
Reversible Redox Processes in Polymer of Unmetalated Salen-Type Ligand: Combined Electrochemical in Situ Studies and Direct Comparison with Corresponding Nickel Metallopolymer


Abstract
:1. Introduction
2. Results and Discussion
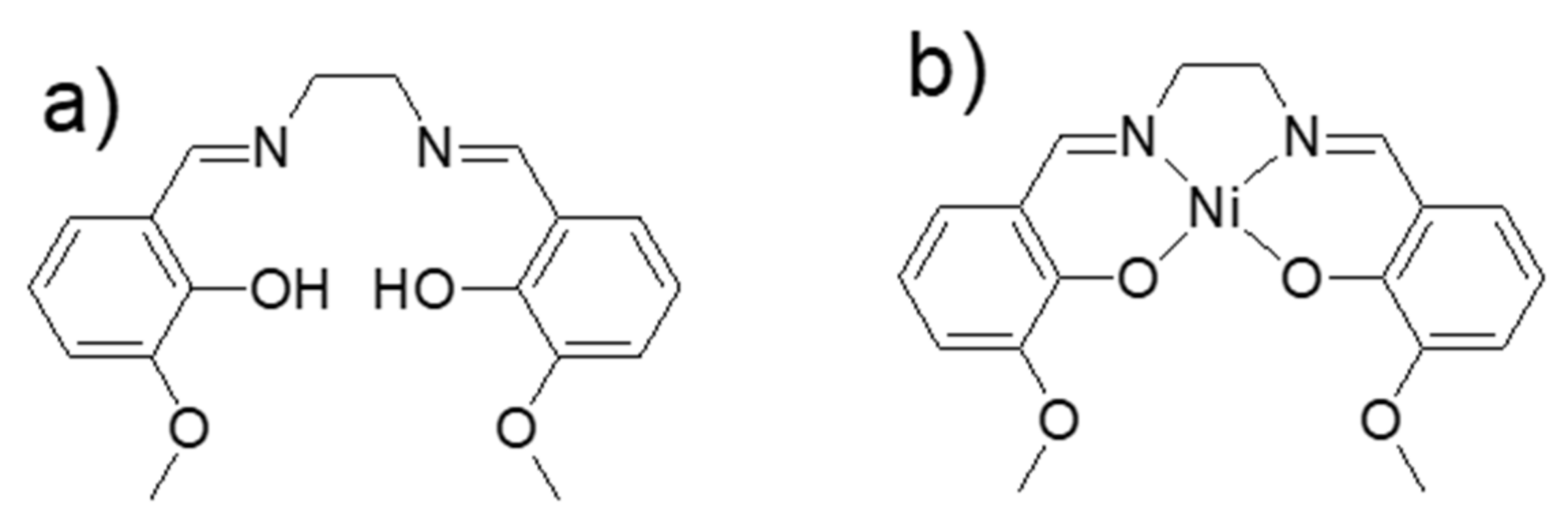
