreicher, C.H.; Penz-Österreicher, M.; Grivennikov, S.I.; Guma, M.; Koltsova, E.K.; Datz, C.; Sasik, R.; Hardiman, G.; Karin, M.; Brenner, D.A. Fibroblast-specific protein 1 identifies an inflammatory subpopulation of macrophages in the liver. Proc. Natl. Acad. Sci. USA 2010, 108, 308–313. [Google Scholar] [CrossRef]
- Leaf, I.A.; Duffield, J.S. What can target kidney fibrosis? Nephrol. Dial. Transplant. 2017, 32, i89–i97. [Google Scholar] [CrossRef]
- Sun, Y. Angiotensin Converting Enzyme and Myofibroblasts during Tissue Repair in the Rat Heart. J. Mol. Cell. Cardiol. 1996, 28, 851–858. [Google Scholar] [CrossRef]
- Badid, C.; Mounier, N.; Costa, A.M.; Desmoulière, A. Role of myofibroblasts during normal tissue repair and excessive scarring: Interest of their assessment in nephropathies. Histol. Histopathol. 2000, 15, 269–280. [Google Scholar] [CrossRef]
- Nishitani, Y.; Iwano, M.; Yamaguchi, Y.; Harada, K.; Nakatani, K.; Akai, Y.; Nishino, T.; Shiiki, H.; Kanauchi, M.; Saito, Y.; et al. Fibroblast-specific protein 1 is a specific prognostic marker for renal survival in patients with IgAN11See Editorial by Bruneval, p. 1366. Kidney Int. 2005, 68, 1078–1085. [Google Scholar] [CrossRef]
- Strutz, F.; Zeisberg, M.; Hemmerlein, B.; Sattler, B.; Hummel, K.; Becker, V.; Müller, G.A. Basic fibroblast growth factor expression is increased in human renal fibrogenesis and may mediate autocrine fibroblast proliferation. Kidney Int. 2000, 57, 1521–1538. [Google Scholar] [CrossRef]
- Margetts, P.J.; Bonniaud, P.; Liu, L.; Hoff, C.M.; Holmes, C.J.; West-Mays, J.A.; Kelly, M.M. Transient Overexpression of TGF-β1 Induces Epithelial Mesenchymal Transition in the Rodent Peritoneum. J. Am. Soc. Nephrol. 2004, 16, 425–436. [Google Scholar] [CrossRef]
- Ng, Y.-Y.; Huang, T.-P.; Yang, W.-C.; Chen, Z.-P.; Yang, A.-H.; Mu, W.; Nikolic-Paterson, D.; Atkins, R.C.; Lan, H.Y. Tubular epithelial-myofibroblast transdifferentiation in progressive tubulointerstitial fibrosis in 5/6 nephrectomized rats. Kidney Int. 1998, 54, 864–876. [Google Scholar] [CrossRef]
- Strutz, F.; Okada, H.; Lo, C.W.; Danoff, T.; Carone, R.L.; Tomaszewski, J.E.; Neilson, E.G. Identification and characterization of a fibroblast marker: FSP1. J. Cell Biol. 1995, 130, 393–405. [Google Scholar] [CrossRef]
- Humphreys, B.D.; Lin, S.-L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate Tracing Reveals the Pericyte and Not Epithelial Origin of Myofibroblasts in Kidney Fibrosis. Am. J. Pathol. 2010, 176, 85–97. [Google Scholar] [CrossRef]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Kusaba, T.; Lalli, M.; Kramann, R.; Kobayashi, A.; Humphreys, B.D. Differentiated kidney epithelial cells repair injured proximal tubule. Proc. Natl. Acad. Sci. USA 2013, 111, 1527–1532. [Google Scholar] [CrossRef]
- Angelotti, M.L.; Ronconi, E.; Ballerini, L.; Peired, A.; Mazzinghi, B.; Sagrinati, C.; Parente, E.; Gacci, M.; Carini, M.; Rotondi, M.; et al. Characterization of Renal Progenitors Committed Toward Tubular Lineage and Their Regenerative Potential in Renal Tubular Injury. Stem Cells 2012, 30, 1714–1725. [Google Scholar] [CrossRef]
- Grácio, P.C.; Gonçalves-Dias, C.; Lopes-Coelho, F.; Monteiro, E.C.; Serpa, J.; da Silva, C.L.; Pereira, S.A. Changes in N-acetyltransferase 8 in kidney tubular cell: Injury, recovery and mesenchymal stromal cell-based therapy. In Proceedings of the 6th IEEE Portuguese Meeting on Bioengineering, ENBENG 2019, Lisbon, Portugal, 22–23 February 2019. [Google Scholar]
- Burns, W.C.; Twigg, S.M.; Forbes, J.; Pete, J.; Tikellis, C.; Thallas-Bonke, V.; Thomas, M.; Cooper, M.E.; Kantharidis, P. Connective Tissue Growth Factor Plays an Important Role in Advanced Glycation End Product–Induced Tubular Epithelial-to-Mesenchymal Transition: Implications for Diabetic Renal Disease. J. Am. Soc. Nephrol. 2006, 17, 2484–2494. [Google Scholar] [CrossRef]
- Yang, J.; Liu, Y. Dissection of Key Events in Tubular Epithelial to Myofibroblast Transition and Its Implications in Renal Interstitial Fibrosis. Am. J. Pathol. 2001, 159, 1465–1475. [Google Scholar] [CrossRef]
- Gagliardini, E.; Benigni, A. Role of anti-TGF-β antibodies in the treatment of renal injury. Cytokine Growth Factor Rev. 2006, 17, 89–96. [Google Scholar] [CrossRef]
- Klahr, S.; Morrissey, J.J. The role of vasoactive compounds, growth factors and cytokines in the progression of renal disease. Kidney Int. 2000, 57, S7–S14. [Google Scholar] [CrossRef]
- Desmoulière, A.; Geinoz, A.; Gabbiani, F. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 1993, 122, 103–111. [Google Scholar] [CrossRef]
- Rodriguezpena, A.B.; Prieto, M.; Duwel, A.; Rivas, J.V.; Eleno, N.; Pérez-Barriocanal, F.; Arévalo, M.; Smith, J.D.; Vary, C.P.H.; Bernabeu, C.; et al. Up-regulation of endoglin, a TGF-β-binding protein, in rats with experimental renal fibrosis induced by renal mass reduction. Nephrol. Dial. Transplant. 2001, 16, 34–39. [Google Scholar] [CrossRef]
- McCarty, M.F. Adjuvant strategies for prevention of glomerulosclerosis. Med Hypotheses 2006, 67, 1277–1296. [Google Scholar] [CrossRef]
- Meran, S.; Steadman, R. Fibroblasts and myofibroblasts in renal fibrosis. Int. J. Exp. Pathol. 2011, 92, 158–167. [Google Scholar] [CrossRef]
- Border, W.A.; Noble, N.A. Interactions of Transforming Growth Factor-β and Angiotensin II in Renal Fibrosis. Hypertension 1998, 31, 181–188. [Google Scholar] [CrossRef]
- Li, J.H.; Huang, X.R.; Zhu, H.; Oldfield, M.; Cooper, M.; Truong, L.D.; Johnson, R.J.; Lan, H.Y. Advanced glycation end products activate Smad signaling via TGF-β-dependent and -independent mechanisms: Implications for diabetic renal and vascular disease. FASEB J. 2003, 18, 176–178. [Google Scholar] [CrossRef]
- Wang, W.; Huang, X.R.; Canlas, E.; Oka, K.; Truong, L.D.; Deng, C.; Bhowmick, N.A.; Ju, W.; Bottinger, E.P.; Lan, H.Y. Essential Role of Smad3 in Angiotensin II–Induced Vascular Fibrosis. Circ. Res. 2006, 98, 1032–1039. [Google Scholar] [CrossRef]
- Chung, A.C.; Zhang, H.; Kong, Y.-Z.; Tan, J.-J.; Huang, X.R.; Kopp, J.B.; Lan, H.Y. Advanced Glycation End-Products Induce Tubular CTGFviaTGF-β–Independent Smad3 Signaling. J. Am. Soc. Nephrol. 2009, 21, 249–260. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, S.; Liu, S.; Li, C.; Wang, J. Role of Smad signaling in kidney disease. Int. Urol. Nephrol. 2015, 47, 1965–1975. [Google Scholar] [CrossRef]
- Whitman, M. Smads and early developmental signaling by the TGFβ superfamily. Genes Dev. 1998, 12, 2445–2462. [Google Scholar] [CrossRef]
- Moustakas, A.; Souchelnytskyi, S.; Heldin, C.H. Smad regulation in TGF-beta signal transduction. J. Cell Sci. 2001, 114. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.; Feng, X.-H. Transcriptional Activators of TGF-β Responses: Smads. Cell 1998, 95, 737–740. [Google Scholar] [CrossRef]
- Fujimoto, M.; Maezawa, Y.; Yokote, K.; Joh, K.; Kobayashi, K.; Kawamura, H.; Nishimura, M.; Roberts, A.B.; Saito, Y.; Mori, S. Mice lacking Smad3 are protected against streptozotocin-induced diabetic glomerulopathy. Biochem. Biophys. Res. Commun. 2003, 305, 1002–1007. [Google Scholar] [CrossRef]
- Liu, Z.; Huang, X.R.; Lan, H.Y. Smad3 mediates ANG II-induced hypertensive kidney disease in mice. Am. J. Physiol. Physiol. 2012, 302, F986–F997. [Google Scholar] [CrossRef]
- Sato, M.; Muragaki, Y.; Saika, S.; Roberts, A.B.; Ooshima, A. Targeted disruption of TGF-β1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Investig. 2003, 112, 1486–1494. [Google Scholar] [CrossRef]
- Zhou, L.; Fu, P.; Huang, X.R.; Liu, F.; Chung, A.C.K.; Lai, K.N.; Lan, H.Y. Mechanism of chronic aristolochic acid nephropathy: Role of Smad3. Am. J. Physiol. Physiol. 2010, 298, F1006–F1017. [Google Scholar] [CrossRef]
- Meng, X.M.; Huang, X.R.; Chung, A.C.; Qin, W.; Shao, X.; Igarashi, P.; Ju, W.; Bottinger, E.P.; Lan, H.Y. Smad2 Protects against TGF-β/Smad3-Mediated Renal Fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1477–1487. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Moustakas, A. Role of Smads in TGFβ signaling. Cell Tissue Res. 2011, 347, 21–36. [Google Scholar] [CrossRef]
- Zeisberg, M.; Hanai, J.-I.; Sugimoto, H.; Mammoto, T.; Charytan, D.; Strutz, F.; Kalluri, R. BMP-7 counteracts TGF-β1–induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003, 9, 964–968. [Google Scholar] [CrossRef]