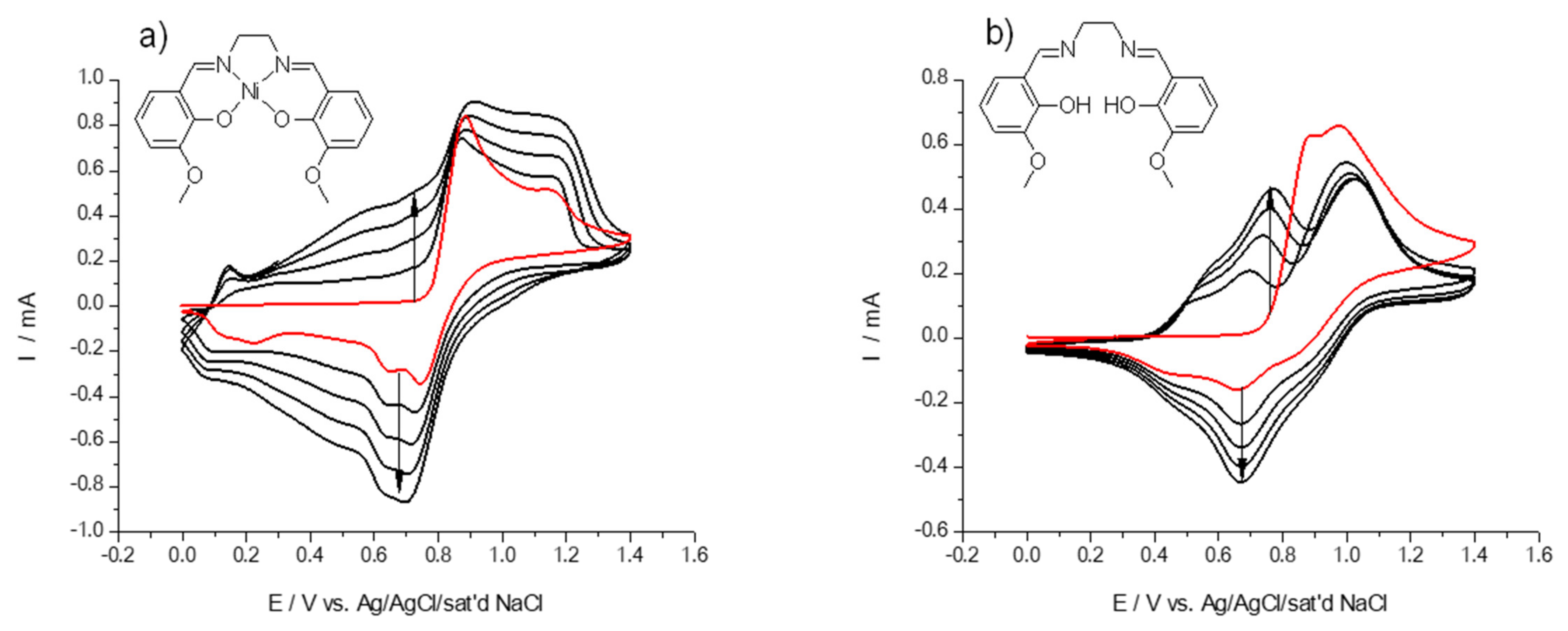
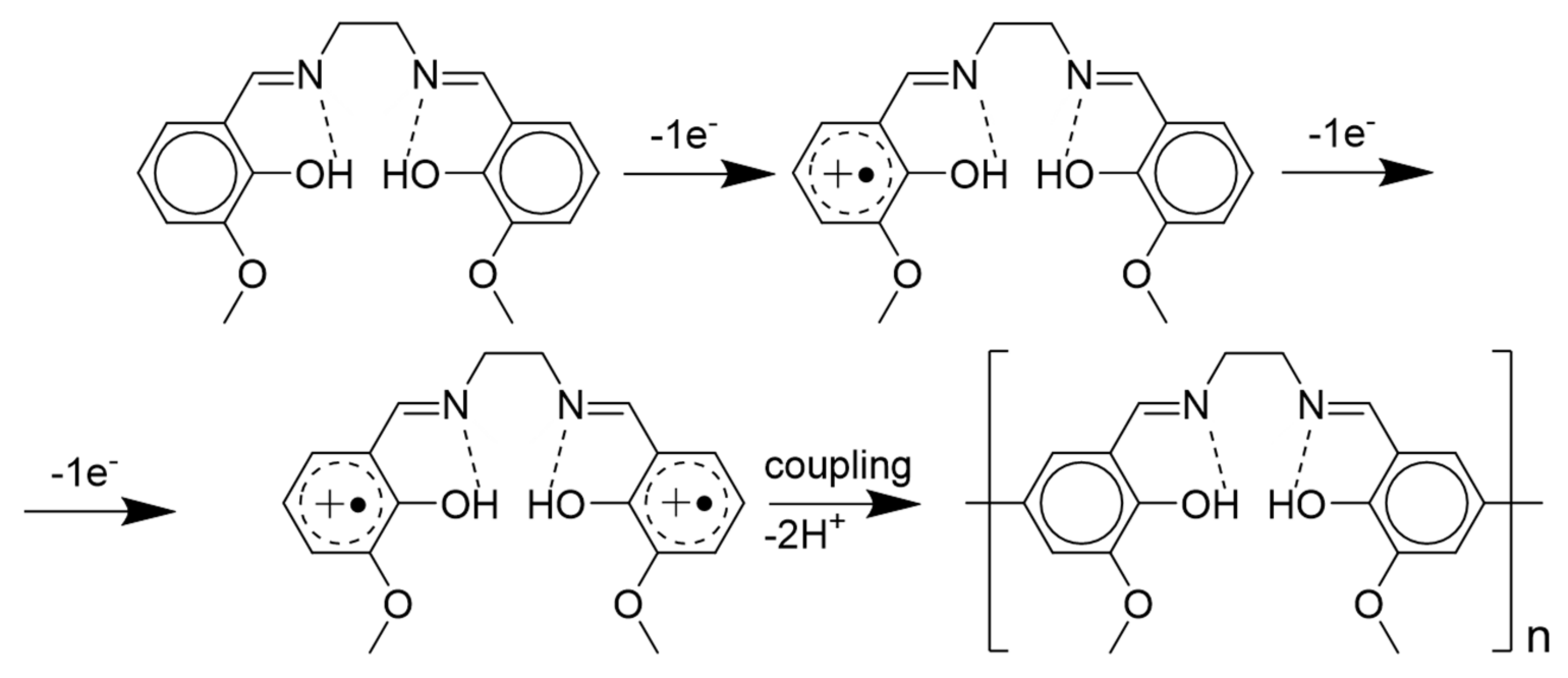
2.1. Electrochemical Polymerization of the Ligand H2(3-MeOSalen) and the Corresponding Nickel Complex Ni(3-MeOSalen)
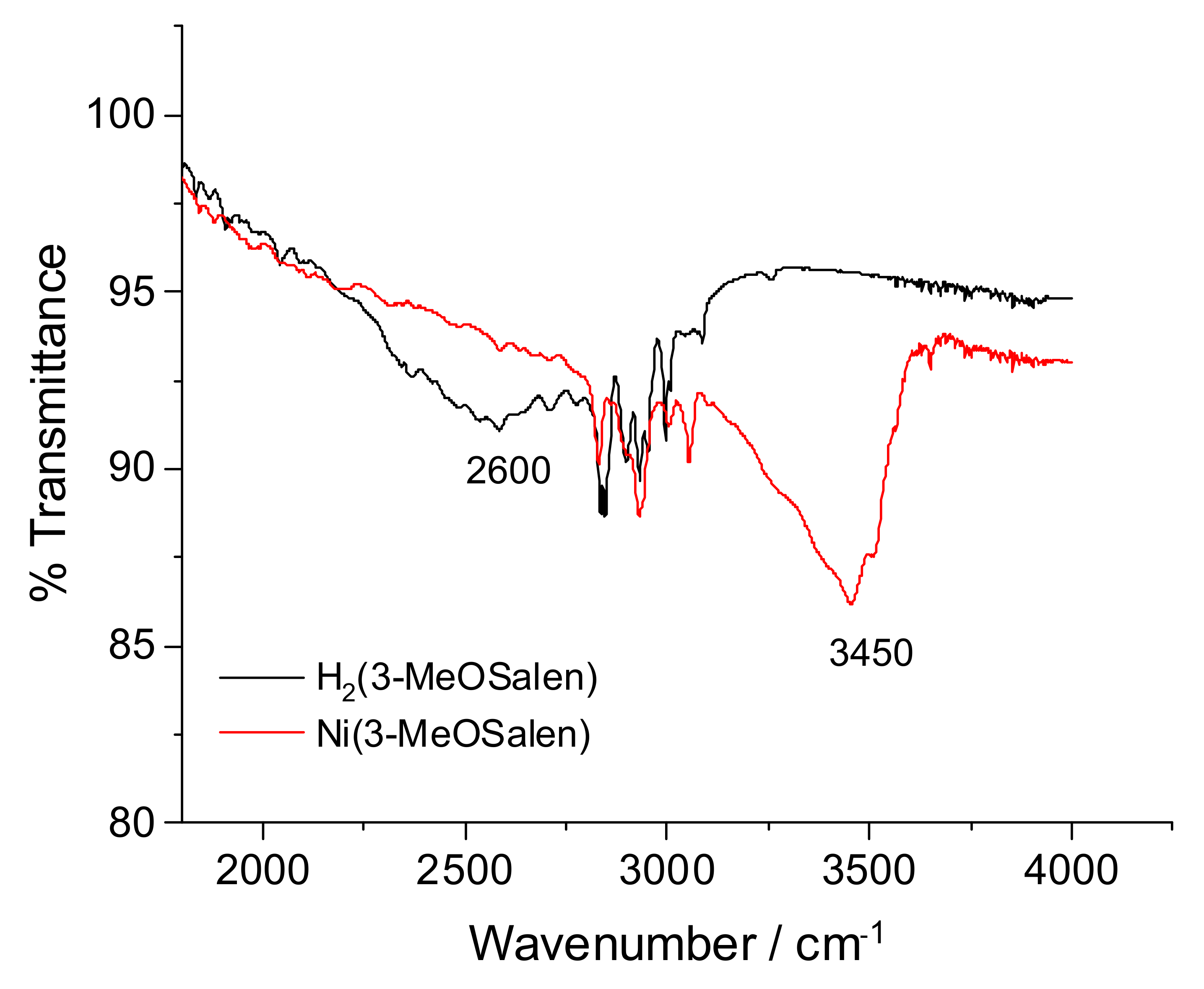
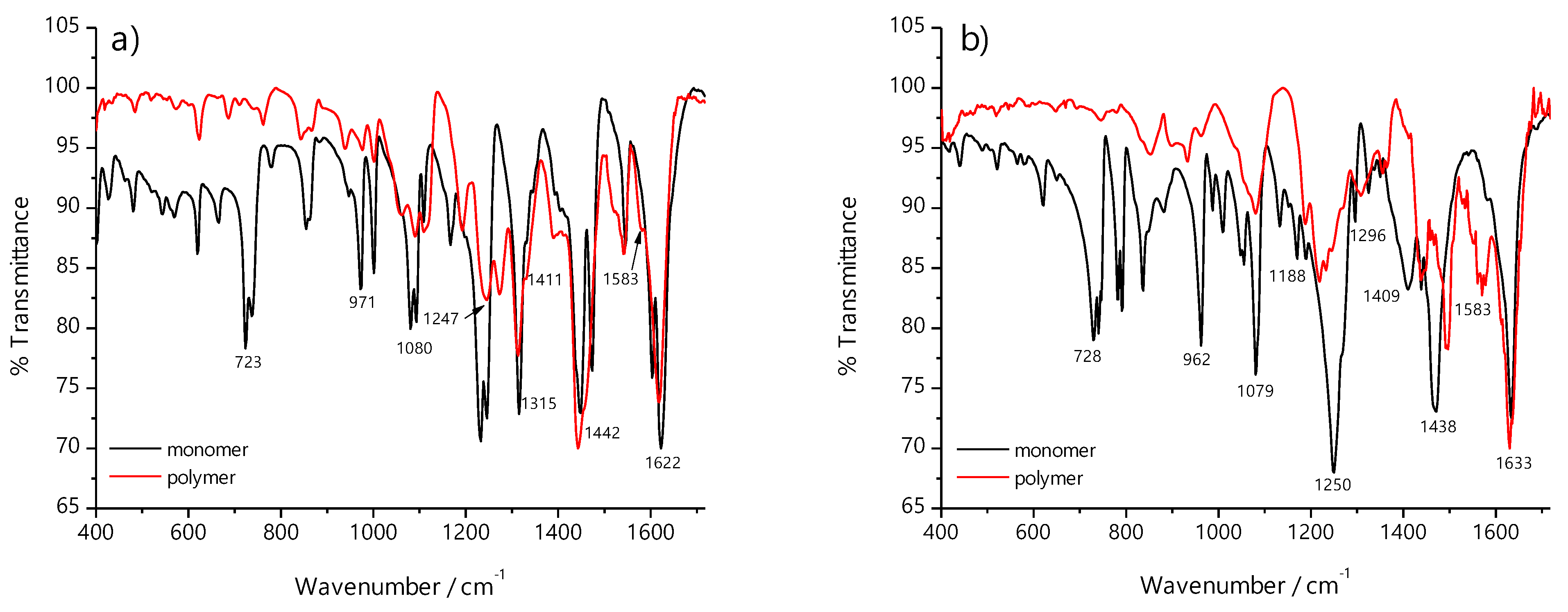