- Motazed, R.; Colville-Nash, P.; Kwan, J.T.C.; Dockrell, M.E.C. BMP-7 and Proximal Tubule Epithelial Cells: Activation of Multiple Signaling Pathways Reveals a Novel Anti-fibrotic Mechanism. Pharm. Res. 2008, 25, 2440–2446. [Google Scholar] [CrossRef]
- Luo, D.D.; Phillips, A.; Fraser, D. Bone Morphogenetic Protein-7 Inhibits Proximal Tubular Epithelial Cell Smad3 Signaling via Increased SnoN Expression. Am. J. Pathol. 2010, 176, 1139–1147. [Google Scholar] [CrossRef]
- Wang, S.-N.; Lapage, J.; Hirschberg, R. Loss of Tubular Bone Morphogenetic Protein—7 in Diabetic Nephropathy. J. Am. Soc. Nephrol. 2001, 12, 2392–2399. [Google Scholar] [CrossRef]
- Zeisberg, M.; Bottiglio, C.; Kumar, N.; Maeshima, Y.; Strutz, F.; Müller, G.A.; Kalluri, R. Bone morphogenic protein-7 inhibits progression of chronic renal fibrosis associated with two genetic mouse models. Am. J. Physiol. Physiol. 2003, 285, F1060–F1067. [Google Scholar] [CrossRef]
- Yamada, S.; Nakamura, J.; Asada, M.; Takase, M.; Matsusaka, T.; Iguchi, T.; Yamada, R.; Tanaka, M.; Higashi, A.Y.; Okuda, T.; et al. Twisted Gastrulation, a BMP Antagonist, Exacerbates Podocyte Injury. PLoS ONE 2014, 9, e89135. [Google Scholar] [CrossRef]
- Kobori, H.; Nangaku, M.; Navar, L.G.; Nishiyama, A. The Intrarenal Renin-Angiotensin System: From Physiology to the Pathobiology of Hypertension and Kidney Disease. Pharmacol. Rev. 2007, 59, 251–287. [Google Scholar] [CrossRef]
- Chen, D.-Q.; Cao, G.; Chen, H.; Liu, D.; Su, W.; Yu, X.-Y.; Vaziri, N.D.; Liu, X.-H.; Bai, X.; Zhang, L.; et al. Gene and protein expressions and metabolomics exhibit activated redox signaling and wnt/β-catenin pathway are associated with metabolite dysfunction in patients with chronic kidney disease. Redox Biol. 2017, 12, 505–521. [Google Scholar] [CrossRef]
- Yang, T.; Xu, C. Physiology and Pathophysiology of the Intrarenal Renin-Angiotensin System: An Update. J. Am. Soc. Nephrol. 2017, 28, 1040–1049. [Google Scholar] [CrossRef]
- Edeling, M.; Ragi, G.; Huang, S.; Pavenstädt, H.; Susztak, K. Developmental signalling pathways in renal fibrosis: The roles of Notch, Wnt and Hedgehog. Nat. Rev. Nephrol. 2016, 12, 426–439. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, Y. Wnt/β-catenin signaling and renin–angiotensin system in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2016, 25, 100–106. [Google Scholar] [CrossRef]
- Turner, A.J.; Hooper, N. The angiotensin–converting enzyme gene family: Genomics and pharmacology. Trends Pharmacol. Sci. 2002, 23, 177–183. [Google Scholar] [CrossRef]
- D’Agati, V.D.; Chagnac, A.; de Vries, A.; Levi, M.; Porrini, E.; Herman-Edelstein, M.; Praga, M. Obesity-related glomerulopathy: Clinical and pathologic characteristics and pathogenesis. Nat. Rev. Nephrol. 2016, 12, 453–471. [Google Scholar] [CrossRef]
- Chen, L.; Chen, D.-Q.; Wang, M.; Liu, D.; Chen, H.; Dou, F.; Vaziri, N.D.; Zhao, Y.-Y. Role of RAS/Wnt/β-catenin axis activation in the pathogenesis of podocyte injury and tubulo-interstitial nephropathy. Chem. Interactions 2017, 273, 56–72. [Google Scholar] [CrossRef]
- Urushihara, M.; Kagami, S. Role of the intrarenal renin–angiotensin system in the progression of renal disease. Pediatr. Nephrol. 2016, 32, 1471–1479. [Google Scholar] [CrossRef]
- Strutz, F.; Zeisberg, M.; Ziyadeh, F.N.; Yang, C.Q.; Kalluri, R.; Muller, G.A.; Neilson, E.G. Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int. 2002, 61, 1714–1728. [Google Scholar] [CrossRef]
- Lipson, K.; Wong, C.; Teng, Y.; Spong, S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair 2012, 5, S24. [Google Scholar] [CrossRef]
- Gupta, S.; Clarkson, M.R.; Duggan, J.; Brady, H.R. Connective tissue growth factor: Potential role in glomerulosclerosis and tubulointerstitial fibrosis. Kidney Int. 2000, 58, 1389–1399. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Kim, S.I.; Choi, M.E. Therapeutic targets for treating fibrotic kidney diseases. Transl. Res. 2014, 165, 512–530. [Google Scholar] [CrossRef]
- Wang, S.; Denichilo, M.; Brubaker, C.; Hirschberg, R. Connective tissue growth factor in tubulointerstitial injury of diabetic nephropathy. Kidney Int. 2001, 60, 96–105. [Google Scholar] [CrossRef]
- Adler, S.G.; Schwartz, S.; Williams, M.E.; Arauz-Pacheco, C.; Bolton, W.K.; Lee, T.; Li, N.; Neff, T.B.; Urquilla, P.R.; Sewell, K.L. Phase 1 Study of Anti-CTGF Monoclonal Antibody in Patients with Diabetes and Microalbuminuria. Clin. J. Am. Soc. Nephrol. 2010, 5, 1420–1428. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Berlin, J.D.; Cosaert, J.; Kauh, J.; Chan, E.; Piha-Paul, S.A.; Amaya, A.; Tang, S.; Driscoll, K.; Kimbung, R.; et al. A phase 1 study of anti-TGFβ receptor type-II monoclonal antibody LY3022859 in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2017, 79, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Voelker, J.; Berg, P.; Sheetz, M.; Duffin, K.; Shen, T.; Moser, B.; Greene, T.; Blumenthal, S.S.; Rychlik, I.; Yagil, Y.; et al. Anti–TGF-β1 Antibody Therapy in Patients with Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016, 28, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qu, X.; Yao, J.; Caruana, G.; Ricardo, S.D.; Yamamoto, Y.; Yamamoto, H.; Bertram, J.F. Blockade of Endothelial-Mesenchymal Transition by a Smad3 Inhibitor Delays the Early Development of Streptozotocin-Induced Diabetic Nephropathy. Diabetes 2010, 59, 2612–2624. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Chen, D.-Q.; Chen, L.; Cao, G.; Zhao, H.; Liu, D.; Vaziri, N.D.; Guo, Y.; Zhao, Y.-Y. Novel inhibitors of the cellular renin-angiotensin system components, poricoic acids, target Smad3 phosphorylation and Wnt/β-catenin pathway against renal fibrosis. J. Cereb. Blood Flow Metab. 2018, 175, 2689–2708. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, S.V.; Ferreira, A.J.; Kitten, G.T.; da Silveira, K.D.; da Silva, D.A.; Santos, S.H.; Gava, E.; Castro, C.H.; Magalhães, J.A.; da Mota, R.K.; et al. Genetic deletion of the angiotensin-(1–7) receptor Mas leads to glomerular hyperfiltration and microalbuminuria. Kidney Int. 2009, 75, 1184–1193. [Google Scholar] [CrossRef]
- Lely, A.T.; Hamming, I.; van Goor, H.; Navis, G.J. Renal ACE2 expression in human kidney disease. J. Pathol. 2004, 204, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.W.; Oudit, G.Y.; Reich, H.; Kassiri, Z.; Zhou, J.; Liu, Q.C.; Backx, P.H.; Penninger, J.M.; Herzenberg, A.M.; Scholey, J.W. Loss of Angiotensin-Converting Enzyme-2 (Ace2) Accelerates Diabetic Kidney Injury. Am. J. Pathol. 2007, 171, 438–451. [Google Scholar] [CrossRef]
- Mizuiri, S.; Aoki, T.; Hemmi, H.; Arita, M.; Sakai, K.; Aikawa, A. Urinary ACE2 in patients with CKD. Nephrology 2011. [Google Scholar] [CrossRef]
- Wysocki, J.; Goodling, A.; Burgaya, M.; Whitlock, K.; Ruzinski, J.; Batlle, D.; Afkarian, M. Urine RAS components in mice and people with type 1 diabetes and chronic kidney disease. Am. J. Physiol. Physiol. 2017, 313, F487–F494. [Google Scholar] [CrossRef]
- Zhong, J.; Guo, D.; Chen, C.B.; Wang, W.; Schuster, M.; Loibner, H.; Penninger, J.; Scholey, J.W.; Kassiri, Z.; Oudit, G.Y. Prevention of Angiotensin II–Mediated Renal Oxidative Stress, Inflammation, and Fibrosis by Angiotensin-Converting Enzyme 2. Hypertension 2011, 57, 314–322. [Google Scholar] [CrossRef]
- Wysocki, J.; Ye, M.; Rodriguez, E.; González-Pacheco, F.R.; Barrios, C.; Evora, K.; Schuster, M.; Loibner, H.; Brosnihan, K.B.; Ferrario, C.M.; et al. Targeting the Degradation of Angiotensin II With Recombinant Angiotensin-Converting Enzyme 2. Hypertension 2010, 55, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Oudit, G.Y.; Liu, G.C.; Zhong, J.; Basu, R.; Chow, F.L.; Zhou, J.; Loibner, H.; Janzek, E.; Schuster, M.; Penninger, J.M.; et al. Human Recombinant ACE2 Reduces the Progression of Diabetic Nephropathy. Diabetes 2009, 59, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Haschke, M.; Schuster, M.; Poglitsch, M.; Loibner, H.; Salzberg, M.; Bruggisser, M.; Penninger, J.; Krähenbühl, S. Pharmacokinetics and Pharmacodynamics of Recombinant Human Angiotensin-Converting Enzyme 2 in Healthy Human Subjects. Clin. Pharmacokinet. 2013, 52, 783–792. [Google Scholar] [CrossRef]