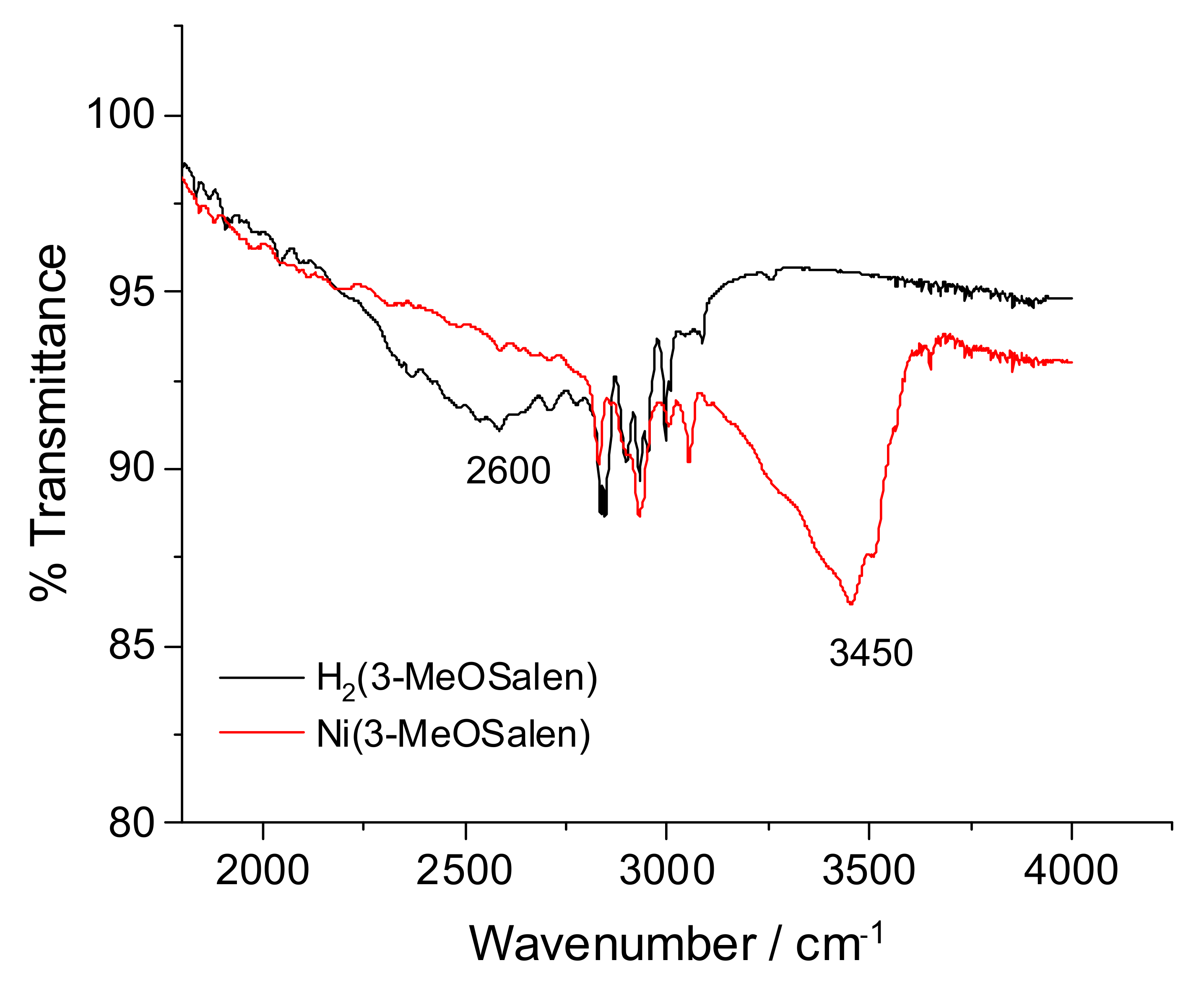
2.2. FTIR Spectroscopy
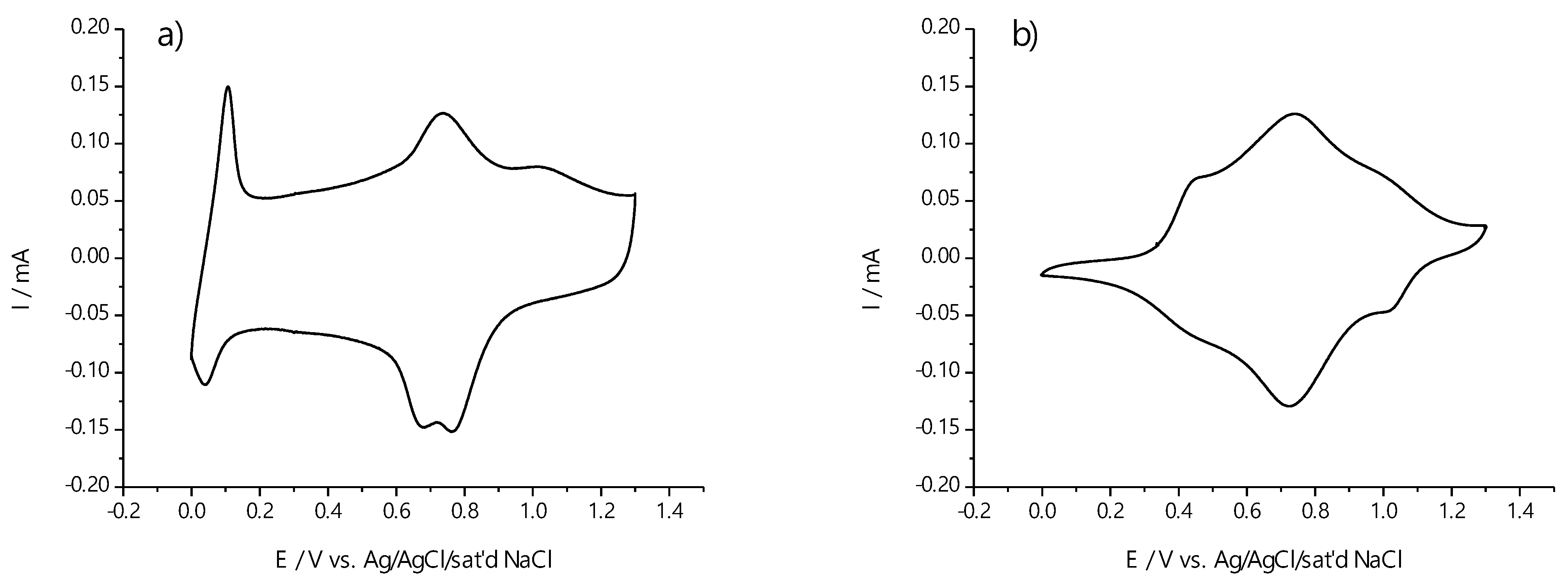
2.3. Electrochemical Behavior of Polymer Films Poly-Ni(3-MeOSalen) and Poly-H2(3-MeOSalen)
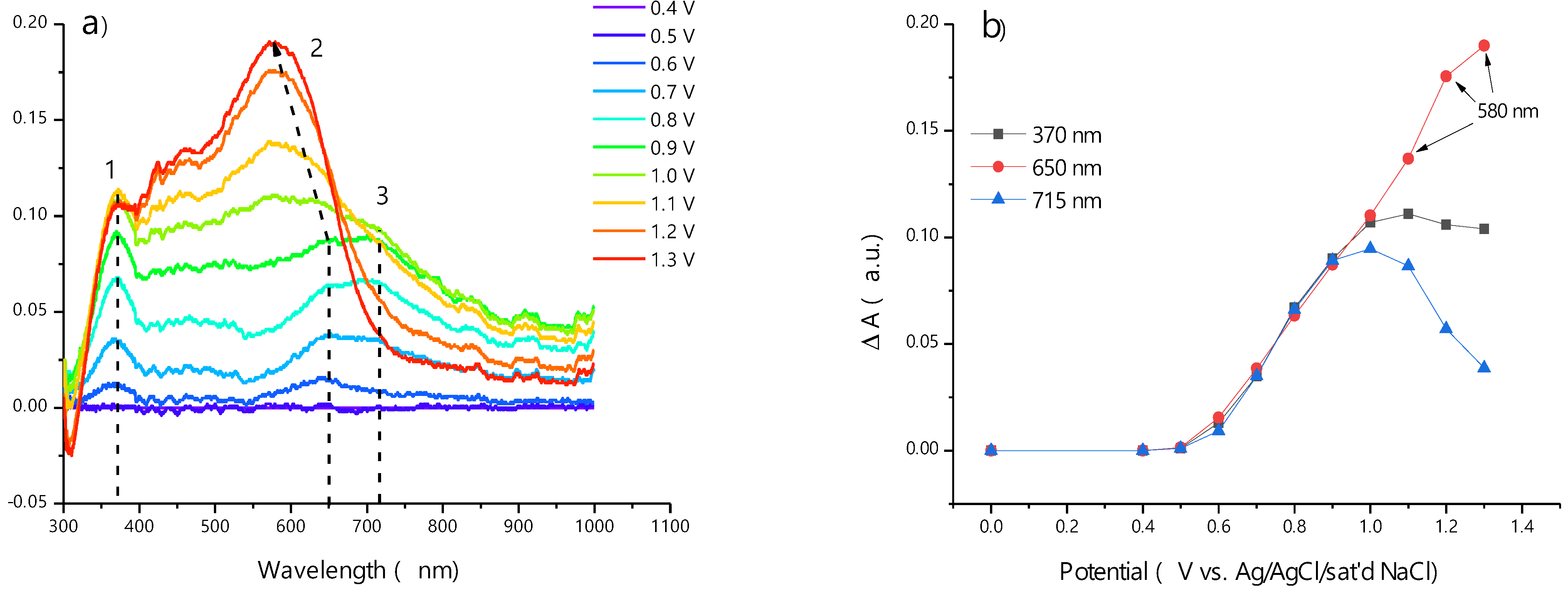