- Williams, V.R.; Scholey, J.W. Angiotensin-converting enzyme 2 and renal disease. Curr. Opin. Nephrol. Hypertens. 2018, 27, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Kruger, B.; Krick, S.; Dhillon, N.; Lerner, S.M.; Ames, S.; Bromberg, J.S.; Lin, M.; Walsh, L.; Vella, J.; Fischereder, M.; et al. Donor Toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proc. Natl. Acad. Sci. USA 2009, 106, 3390–3395. [Google Scholar] [CrossRef]
- Agrawal, S.; Tripathi, G.; Rangaswamy, D.; Borkar, M.; Prasad, N.; Sharma, R.K.; Sankhwar, S.N. Interleukin-1 gene cluster variants in hemodialysis patients with end stage renal disease: An association and meta-analysis. Indian J. Nephrol. 2015, 25, 34–42. [Google Scholar] [CrossRef]
- Ho, J.E.; Chen, W.-Y.; Chen, M.-H.; Larson, M.G.; McCabe, E.L.; Cheng, S.; Ghorbani, A.; Coglianese, E.; Emilsson, V.; Johnson, A.D.; et al. Common genetic variation at the IL1RL1 locus regulates IL-33/ST2 signaling. J. Clin. Investig. 2013, 123, 4208–4218. [Google Scholar] [CrossRef]
- Park, J.S.; Choi, H.-I.; Kim, D.-H.; Kim, C.S.; Bae, E.H.; Ma, S.K.; Kim, S.W. RON Receptor Tyrosine Kinase Regulates Epithelial Mesenchymal Transition and the Expression of Pro-Fibrotic Markers via Src/Smad Signaling in HK-2 and NRK49F Cells. Int. J. Mol. Sci. 2019, 20, 5489. [Google Scholar] [CrossRef]
- Floege, J.; Eitner, F.; Alpers, C.E. A New Look at Platelet-Derived Growth Factor in Renal Disease. J. Am. Soc. Nephrol. 2007, 19, 12–23. [Google Scholar] [CrossRef]
- Chen, J.; Chen, J.-K.; Nagai, K.; Plieth, D.; Tan, M.; Lee, T.-C.; Threadgill, D.; Neilson, E.G.; Harris, R.C. EGFR Signaling Promotes TGFβ-Dependent Renal Fibrosis. J. Am. Soc. Nephrol. 2011, 23, 215–224. [Google Scholar] [CrossRef]
- Campistol, J.M.; Iñigo, P.; Jiménez, W.; Lario, S.; Clesca, P.H.; Oppenheimer, F.; Rivera, F. Losartan decreases plasma levels of TGF-β1 in transplant patients with chronic allograft nephropathy. Kidney Int. 1999, 56, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Eltayeb, B.O.; McGowan, T.A.; Dunn, S.R.; Alzahabi, B.; Rohde, R.; Ziyadeh, F.N.; Lewis, E.J. Captopril-induced reduction of serum levels of transforming growth Factor-β1 correlates with long-term renoprotection in insulin-dependent diabetic patients. Am. J. Kidney Dis. 1999, 34, 818–823. [Google Scholar] [CrossRef]
- Morizane, R.; Bonventre, J.V. Kidney Organoids: A Translational Journey. Trends Mol. Med. 2017, 23, 246–263. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Kwan, J.Y.Y.; Yip, K.; Liu, P.P.; Liu, F.-F. Targeting metabolic dysregulation for fibrosis therapy. Nat. Rev. Drug Discov. 2019, 19, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Jiang, L.; Xu, J.; Bai, F.; Zhou, Y.; Yuan, Q.; Luo, J.; Zen, K.; Yang, J. Inhibiting aerobic glycolysis suppresses renal interstitial fibroblast activation and renal fibrosis. Am. J. Physiol. Physiol. 2017, 313, F561–F575. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. The Ascending GLP-1 Road From Clinical Safety to Reduction of Cardiovascular Complications. Diabetes 2018, 67, 1710–1719. [Google Scholar] [CrossRef]
- De Lucas, M.D.G.; Bueno, B.A.; Sierra, J.O. Liraglutide preserves renal function in overweight diabetic patients with stage 3 chronic kidney disease. Eur. J. Intern. Med. 2017, 44, e28–e29. [Google Scholar] [CrossRef]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef]
- Jia, Y.; Zheng, Z.; Guan, M.; Zhang, Q.; Li, Y.; Wang, L.; Xue, Y. Exendin-4 ameliorates high glucose-induced fibrosis by inhibiting the secretion of miR-192 from injured renal tubular epithelial cells. Exp. Mol. Med. 2018, 50, 1–13. [Google Scholar] [CrossRef]
- Kawanami, D.; Takashi, Y. GLP-1 Receptor Agonists in Diabetic Kidney Disease: From Clinical Outcomes to Mechanisms. Front. Pharmacol. 2020, 11, 967. [Google Scholar] [CrossRef]
- Vallon, V. The Mechanisms and Therapeutic Potential of SGLT2 Inhibitors in Diabetes Mellitus. Annu. Rev. Med. 2015, 66, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Parving, H.-H.; Lambers-Heerspink, H.; De Zeeuw, D. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1799–1802. [Google Scholar] [CrossRef]
- Vallon, V.; Richter, K.; Blantz, R.C.; Thomson, S.; Osswald, H. Glomerular hyperfiltration in experimental diabetes mellitus: Potential role of tubular reabsorption. J. Am. Soc. Nephrol. 1999, 10. [Google Scholar] [CrossRef]
- Gagnon, L.; Leduc, M.; Thibodeau, J.-F.; Zhang, M.-Z.; Grouix, B.; Sarra-Bournet, F.; Gagnon, W.; Hince, K.; Tremblay, M.; Geerts, L.; et al. A Newly Discovered Antifibrotic Pathway Regulated by Two Fatty Acid Receptors. Am. J. Pathol. 2018, 188, 1132–1148. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.-A.; Han, S.H.; Chinga, F.; Park, A.S.D.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Yanagisawa, M.; Kurihara, H.; Kimura, S.; Tomobe, Y.; Kobayashi, M.; Mitsui, Y.; Yazaki, Y.; Goto, K.; Masaki, T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988, 332, 411–415. [Google Scholar] [CrossRef]
- Hocher, B.; Thöne-Reineke, C.; Rohmeiss, P.; Schmager, F.; Slowinski, T.; Burst, V.; Siegmund, F.; Quertermous, T.; Bauer, C.; Neumayer, H.H.; et al. Endothelin-1 transgenic mice develop glomerulosclerosis, interstitial fibrosis, and renal cysts but not hypertension. J. Clin. Investig. 1997, 99, 1380–1389. [Google Scholar] [CrossRef]
- Simonson, M.S.; Ismail-Beigi, F. Endothelin-1 Increases Collagen Accumulation in Renal Mesangial Cells by Stimulating a Chemokine and Cytokine Autocrine Signaling Loop. J. Biol. Chem. 2011, 286, 11003–11008. [Google Scholar] [CrossRef]
- Shin, S.-J.; Lee, Y.-J.; Tsai, J.-H. The correlation of plasma and urine endothelin-1 with the severity of nephropathy in Chinese patients with Type 2 diabetes. Scand. J. Clin. Lab. Investig. 1996, 56, 571–576. [Google Scholar] [CrossRef]
- Sasser, J.M.; Sullivan, J.C.; Hobbs, J.L.; Yamamoto, T.; Pollock, D.M.; Carmines, P.K.; Pollock, J.S. Endothelin A Receptor Blockade Reduces Diabetic Renal Injuryviaan Anti-Inflammatory Mechanism. J. Am. Soc. Nephrol. 2006, 18, 143–154. [Google Scholar] [CrossRef]
- Gagliardini, E.; Corna, D.; Zoja, C.; Sangalli, F.; Carrara, F.; Rossi, M.; Conti, S.; Rottoli, D.; Longaretti, L.; Remuzzi, A.; et al. Unlike each drug alone, lisinopril if combined with avosentan promotes regression of renal lesions in experimental diabetes. Am. J. Physiol. Physiol. 2009, 297, F1448–F1456. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.F.E.; Green, D.; Jamerson, K.; Ruilope, L.M.; Kuranoff, S.J.; Littke, T.; Viberti, G.; for the ASCEND Study Group. Avosentan for Overt Diabetic Nephropathy. J. Am. Soc. Nephrol. 2010, 21, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Deng, Y.; Chen, Y.; Chuang, P.Y.; He, J.C. Therapeutic use of traditional Chinese herbal medications for chronic kidney diseases. Kidney Int. 2013, 84, 1108–1118. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Gong, W.; Chen, Z.; Huang, J.; Chen, Q.; Huang, H.; Zhao, C. Emodin self-emulsifying platform ameliorates the expression of FN, ICAM-1 and TGF-β1 in AGEs-induced glomerular mesangial cells by promoting absorption. Eur. J. Pharm. Sci. 2017, 99, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Wu, G.; Song, Y.; Wang, L.; Tu, L.; Zhang, L.; Zhang, C. Celastrol-Induced Suppression of the MiR-21/ERK Signalling Pathway Attenuates Cardiac Fibrosis and Dysfunction. Cell. Physiol. Biochem. 2016, 38, 1928–1938. [Google Scholar] [CrossRef]
- Divya, T.; Sureshkumar, A.; Sudhandiran, G. Autophagy induction by celastrol augments protection against bleomycin-induced experimental pulmonary fibrosis in rats: Role of adaptor protein p62/ SQSTM1. Pulm. Pharmacol. Ther. 2017, 45, 47–61. [Google Scholar] [CrossRef]
- Tang, M.; Cao, X.; Zhang, K.; Li, Y.; Zheng, Q.-Y.; Li, G.-Q.; He, Q.-H.; Li, S.-J.; Xu, G.-L.; Zhang, K.-Q. Celastrol alleviates renal fibrosis by upregulating cannabinoid receptor 2 expression. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Ning, G.; Deb, D.K.; Kong, J.; Li, Y.C. Combination therapy with AT1 blocker and vitamin D analog markedly ameliorates diabetic nephropathy: Blockade of compensatory renin increase. Proc. Natl. Acad. Sci. USA 2008, 105, 15896–15901. [Google Scholar] [CrossRef]
- Wang, Y.; Deb, D.K.; Zhang, Z.; Sun, T.; Liu, W.; Yoon, D.; Kong, J.; Chen, Y.; Chang, A.; Li, Y.C. Vitamin D Receptor Signaling in Podocytes Protects against Diabetic Nephropathy. J. Am. Soc. Nephrol. 2012, 23, 1977–1986. [Google Scholar] [CrossRef]
- Xiong, M.; Gong, J.; Liu, Y.; Xiang, R.; Tan, X. Loss of vitamin D receptor in chronic kidney disease: A potential mechanism linking inflammation to epithelial-to-mesenchymal transition. Am. J. Physiol. Physiol. 2012, 303, F1107–F1115. [Google Scholar] [CrossRef]
- Nakai, K.; Fujii, H.; Kono, K.; Goto, S.; Kitazawa, R.; Kitazawa, S.; Hirata, M.; Shinohara, M.; Fukagawa, M.; Nishi, S. Vitamin D Activates the Nrf2-Keap1 Antioxidant Pathway and Ameliorates Nephropathy in Diabetic Rats. Am. J. Hypertens. 2013, 27, 586–595. [Google Scholar] [CrossRef] [PubMed]
- de Zeeuw, D.; Agarwal, R.; Amdahl, M.; Audhya, P.; Coyne, D.; Garimella, T.; Parving, H.-H.; Pritchett, Y.; Remuzzi, G.; Ritz, E.; et al. Selective vitamin D receptor activation with paricalcitol for reduction of albuminuria in patients with type 2 diabetes (VITAL study): A randomised controlled trial. Lancet 2010, 376, 1543–1551. [Google Scholar] [CrossRef]
- Yu, G.; Tzouvelekis, A.; Wang, R.; Herazo-Maya, J.D.; Ibarra, G.H.; Srivastava, A.; Werneck-De-Castro, J.P.; DeIuliis, G.; Ahangari, F.; Woolard, T.; et al. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat. Med. 2017, 24, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Merino, E.; Orozco, R.M.; Ruíz-Llorente, L.; Martínez-Iglesias, O.A.; Velasco-Martín, J.P.; Montero-Pedrazuela, A.; Fanjul-Rodríguez, L.; Contreras-Jurado, C.; Regadera, J.; Aranda, A. Thyroid hormones inhibit TGF-β signaling and attenuate fibrotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E3451–E3460. [Google Scholar] [CrossRef]
- Lu, X.; Chen, Z.; Liang, H.; Li, Z.; Zou, X.; Luo, H.; Guo, W.; Xu, L. Thyroid hormone inhibits TGFβ1 induced renal tubular epithelial to mesenchymal transition by increasing miR34a expression. Cell. Signal. 2013, 25, 1949–1954. [Google Scholar] [CrossRef] [PubMed]
- Morishita, Y.; Yoshizawa, H.; Watanabe, M.; Ishibashi, K.; Muto, S.; Kusano, E.; Nagata, D. siRNAs targeted to Smad4 prevent renal fibrosis in vivo. Sci. Rep. 2014, 4, 6424. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Chung, A.C.K.; Chen, H.Y.; Meng, X.-M.; Lan, H.Y. Smad3-Mediated Upregulation of miR-21 Promotes Renal Fibrosis. J. Am. Soc. Nephrol. 2011, 22, 1668–1681. [Google Scholar] [CrossRef]
- Qin, W.; Chung, A.C.; Huang, X.R.; Meng, X.-M.; Hui, D.; Yu, C.-M.; Sung, J.J.Y.; Lan, H.Y. TGF-β/Smad3 Signaling Promotes Renal Fibrosis by Inhibiting miR-29. J. Am. Soc. Nephrol. 2011, 22, 1462–1474. [Google Scholar] [CrossRef]
- Chung, A.C.; Huang, X.R.; Meng, X.; Lan, H.Y. miR-192 Mediates TGF-β/Smad3-Driven Renal Fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Zarjou, A.; Yang, S.; Abraham, E.; Agarwal, A.; Liu, G. Identification of a microRNA signature in renal fibrosis: Role of miR-21. Am. J. Physiol. Physiol. 2011, 301, F793–F801. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Jha, J.C.; Hagiwara, S.; McClelland, A.D.; Jandeleit-Dahm, K.; Thomas, M.C.; Cooper, M.E.; Kantharidis, P. Transforming growth factor-β1-mediated renal fibrosis is dependent on the regulation of transforming growth factor receptor 1 expression by let-7b. Kidney Int. 2014, 85, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yao, K.; Huuskes, B.M.; Shen, H.-H.; Zhuang, J.; Godson, C.; Brennan, E.P.; Wilkinson-Berka, J.L.; Wise, A.F.; Ricardo, S.D. Mesenchymal Stem Cells Deliver Exogenous MicroRNA-let7c via Exosomes to Attenuate Renal Fibrosis. Mol. Ther. 2016, 24, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Franssen, E.J.F.; Moolenaar, F.; De Zeeuw, D.; Meijer, D.K.F. Low Molecular Weight Proteins as Carriers for Renal Drug Targeting: Naproxen Coupled to Lysozyme via the Spacer L-Lactic Acid. Pharm. Res. 1993, 10, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Sun, X.; Zhang, Z. Kidney–targeted drug delivery systems. Acta Pharm. Sin. B 2014, 4, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Haverdings, R.F.G.; Haas, M.; Greupink, A.R.; Devries, P.A.M.; Moolenaar, F.; de Zeeuw, D.; Meijer, D.K.F. Potentials and Limitations of the Low-Molecular-Weight Protein Lysozyme as a Carrier for Renal Drug Targeting. Ren. Fail. 2001, 23, 397–409. [Google Scholar] [CrossRef]
- Park, J.; Hwang, S.; Han, H. Delayed Treatment With Human Umbilical Cord Blood-Derived Stem Cells Attenuates Diabetic Renal Injury. Transplant. Proc. 2012, 44, 1123–1126. [Google Scholar] [CrossRef]
- Lim, A. Diabetic nephropathy – complications and treatment. Int. J. Nephrol. Renov. Dis. 2014, 7, 361–381. [Google Scholar] [CrossRef]
- De Caestecker, M.; Harris, R. Translating Knowledge Into Therapy for Acute Kidney Injury. Semin. Nephrol. 2018, 38, 88–97. [Google Scholar] [CrossRef]
- Sabbisetti, V.S.; Waikar, S.S.; Antoine, D.J.; Smiles, A.; Wang, C.; Ravisankar, A.; Ito, K.; Sharma, S.; Ramadesikan, S.; Lee, M.; et al. Blood Kidney Injury Molecule-1 Is a Biomarker of Acute and Chronic Kidney Injury and Predicts Progression to ESRD in Type I Diabetes. J. Am. Soc. Nephrol. 2014, 25, 2177–2186. [Google Scholar] [CrossRef]
- Nielsen, S.E.; Andersen, S.; Zdunek, D.; Hess, G.; Parving, H.-H.; Rossing, P. Tubular markers do not predict the decline in glomerular filtration rate in type 1 diabetic patients with overt nephropathy. Kidney Int. 2011, 79, 1113–1118. [Google Scholar] [CrossRef]
- Lacquaniti, A.; Donato, V.; Pintaudi, B.; Di Vieste, G.; Chirico, V.; Buemi, A.; Di Benedetto, A.; Arena, A.; Buemi, M. “Normoalbuminuric” diabetic nephropathy: Tubular damage and NGAL. Geol. Rundsch. 2013, 50, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Panduru, N.M.; Forsblom, C.; Saraheimo, M.; Thorn, L.; Bierhaus, A.; Humpert, P.M.; Groop, P.-H.; on behalf of the FinnDiane Study Group. Urinary Liver-Type Fatty Acid–Binding Protein and Progression of Diabetic Nephropathy in Type 1 Diabetes. Diabetes Care 2013, 36, 2077–2083. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Song, S.H.; Kim, I.J.; Jeon, Y.K.; Kim, B.H.; Kwak, I.S.; Lee, E.K.; Kim, Y.K. Urinary Cystatin C and Tubular Proteinuria Predict Progression of Diabetic Nephropathy. Diabetes Care 2013, 36, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Pebay-Peyroula, E.; Dahout-Gonzalez, C.; Kahn, R.; Trézéguet, V.; Lauquin, G.J.-M.; Brandolin, G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 2003, 426, 39–44. [Google Scholar] [CrossRef]
- Forbes, J.M. Mitochondria–Power Players in Kidney Function? Trends Endocrinol. Metab. 2016, 27, 441–442. [Google Scholar] [CrossRef] [PubMed]
- Ralto, K.M.; Rhee, E.P.; Parikh, S.M. NAD+ homeostasis in renal health and disease. Nat. Rev. Nephrol. 2019, 16, 99–111. [Google Scholar] [CrossRef]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Wang, Z.; Ying, Z.; Bosy-Westphal, A.; Zhang, J.; Schautz, B.; Later, W.; Heymsfield, S.B.; Müller, M.J. Specific metabolic rates of major organs and tissues across adulthood: Evaluation by mechanistic model of resting energy expenditure. Am. J. Clin. Nutr. 2010, 92, 1369–1377. [Google Scholar] [CrossRef]
- Rich, P. The molecular machinery of Keilin’s respiratory chain. Biochem. Soc. Trans. 2003, 31, 1095–1105. [Google Scholar] [CrossRef]
- Simon, N.; Hertig, A. Alteration of Fatty Acid Oxidation in Tubular Epithelial Cells: From Acute Kidney Injury to Renal Fibrogenesis. Front. Med. 2015, 2, 52. [Google Scholar] [CrossRef]
- Fornoni, A.; Merscher, S.; Kopp, J. Lipid biology of the podocyte—new perspectives offer new opportunities. Nat. Rev. Nephrol. 2014, 10, 379–388. [Google Scholar] [CrossRef]
- Kampe, K.; Sieber, J.; Orellana, J.M.; Mundel, P.; Jehle, A.W. Susceptibility of podocytes to palmitic acid is regulated by fatty acid oxidation and inversely depends on acetyl-CoA carboxylases 1 and 2. Am. J. Physiol. Physiol. 2014, 306, F401–F409. [Google Scholar] [CrossRef]