2.4. In Situ Electronic Spectra of Poly-Ni(3-MeOSalen) and Poly-H2(3-MeOSalen) Films
2.5. The Morphology of the Films
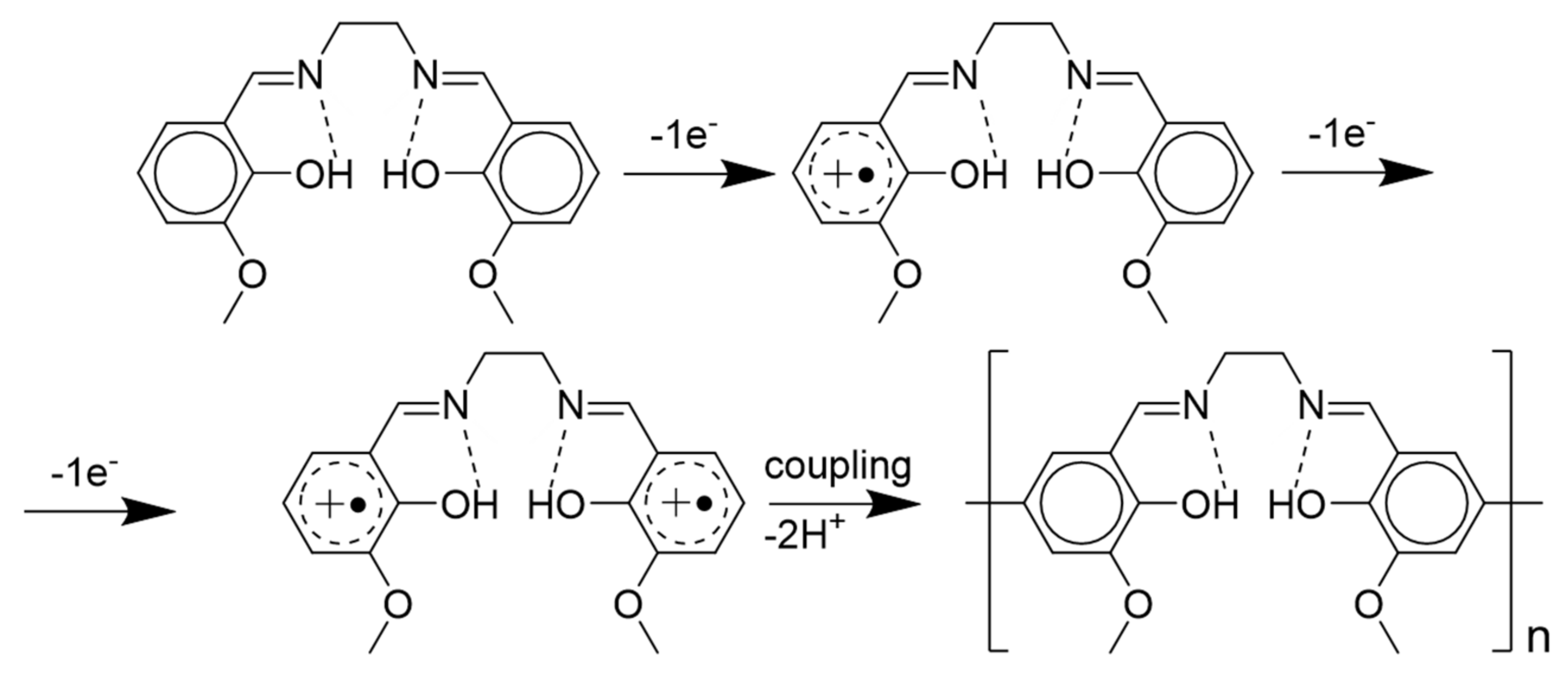