- Chung, J.-J.; Huber, T.B.; Gödel, M.; Jarad, G.; Hartleben, B.; Kwoh, C.; Keil, A.; Karpitskiy, A.; Hu, J.; Huh, C.J.; et al. Albumin-associated free fatty acids induce macropinocytosis in podocytes. J. Clin. Investig. 2015, 125, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Baldi, S.; Frascerra, S.; Astiarraga, B.; Heise, T.; Bizzotto, R.; Mari, A.; Pieber, T.R.; Muscelli, E. Shift to Fatty Substrate Utilization in Response to Sodium–Glucose Cotransporter 2 Inhibition in Subjects Without Diabetes and Patients With Type 2 Diabetes. Diabetes 2016, 65, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Rosca, M.G.; Vazquez, E.J.; Chen, Q.; Kerner, J.; Kern, T.S.; Hoppel, C.L. Oxidation of Fatty Acids Is the Source of Increased Mitochondrial Reactive Oxygen Species Production in Kidney Cortical Tubules in Early Diabetes. Diabetes 2012, 61, 2074–2083. [Google Scholar] [CrossRef] [PubMed]
- Stadler, K.; Goldberg, I.J.; Susztak, K. The Evolving Understanding of the Contribution of Lipid Metabolism to Diabetic Kidney Disease. Curr. Diabetes Rep. 2015, 15, 1–8. [Google Scholar] [CrossRef]
- Guder, W.G.; Wagner, S.; Wirthensohn, G. Metabolic fuels along the nephron: Pathways and intracellular mechanisms of interaction. Kidney Int. 1986, 29, 41–45. [Google Scholar] [CrossRef]
- Harris, S.I.; Balaban, R.S.; Barrett, L.; Mandel, L.J. Mitochondrial respiratory capacity and Na+- and K+-dependent aden-osine triphosphatase-mediated ion transport in the intact renal cell. J. Biol. Chem. 1981, 256, 10319–10328. [Google Scholar] [CrossRef]
- Uchida, S.; Endou, H. Substrate specificity to maintain cellular ATP along the mouse nephron. Am. J. Physiol. Physiol. 1988, 255, F977–F983. [Google Scholar] [CrossRef]
- Ikeda, T.; Ishimura, M.; Terasawa, H.; Ochi, H.; Ohtani, I.; Fujiyama, K.; Hoshino, T.; Tanaka, Y.; Mashiba, H. Uptake of Ketone Bodies in Perfused Hindquarter and Kidney of Starved, Thyrotoxic, and Diabetic Rats. Exp. Biol. Med. 1993, 203, 55–59. [Google Scholar] [CrossRef]
- Krebs, H.A.; Yoshida, T. Renal Gluconeogenesis. 2. the Gluconeogenic Capacity of the Kidney Cortex of Various Species. Biochem. J. 1963, 89, 398–400. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Stumvoll, M.; Welle, S.; Woerle, H.J.; Haymond, M.; Gerich, J. Relative importance of liver, kidney, and substrates in epinephrine-induced increased gluconeogenesis in humans. Am. J. Physiol. Metab. 2003, 285, E819–E826. [Google Scholar] [CrossRef] [PubMed]
- Stumvoll, M.; Meyer, C.; Perriello, G.; Kreider, M.; Welle, S.; Gerich, J. Human kidney and liver gluconeogenesis: Evidence for organ substrate selectivity. Am. J. Physiol. Metab. 1998, 274, E817–E826. [Google Scholar] [CrossRef] [PubMed]
- Gerich, J.E.; Meyer, C.; Woerle, H.J.; Stumvoll, M. Renal Gluconeogenesis. Diabetes Care 2001, 24, 382–391. [Google Scholar] [CrossRef]
- Friedrichs, D. On the stimulation of gluconeogenesis by l-Lysine in isolated rat kidney cortex tubules. Biochim. Biophys. Acta (BBA)-Gen. Subj. 1975, 392, 255–270. [Google Scholar] [CrossRef]
- Stumvoll, M.; Perriello, G.; Meyer, C.; Gerich, J. Role of glutamine in human carbohydrate metabolism in kidney and other tissues. Kidney Int. 1999, 55, 778–792. [Google Scholar] [CrossRef]
- Forbes, J.M.; Thorburn, D.R. Mitochondrial dysfunction in diabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 291–312. [Google Scholar] [CrossRef]
- Ding, Y.; Choi, M.E. Autophagy in diabetic nephropathy. J. Endocrinol. 2014, 224, R15–R30. [Google Scholar] [CrossRef]
- Duann, P.; Lin, P.-H. Mitochondria Damage and Kidney Disease. In Mitochondrial Dynamics in Cardiovascular Medicine; Advances in Experimental Medicine and Biology; Springer International Publishing: New York, NY, USA, 2017; Volume 982, pp. 529–551. [Google Scholar] [CrossRef]
- Wang, K.; Kestenbaum, B. Proximal Tubular Secretory Clearance. Clin. J. Am. Soc. Nephrol. 2018, 13, 1291–1296. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.C.; Coughlan, M. Mitochondrial dysfunction and mitophagy: The beginning and end to diabetic nephropathy? J. Cereb. Blood Flow Metab. 2014, 171, 1917–1942. [Google Scholar] [CrossRef]
- Tang, C.; Dong, Z. Mitochondria in Kidney Injury: When the Power Plant Fails. J. Am. Soc. Nephrol. 2016, 27, 1869–1872. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.-E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A Mitochondrial Protein Compendium Elucidates Complex I Disease Biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, P.M. Renal Oxygen Delivery: Matching Delivery to Metabolic Demand. Clin. Exp. Pharmacol. Physiol. 2006, 33, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Soltoff, S.P. ATP and the Regulation of Renal Cell Function. Annu. Rev. Physiol. 1986, 48, 9–31. [Google Scholar] [CrossRef] [PubMed]
- Chaiyarit, S.; Thongboonkerd, V. Mitochondrial Dysfunction and Kidney Stone Disease. Front. Physiol. 2020, 11, 566506. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Thorburn, D.R.; Penfold, S.A.; Laskowski, A.; Harcourt, B.E.; Sourris, K.C.; Tan, A.L.; Fukami, K.; Thallas-Bonke, V.; Nawroth, P.P.; et al. RAGE-Induced Cytosolic ROS Promote Mitochondrial Superoxide Generation in Diabetes. J. Am. Soc. Nephrol. 2009, 20, 742–752. [Google Scholar] [CrossRef]
- Peng, M.; Jarett, L.; Meade, R.; Madaio, M.P.; Hancock, W.W.; George, A.L.; Neilson, E.G.; Gasser, D.L. Mutant prenyltransferase-like mitochondrial protein (PLMP) and mitochondrial abnormalities in kd/kd mice. Kidney Int. 2004, 66, 20–28. [Google Scholar] [CrossRef]
- Kaneda, K.; Iwao, J.; Sakata, N.; Takebayashi, S. Correlation between MitochondriaI Enlargement in RenaI Proximal Tubules and Microalbuminuria in Rats with Early Streptozotocin-induced Diabetes. Acta Pathol. Jpn. 1992, 42, 855–860. [Google Scholar] [CrossRef]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2018, 34, 975–991. [Google Scholar] [CrossRef]
- Ling, X.C.; Kuo, K.-L. Oxidative stress in chronic kidney disease. Ren. Replace. Ther. 2018, 4, 53. [Google Scholar] [CrossRef]
- Podkowińska, A.; Formanowicz, D. Chronic Kidney Disease as Oxidative Stress- and Inflammatory-Mediated Cardiovascular Disease. Antioxidants 2020, 9, 752. [Google Scholar] [CrossRef] [PubMed]
- Quoilin, C.; Mouithys-Mickalad, A.; Lécart, S.; Fontaine-Aupart, M.-P.; Hoebeke, M. Evidence of oxidative stress and mitochondrial respiratory chain dysfunction in an in vitro model of sepsis-induced kidney injury. Biochim. Biophys. Acta 2014, 1837, 1790–1800. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.B.; Yu, M.-R.; Yang, Y.; Jiang, Z.; Ha, H. Reactive Oxygen Species-Regulated Signaling Pathways in Diabetic Nephropathy. J. Am. Soc. Nephrol. 2003, 14, S241–S245. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Xu, X.; Zhao, C.; Zhao, M.; Wang, H.; Zhang, B.; Wang, N.; Mao, H.; Zhang, A.; Xing, C. The roles of oxidative stress, endoplasmic reticulum stress, and autophagy in aldosterone/mineralocorticoid receptor-induced podocyte injury. Lab. Investig. 2015, 95, 1374–1386. [Google Scholar] [CrossRef] [PubMed]
- Mehr, A.P.; Tran, M.; Ralto, K.M.; Leaf, D.; Washco, V.; Messmer, J.; Lerner, A.; Kher, A.; Kim, S.H.; Khoury, C.C.; et al. De novo NAD+ biosynthetic impairment in acute kidney injury in humans. Nat. Med. 2018, 24, 1351–1359. [Google Scholar] [CrossRef]
- Zheng, M.; Cai, J.; Liu, Z.; Shu, S.; Wang, Y.; Tang, C.; Dong, Z. Nicotinamide reduces renal interstitial fibrosis by suppressing tubular injury and inflammation. J. Cell. Mol. Med. 2019, 23, 3995–4004. [Google Scholar] [CrossRef]
- Feng, Y.; Ren, J.; Gui, Y.; Wei, W.; Shu, B.; Lu, Q.; Xue, X.; Sun, X.; He, W.; Yang, J.; et al. Wnt/β-Catenin–Promoted Macrophage Alternative Activation Contributes to Kidney Fibrosis. J. Am. Soc. Nephrol. 2017, 29, 182–193. [Google Scholar] [CrossRef]
- Zhou, L.L.; Cao, W.; Xie, C.; Tian, J.; Zhou, Z.; Zhou, Q.; Zhu, P.; Li, A.; Liu, Y.; Miyata, T.; et al. The receptor of advanced glycation end products plays a central role in advanced oxidation protein products-induced podocyte apoptosis. Kidney Int. 2012, 82, 759–770. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Ishimoto, Y.; Inagi, R. Mitochondria: A therapeutic target in acute kidney injury. Nephrol. Dial. Transplant. 2015, 31, 1062–1069. [Google Scholar] [CrossRef]
- Emma, F.; Montini, G.; Parikh, S.M.; Salviati, L. Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat. Rev. Nephrol. 2016, 12, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.; Tam, D.; Bardia, A.; Bhasin, M.; Rowe, G.C.; Kher, A.; Zsengeller, Z.K.; Akhavan-Sharif, M.R.; Khankin, E.V.; Saintgeniez, M.; et al. PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Investig. 2011, 121, 4003–4014. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Hsu, C.-C.; Hamm, G.; Darshi, M.; Diamond-Stanic, M.; Declèves, A.-E.; Slater, L.; Pennathur, S.; Stauber, J.; Dorrestein, P.C.; et al. Mass Spectrometry Imaging Reveals Elevated Glomerular ATP/AMP in Diabetes/obesity and Identifies Sphingomyelin as a Possible Mediator. eBioMedicine 2016, 7, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Czajka, A.; Malik, A.N. Hyperglycemia induced damage to mitochondrial respiration in renal mesangial and tubular cells: Implications for diabetic nephropathy. Redox Biol. 2016, 10, 100–107. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Higgins, G.C.; Nguyen, T.-V.; Penfold, S.A.; Thallas-Bonke, V.; Tan, S.M.; Ramm, G.; Van Bergen, N.J.; Henstridge, D.C.; Sourris, K.C.; et al. Deficiency in Apoptosis-Inducing Factor Recapitulates Chronic Kidney Disease via Aberrant Mitochondrial Homeostasis. Diabetes 2016, 65, 1085–1098. [Google Scholar] [CrossRef]
- Oyarzún, C.; Salinas, C.; Gómez, D.; Jaramillo, K.; Pérez, G.; Alarcón, S.; Podestá, L.; Flores, C.; Quezada, C.; Martín, R.S. Increased levels of adenosine and ecto 5′-nucleotidase (CD73) activity precede renal alterations in experimental diabetic rats. Biochem. Biophys. Res. Commun. 2015, 468, 354–359. [Google Scholar] [CrossRef]
- Smith, J.A.; Stallons, L.J.; Schnellmann, R.G. Renal cortical hexokinase and pentose phosphate pathway activation through the EGFR/Akt signaling pathway in endotoxin-induced acute kidney injury. Am. J. Physiol. Physiol. 2014, 307, F435–F444. [Google Scholar] [CrossRef]
- Zager, R.A.; Johnson, A.C.M.; Becker, K. Renal Cortical Pyruvate Depletion during AKI. J. Am. Soc. Nephrol. 2014, 25, 998–1012. [Google Scholar] [CrossRef]
- Eklund, T.; Wahlberg, J.; Ungerstedt, U.; Hillered, L. Interstitial lactate, inosine and hypoxanthine in rat kidney during normothermic ischaemia and recirculation. Acta Physiol. Scand. 1991, 143, 279–286. [Google Scholar] [CrossRef]
- Ozawa, S.; Ueda, S.; Imamura, H.; Mori, K.; Asanuma, K.; Yanagita, M.; Nakagawa, T. Glycolysis, but not Mitochondria, responsible for intracellular ATP distribution in cortical area of podocytes. Sci. Rep. 2015, 5, 18575. [Google Scholar] [CrossRef]
- Fink, B.D.; Herlein, J.A.; O’Malley, Y.; Sivitz, W.I. Endothelial Cell and Platelet Bioenergetics: Effect of Glucose and Nutrient Composition. PLoS ONE 2012, 7, e39430. [Google Scholar] [CrossRef] [PubMed]
- Sas, K.M.; Kayampilly, P.; Byun, J.; Nair, V.; Hinder, L.M.; Hur, J.; Zhang, H.; Lin, C.; Qi, N.R.; Michailidis, G.; et al. Tissue-specific metabolic reprogramming drives nutrient flux in diabetic complications. JCI Insight 2016, 1, e86976. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, Y.; Long, J.; Wang, J.; Haudek, S.B.; Overbeek, P.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Mitochondrial Fission Triggered by Hyperglycemia Is Mediated by ROCK1 Activation in Podocytes and Endothelial Cells. Cell Metab. 2012, 15, 186–200. [Google Scholar] [CrossRef]
- Sharma, K.; Karl, B.; Mathew, A.V.; Gangoiti, J.A.; Wassel, C.L.; Saito, R.; Pu, M.; Sharma, S.; You, Y.-H.; Wang, L.; et al. Metabolomics Reveals Signature of Mitochondrial Dysfunction in Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2013, 24, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Heeringa, S.F.; Chernin, G.; Chaki, M.; Zhou, W.; Sloan, A.J.; Ji, Z.; Xie, L.X.; Salviati, L.; Hurd, T.W.; Vega-Warner, V.; et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J. Clin. Investig. 2011, 121, 2013–2024. [Google Scholar] [CrossRef]
- Valnot, I.; Von Kleist-Retzow, J.-C.; Barrientos, A.; Gorbatyuk, M.; Taanman, J.-W.; Mehaye, B.; Rustin, P.; Tzagoloff, A.; Munnich, A.; Rotig, A. A mutation in the human heme A:farnesyltransferase gene (COX10) causes cytochrome c oxidase deficiency. Hum. Mol. Genet. 2000, 9, 1245–1249. [Google Scholar] [CrossRef]
- Peng, M.; Falk, M.J.; Haase, V.H.; King, R.; Polyak, E.; Selak, M.; Yudkoff, M.; Hancock, W.W.; Meade, R.; Saiki, R.; et al. Primary Coenzyme Q Deficiency in Pdss2 Mutant Mice Causes Isolated Renal Disease. PLoS Genet. 2008, 4, e1000061. [Google Scholar] [CrossRef]
- Bourdon, A.; Minai, L.; Serre, V.; Jais, J.-P.; Sarzi, E.; Aubert, S.; Chrétien, D.; De Lonlay, P.; Paquis-Flucklinger, V.; Arakawa, H.; et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat. Genet. 2007, 39, 776–780. [Google Scholar] [CrossRef]
- Tucker, E.J.; Compton, A.G.; Calvo, S.E.; Thorburn, D.R. The molecular basis of human complex I deficiency. IUBMB Life 2011, 63, 669–677. [Google Scholar] [CrossRef]
- Forbes, J.M.; Ke, B.-X.; Nguyen, T.-V.; Henstridge, D.C.; Penfold, S.A.; Laskowski, A.; Sourris, K.C.; Groschner, L.N.; Cooper, M.E.; Thorburn, D.R.; et al. Deficiency in Mitochondrial Complex I Activity Due to Ndufs6 Gene Trap Insertion Induces Renal Disease. Antioxid. Redox Signal. 2013, 19, 331–343. [Google Scholar] [CrossRef]
- Hall, A.M.; Unwin, R.J. The Not So ‘Mighty Chondrion’: Emergence of Renal Diseases due to Mitochondrial Dysfunction. Nephron Physiol. 2007, 105, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rosca, M.G.; Mustata, T.G.; Kinter, M.T.; Ozdemir, A.M.; Kern, T.S.; Szweda, L.I.; Brownlee, M.; Monnier, V.M.; Weiss, M.F. Glycation of mitochondrial proteins from diabetic rat kidney is associated with excess superoxide formation. Am. J. Physiol. Ren. Physiol. 2005, 289, F420–F430. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, M.T.; Nguyen, T.-V.; Penfold, S.A.; Higgins, G.C.; Thallas-Bonke, V.; Tan, S.M.; Van Bergen, N.J.; Sourris, K.C.; Harcourt, B.E.; Thorburn, D.R.; et al. Mapping time-course mitochondrial adaptations in the kidney in experimental diabetes. Clin. Sci. 2016, 130, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2009, 107, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Caramori, M.L.; Kim, Y.; Goldfine, A.B.; Moore, J.H.; Rich, S.S.; Mychaleckyj, J.C.; Kirkpatrick, D.; Nickerson, H.; Krolewski, A.S.; Mauer, M. Differential Gene Expression in Diabetic Nephropathy in Individuals With Type 1 Diabetes. J. Clin. Endocrinol. Metab. 2015, 100, E876–E882. [Google Scholar] [CrossRef][Green Version]
- Han, D.; Williams, E.; Cadenas, E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem. J. 2001, 353, 411–416. [Google Scholar] [CrossRef]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M. Topology of Superoxide Production from Different Sites in the Mitochondrial Electron Transport Chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef]
- Quinzii, C.M.; Lopez, L.C.; Gilkerson, R.W.; Dorado, B.; Coku, J.; Naini, A.B.; Lagier-Tourenne, C.; Schuelke, M.; Salviati, L.; Carrozzo, R.; et al. Reactive oxygen species, oxidative stress, and cell death correlate with level of CoQ 10 deficiency. FASEB J. 2010, 24, 3733–3743. [Google Scholar] [CrossRef]
- Ozaltin, F. Primary coenzyme Q10 (CoQ10) deficiencies and related nephropathies. Pediatr. Nephrol. 2013, 29, 961–969. [Google Scholar] [CrossRef]
- Diomedi-Camassei, F.; Di Giandomenico, S.; Santorelli, F.M.; Caridi, G.; Piemonte, F.; Montini, G.; Ghiggeri, G.M.; Murer, L.; Barisoni, L.; Pastore, A.; et al. COQ2 Nephropathy: A Newly Described Inherited Mitochondriopathy with Primary Renal Involvement. J. Am. Soc. Nephrol. 2007, 18, 2773–2780. [Google Scholar] [CrossRef]
- López, L.C.; Schuelke, M.; Quinzii, C.M.; Kanki, T.; Rodenburg, R.J.; Naini, A.; DiMauro, S.; Hirano, M. Leigh Syndrome with Nephropathy and CoQ10 Deficiency Due to decaprenyl diphosphate synthase subunit 2 (PDSS2) Mutations. Am. J. Hum. Genet. 2006, 79, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Salviati, L.; Sacconi, S.; Murer, L.; Zacchello, G.; Franceschini, L.; Laverda, A.M.; Basso, G.; Quinzii, C.; Angelini, C.; Hirano, M.; et al. Infantile encephalomyopathy and nephropathy with CoQ10 deficiency: A CoQ10-responsive condition. Neurology 2005, 65, 606–608. [Google Scholar] [CrossRef] [PubMed]
- Granata, S.; Zaza, G.; Simone, S.; Villani, G.; Latorre, D.; Pontrelli, P.; Carella, M.; Schena, F.P.; Grandaliano, G.; Pertosa, G. Mitochondrial dysregulation and oxidative stress in patients with chronic kidney disease. BMC Genom. 2009, 10, 388. [Google Scholar] [CrossRef] [PubMed]
- McClelland, A.D.; Herman-Edelstein, M.; Komers, R.; Jha, J.C.; Winbanks, C.E.; Hagiwara, S.; Gregorevic, P.; Kantharidis, P.; Cooper, M.E. miR-21 promotes renal fibrosis in diabetic nephropathy by targeting PTEN and SMAD7. Clin. Sci. 2015, 129, 1237–1249. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Jeninga, E.H.; Schoonjans, K.; Auwerx, J. Reversible acetylation of PGC-1: Connecting energy sensors and effectors to guarantee metabolic flexibility. Oncogene 2010, 29, 4617–4624. [Google Scholar] [CrossRef]
- Xiao, L.; Zhu, X.; Yang, S.; Liu, F.; Zhou, Z.; Zhan, M.; Xie, P.; Zhang, D.; Li, J.; Song, P.; et al. Rap1 Ameliorates Renal Tubular Injury in Diabetic Nephropathy. Diabetes 2014, 63, 1366–1380. [Google Scholar] [CrossRef]
- Rasbach, K.A.; Schnellmann, R.G.; Rasbach, K.A.; Schnellmann, R.G. Signaling of Mitochondrial Biogenesis following Oxidant Injury. J. Biol. Chem. 2007, 282, 2355–2362. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Romanello, V.; Sandri, M. Mitochondrial Quality Control and Muscle Mass Maintenance. Front. Physiol. 2016, 6, 422. [Google Scholar] [CrossRef]
- Mourier, A.; Motori, E.; Brandt, T.; Lagouge, M.; Atanassov, I.; Galinier, A.; Rappl, G.; Brodesser, S.; Hultenby, K.; Dieterich, C.; et al. Mitofusin 2 is required to maintain mitochondrial coenzyme Q levels. J. Cell Biol. 2015, 208, 429–442. [Google Scholar] [CrossRef]
- Morigi, M.; Perico, L.; Rota, C.; Longaretti, L.; Conti, S.; Rottoli, D.; Novelli, R.; Remuzzi, G.; Benigni, A. Sirtuin 3–dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Investig. 2015, 125, 715–726. [Google Scholar] [CrossRef]
- Madsen-Bouterse, S.; Zhong, Q.; Mohammad, G.; Ho, Y.-S.; Kowluru, R.A. Oxidative damage of mitochondrial DNA in diabetes and its protection by manganese superoxide dismutase. Free Radic. Res. 2010, 44, 313–321. [Google Scholar] [CrossRef]
- Ducasa, G.M.; Mitrofanova, A.; Fornoni, A. Crosstalk Between Lipids and Mitochondria in Diabetic Kidney Disease. Curr. Diabetes Rep. 2019, 19, 144. [Google Scholar] [CrossRef]
- Forman, H.J.; Davies, K.J.; Ursini, F. How do nutritional antioxidants really work: Nucleophilic tone and para-hormesis versus free radical scavenging in vivo. Free Radic. Biol. Med. 2014, 66, 24–35. [Google Scholar] [CrossRef]
- Stowe, D.F.; Camara, A.K.S. Mitochondrial Reactive Oxygen Species Production in Excitable Cells: Modulators of Mitochondrial and Cell Function. Antioxid. Redox Signal. 2009, 11, 1373–1414. [Google Scholar] [CrossRef]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef]
- Ruiz, S.; Pergola, P.E.; Zager, R.A.; Vaziri, N.D. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 2013, 83, 1029–1041. [Google Scholar] [CrossRef]
- Jiang, T.; Huang, Z.; Lin, Y.; Zhang, Z.; Fang, D.; Zhang, D.D. The Protective Role of Nrf2 in Streptozotocin-Induced Diabetic Nephropathy. Diabetes 2010, 59, 850–860. [Google Scholar] [CrossRef]
- El Gheit, R.A.; Emam, M.N. Targeting heme oxygenase-1 in early diabetic nephropathy in streptozotocin-induced diabetic rats. Physiol. Int. 2016, 103, 413–427. [Google Scholar] [CrossRef]
- Yamawaki, K.; Kanda, H.; Shimazaki, R. Nrf2 activator for the treatment of kidney diseases. Toxicol. Appl. Pharmacol. 2018, 360, 30–37. [Google Scholar] [CrossRef]
- Shin, J.H.; Kim, K.M.; Jeong, J.U.; Shin, J.M.; Kang, J.H.; Bang, K.; Kim, J.-H. Nrf2-Heme Oxygenase-1 Attenuates High-Glucose-Induced Epithelial-to-Mesenchymal Transition of Renal Tubule Cells by Inhibiting ROS-Mediated PI3K/Akt/GSK-3β Signaling. J. Diabetes Res. 2019, 2019, 1–8. [Google Scholar] [CrossRef]
- Chen, Q.; Tao, J.; Xie, X. Astaxanthin Promotes Nrf2/ARE Signaling to Inhibit HG-Induced Renal Fibrosis in GMCs. Mar. Drugs 2018, 16, 117. [Google Scholar] [CrossRef]
- Ali, B.H.; Al-Salam, S.; Al Suleimani, Y.; Al Kalbani, J.; Al Bahlani, S.; Ashique, M.; Manoj, P.; Al Dhahli, B.; Al Abri, N.; Naser, H.T.; et al. Curcumin Ameliorates Kidney Function and Oxidative Stress in Experimental Chronic Kidney Disease. Basic Clin. Pharmacol. Toxicol. 2017, 122, 65–73. [Google Scholar] [CrossRef]
- Sun, L.-N.; Chen, Z.-X.; Liu, X.-C.; Liu, H.-Y.; Guan, G.-J.; Liu, G. Curcumin ameliorates epithelial-to-mesenchymal transition of podocytes in vivo and in vitro via regulating caveolin-1. Biomed. Pharmacother. 2014, 68, 1079–1088. [Google Scholar] [CrossRef]
- Wu, W.; Geng, H.; Liu, Z.; Li, H.; Zhu, Z. Effect of curcumin on rats/mice with diabetic nephropathy: A systematic review and Meta-analysis of randomized controlled trials. J. Tradit. Chin. Med. 2014, 34, 419–429. [Google Scholar] [CrossRef]
- Prabu, S.M.; Muthumani, M. RETRACTED ARTICLE: Silibinin ameliorates arsenic induced nephrotoxicity by abrogation of oxidative stress, inflammation and apoptosis in rats. Mol. Biol. Rep. 2012, 39, 11201–11216. [Google Scholar] [CrossRef]
- Minich, D.M.; Brown, B.I. A Review of Dietary (Phyto)Nutrients for Glutathione Support. Nutrients 2019, 11, 2073. [Google Scholar] [CrossRef]
- Bajic, V.P.; Van Neste, C.; Obradovic, M.; Zafirovic, S.; Radak, D.; Bajic, V.B.; Essack, M.; Isenovic, E.R. Glutathione “Redox Homeostasis” and Its Relation to Cardiovascular Disease. Oxidative Med. Cell. Longev. 2019, 2019, 1–14. [Google Scholar] [CrossRef]
- Meister, A. Glutathione-ascorbic acid antioxidant system in animals. J. Biol. Chem. 1994, 269, 9397–9400. [Google Scholar] [CrossRef]
- Dennis, J.M.; Witting, P.K. Protective role for antioxidants in acute kidney disease. Nutrients 2017, 9, 718. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Superoxide Dismutases: Role in Redox Signaling, Vascular Function, and Diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed]
- Paller, M.S.; Neumann, T.V.; Knobloch, E.; Patten, M. Reactive oxygen species and rat renal epithelial cells during hypoxia and reoxygenation. Kidney Int. 1991, 40, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, U.A.; Haraldsson, G.; Bratell, S.; Sorensen, V.; Akerlund, S.; Pettersson, S.; Schersten, T.; Jonsson, O. ESR-measurement of oxygen radicalsin vivoafter renal ischaemia in the rabbit. Effects of pre-treatment with superoxide dismutase and heparin. Acta Physiol. Scand. 1993, 147, 263–270. [Google Scholar] [CrossRef]
- Paller, M.S.; Hoidal, J.R.; Ferris, T.F. Oxygen free radicals in ischemic acute renal failure in the rat. J. Clin. Investig. 1984, 74, 1156–1164. [Google Scholar] [CrossRef]
- Kim, J.; Seok, Y.M.; Jung, K.-J.; Park, K.M. Reactive oxygen species/oxidative stress contributes to progression of kidney fibrosis following transient ischemic injury in mice. Am. J. Physiol. Physiol. 2009, 297, F461–F470. [Google Scholar] [CrossRef]
- Wang, Z.; Holthoff, J.H.; Seely, K.A.; Pathak, E.; Spencer, H.J.; Gokden, N.; Mayeux, P.R. Development of Oxidative Stress in the Peritubular Capillary Microenvironment Mediates Sepsis-Induced Renal Microcirculatory Failure and Acute Kidney Injury. Am. J. Pathol. 2012, 180, 505–516. [Google Scholar] [CrossRef]
- Quiroz, Y.; Ferrebuz, A.; Vaziri, N.D.; Rodriguez-Iturbe, B. Effect of Chronic Antioxidant Therapy with Superoxide Dismutase-Mimetic Drug, Tempol, on Progression of Renal Disease in Rats with Renal Mass Reduction. Nephron Exp. Nephrol. 2009, 112, e31–e42. [Google Scholar] [CrossRef]
- Dugan, L.L.; You, Y.-H.; Ali, S.S.; Diamond-Stanic, M.; Miyamoto, S.; DeCleves, A.-E.; Andreyev, A.Y.; Quach, T.; Ly, S.; Shekhtman, G.; et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J. Clin. Investig. 2013, 123, 4888–4899. [Google Scholar] [CrossRef]
- Lee, H.C.; Yin, P.H.; Lu, C.Y.; Chi, C.W.; Wei, Y.H. Increase of mitochondria and mitochondrial DNA in response to oxi-dative stress in human cells. Biochem. J. 2000, 348 Pt 2, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K. Mitochondrial Hormesis and Diabetic Complications. Diabetes 2015, 64, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Small, D.M.; Coombes, J.S.; Bennett, N.; Johnson, D.W.; Gobe, G.C. Oxidative stress, anti-oxidant therapies and chronic kidney disease. Nephrology 2012, 17, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Kezic, A.; Spasojevic, I.; Lezaic, V.; Bajcetic, M. Mitochondria-Targeted Antioxidants: Future Perspectives in Kidney Ischemia Reperfusion Injury. Oxidative Med. Cell. Longev. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Snow, B.J.; Rolfe, F.L.; Lockhart, M.M.; Frampton, C.M.; O’Sullivan, J.D.; Fung, V.; Smith, R.A.; Murphy, M.P.; Taylor, K.M.; Protect Study Group. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord. 2010, 25, 1670–1674. [Google Scholar] [CrossRef]
- Chacko, B.K.; Reily, C.; Srivastava, A.; Johnson, M.S.; Ye, Y.; Ulasova, E.; Agarwal, A.; Zinn, K.; Murphy, M.; Kalyanaraman, B.; et al. Prevention of diabetic nephropathy in Ins2+/−AkitaJ mice by the mitochondria-targeted therapy MitoQ. Biochem. J. 2010, 432, 9–19. [Google Scholar] [CrossRef]
- Ward, M.S.; Flemming, N.B.; Gallo, L.A.; Fotheringham, A.; McCarthy, D.A.; Zhuang, A.; Tang, P.H.; Borg, D.J.; Shaw, H.; Harvie, B.; et al. Targeted mitochondrial therapy using MitoQ shows equivalent renoprotection to angiotensin converting enzyme inhibition but no combined synergy in diabetes. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Hou, Y.; Li, S.; Wu, M.; Wei, J.; Ren, Y.; Du, C.; Wu, H.; Han, C.; Duan, H.; Shi, Y. Mitochondria-targeted peptide SS-31 attenuates renal injury via an antioxidant effect in diabetic nephropathy. Am. J. Physiol. Physiol. 2016, 310, F547–F559. [Google Scholar] [CrossRef]
- Karaa, A.; Haas, R.; Goldstein, A.; Vockley, J.; Weaver, W.D.; Cohen, B.H. Randomized dose-escalation trial of elamipretide in adults with primary mitochondrial myopathy. Neurology 2018, 90, e1212–e1221. [Google Scholar] [CrossRef]
- Sun, J.; Zhu, H.; Wang, X.; Gao, Q.; Li, Z.; Huang, H. CoQ10 ameliorates mitochondrial dysfunction in diabetic nephropathy through mitophagy. J. Endocrinol. 2019, 240, 445–465. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Ding, W.; Wang, Y. Mito-TEMPO Alleviates Renal Fibrosis by Reducing Inflammation, Mitochondrial Dysfunction, and Endoplasmic Reticulum Stress. Oxidative Med. Cell. Longev. 2018, 2018, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Patil, N.K.; Parajuli, N.; MacMillan-Crow, L.A.; Mayeux, P.R. Inactivation of renal mitochondrial respiratory complexes and manganese superoxide dismutase during sepsis: Mitochondria-targeted antioxidant mitigates injury. Am. J. Physiol. Physiol. 2014, 306, F734–F743. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Horváth, B.; Zsengellér, Z.; Zielonka, J.; Tanchian, G.; Holovac, E.; Kechrid, M.; Patel, V.; Stillman, I.E.; Parikh, S.M.; et al. Mitochondrial-targeted antioxidants represent a promising approach for prevention of cisplatin-induced nephropathy. Free Radic. Biol. Med. 2012, 52, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Tábara, L.C.; Poveda, J.; Martin-Cleary, C.; Selgas, R.; Arduan, A.O.; Sanchez-Niño, M.D. Mitochondria-targeted therapies for acute kidney injury. Expert Rev. Mol. Med. 2014, 16, e13. [Google Scholar] [CrossRef]
- Doi, K.; Suzuki, Y.; Nakao, A.; Fujita, T.; Noiri, E. Radical scavenger edaravone developed for clinical use ameliorates ischemia/reperfusion injury in rat kidney. Kidney Int. 2004, 65, 1714–1723. [Google Scholar] [CrossRef]
- Tahara, M.; Nakayama, M.; Jin, M.B.; Fujita, M.; Suzuki, T.; Taniguchi, M.; Shimamura, T.; Furukawa, H.; Todo, S. A Radical Scavenger, Edaravone, Protects Canine Kidneys from Ischemia-Reperfusion Injury after 72 Hours of Cold Preservation and Autotransplantation. Transplantation 2005, 80, 213–221. [Google Scholar] [CrossRef]
- Panizo, N.; Rubio-Navarro, A.; Amaro-Villalobos, J.M.; Egido, J.; Moreno, J.A. Molecular Mechanisms and Novel Therapeutic Approaches to Rhabdomyolysis-Induced Acute Kidney Injury. Kidney Blood Press. Res. 2015, 40, 520–532. [Google Scholar] [CrossRef]
- Chalikias, G.; Drosos, I.; Tziakas, D.N. Prevention of Contrast-Induced Acute Kidney Injury: An Update. Cardiovasc. Drugs Ther. 2016, 30, 515–524. [Google Scholar] [CrossRef]
- Koyner, J.L.; Ali, R.S.; Murray, P. Antioxidants. Nephron Exp. Nephrol. 2008, 109, e109–e117. [Google Scholar] [CrossRef]
- Shanu, A.; Groebler, L.; Kim, H.B.; Wood, S.; Weekley, C.M.; Aitken, J.B.; Harris, H.H.; Witting, P.K. Selenium Inhibits Renal Oxidation and Inflammation But Not Acute Kidney Injury in an Animal Model of Rhabdomyolysis. Antioxid. Redox Signal. 2013, 18, 756–769. [Google Scholar] [CrossRef]
- Carr, A.C.; Frei, B. Toward a new recommended dietary allowance for vitamin C based on antioxidant and health effects in humans. Am. J. Clin. Nutr. 1999, 69, 1086–1107. [Google Scholar] [CrossRef] [PubMed]
- Buettner, G. The Pecking Order of Free Radicals and Antioxidants: Lipid Peroxidation, α-Tocopherol, and Ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Keaney, J.F., Jr. Role of Oxidative Modifications in Atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Cullen, J.J.; Buettner, G.R. Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2012, 1826, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Straaten, H.M.O.-V.; Man, A.M.S.-D.; De Waard, M.C. Vitamin C revisited. Crit. Care 2014, 18, 1–13. [Google Scholar] [CrossRef]
- Tu, H.; Li, H.; Wang, Y.; Niyyati, M.; Wang, Y.; Leshin, J.; Levine, M. Low Red Blood Cell Vitamin C Concentrations Induce Red Blood Cell Fragility: A Link to Diabetes Via Glucose, Glucose Transporters, and Dehydroascorbic Acid. eBioMedicine 2015, 2, 1735–1750. [Google Scholar] [CrossRef] [PubMed]
- Groebler, L.K.; Wang, X.S.; Kim, H.B.; Shanu, A.; Hossain, F.; McMahon, A.C.; Witting, P.K. Cosupplementation with a synthetic, lipid-soluble polyphenol and vitamin C inhibits oxidative damage and improves vascular function yet does not inhibit acute renal injury in an animal model of rhabdomyolysis. Free Radic. Biol. Med. 2012, 52, 1918–1928. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Therapeutic Options | Target | Function | Effect | References | |
|---|---|---|---|---|---|
| LY2382770 (phase II study) | TGFβ-1 | Modulator of TGFβ-1 | - | [35] | |
| Barcitinib | JAK-STAT pathway | ↓ JAK-STAT pathway | ↓proteinuria levels | [35] | |
| FG-3019 | CTGF | Modulator of TGFβ-1 | ↓microalbuminuria | [91] | |
| Triterpenoid compounds (PZC, PZD, PZE) | RAS | Smad3 signaling and TGFβ-1 signaling | ↓ TGFβ-1 | [95] | |
| ACE2 | TGFβ-1 with Smad3 signaling | Degrade Angiotensin II | ↓renal fibrosis ↓albuminuria ↓glomerular hypertrophy | [98] | |
| Imatinib | RTK | PDFGR inhibitor | ↓proliferation of fibroblasts | [110] | |
| AG1296 | RTK | FGFR blockage | - | [111] | |
| Losartan | RAS | Angiotensin II | Delay of DN | [112] | |
| Captopril | [113] | ||||
| Liraglitude semaglutide | GLP-1 | insulin secretion and suppression of glucagon | ↓albuminuria ↑GFR | [117,118,119] | |
| Empagliflozin | SGLT2 | SGLT2 inhibition | Delay the advance of renal disease | [123] | |
| PBI-4050 | FFA1R | Anagonist of FFA1R | ↓renal fibrosis | [125] | |
| C75 | CPT1 | CPT1 activator | ↓renal fibrosis | [126] | |
| Atrasentan | ETA blockers | - | ↓renal fibrosis ↓albuminuria | [131] | |
| Avosentan | [132] | ||||
| Emodin | - | - | ↓ fibronectin ↓ TGFβ-1 | [135] | |
| Celastrol | Smad3 | ↑cannabinoid receptor 2 expression | ↓renal fibrosis | [138] | |
| microRNAs | siRNA | Smad4 | Silecing of Smad4 | ↓fibrosis ↓ α-SMA expression | [147] |
| miR-21 miR-29 miR-192 | TGF-β1/SMAD signaling | Smad3 inhibitors | ↓collagen deposition ↓ fibronectin ↓α-SMA expression | [148,149,150] | |
| miRlet7c | - | - | ↓fibrosis ↓ TGFβ-1 ↓MMP-9 | [153,154] | |
| hUCB-SC | - | - | ↓ fibronectin ↓α-SMA expression ↓proteinuria levels ↓E-cadherin | [157] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braga, P.C.; Alves, M.G.; Rodrigues, A.S.; Oliveira, P.F. Mitochondrial Pathophysiology on Chronic Kidney Disease. Int. J. Mol. Sci. 2022, 23, 1776. https://doi.org/10.3390/ijms23031776
Braga PC, Alves MG, Rodrigues AS, Oliveira PF. Mitochondrial Pathophysiology on Chronic Kidney Disease. International Journal of Molecular Sciences. 2022; 23(3):1776. https://doi.org/10.3390/ijms23031776
Chicago/Turabian StyleBraga, Patrícia C., Marco G. Alves, Anabela S. Rodrigues, and Pedro F. Oliveira. 2022. "Mitochondrial Pathophysiology on Chronic Kidney Disease" International Journal of Molecular Sciences 23, no. 3: 1776. https://doi.org/10.3390/ijms23031776
APA StyleBraga, P. C., Alves, M. G., Rodrigues, A. S., & Oliveira, P. F. (2022). Mitochondrial Pathophysiology on Chronic Kidney Disease. International Journal of Molecular Sciences, 23(3), 1776. https://doi.org/10.3390/ijms23031776