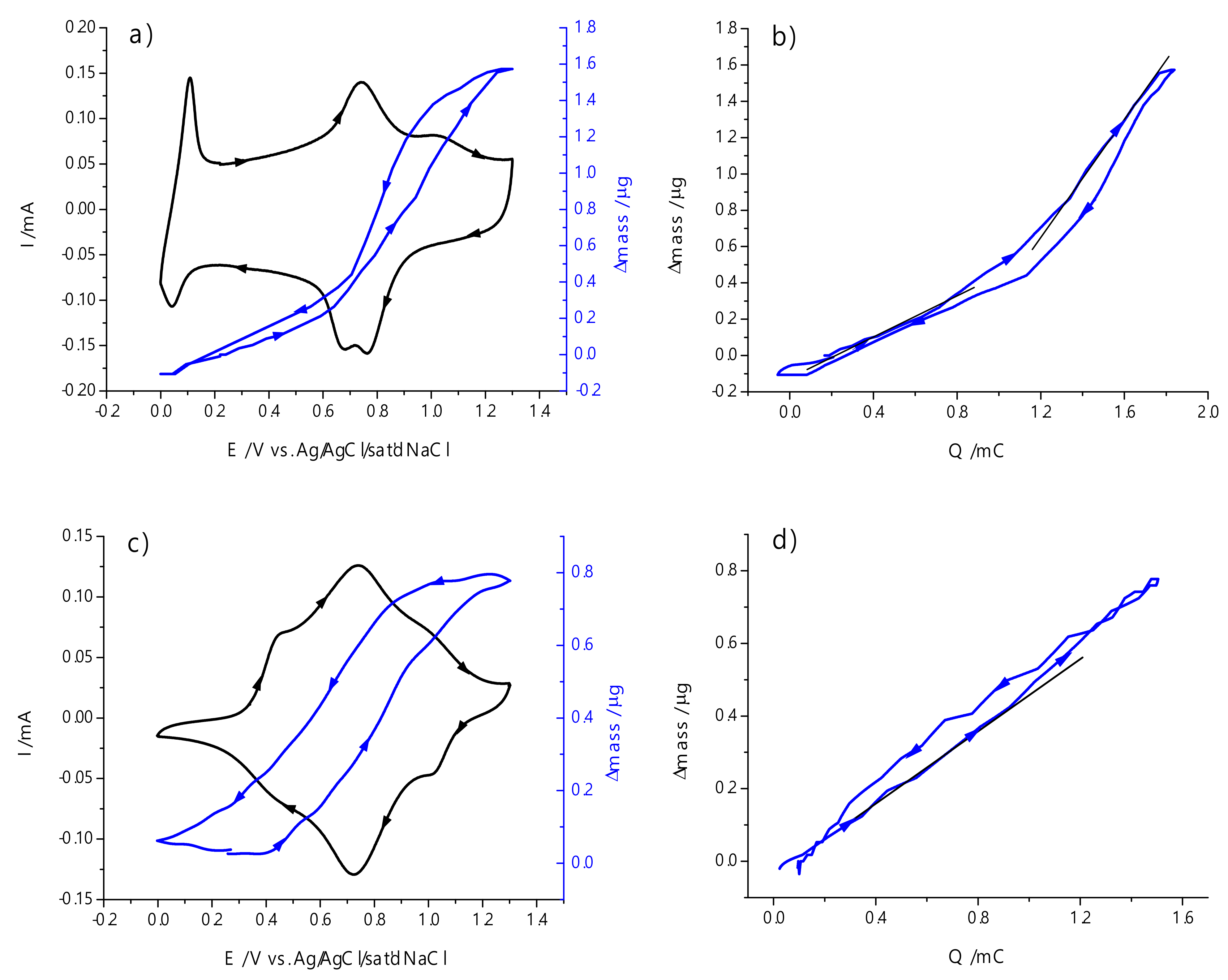
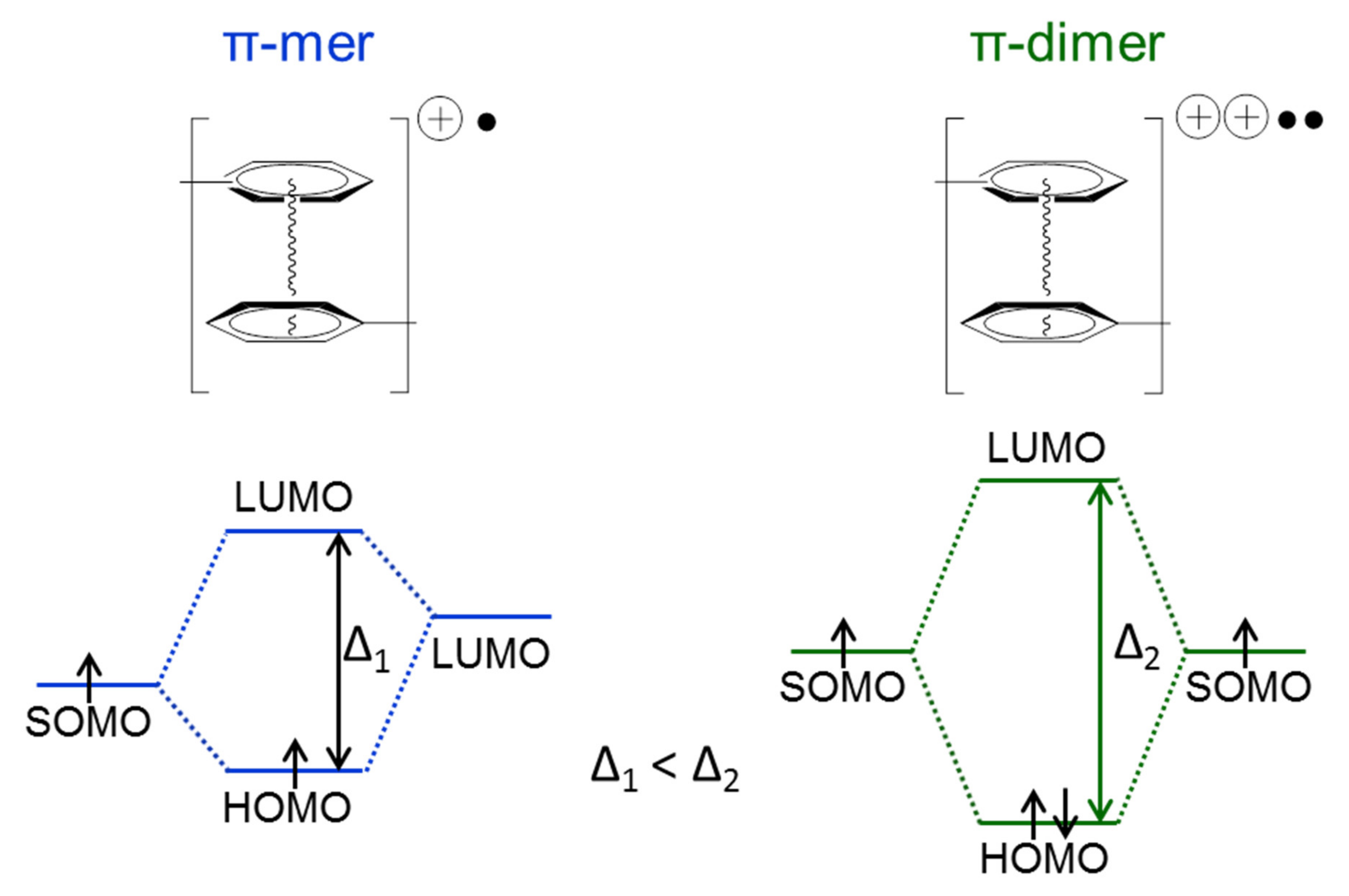
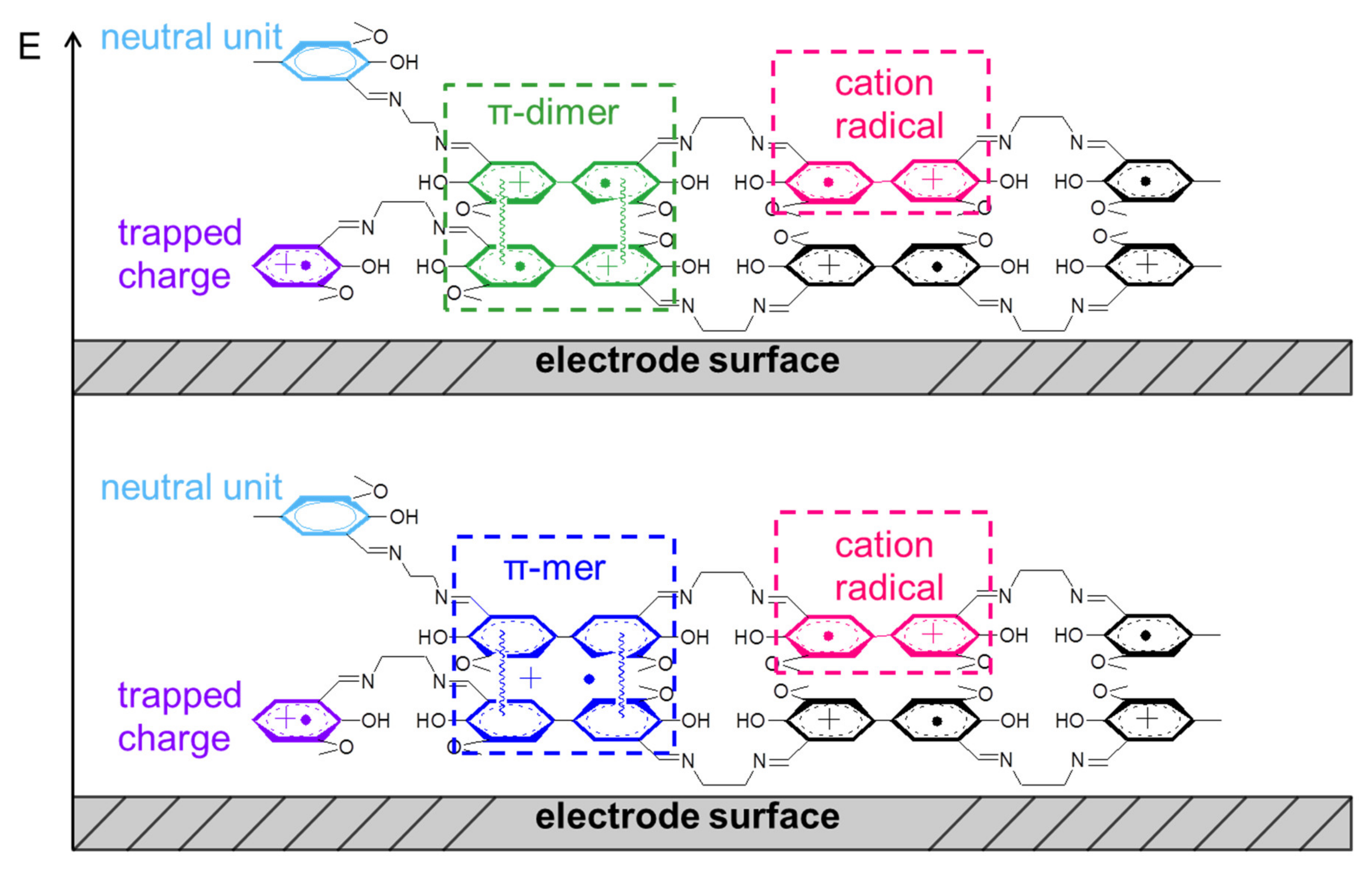
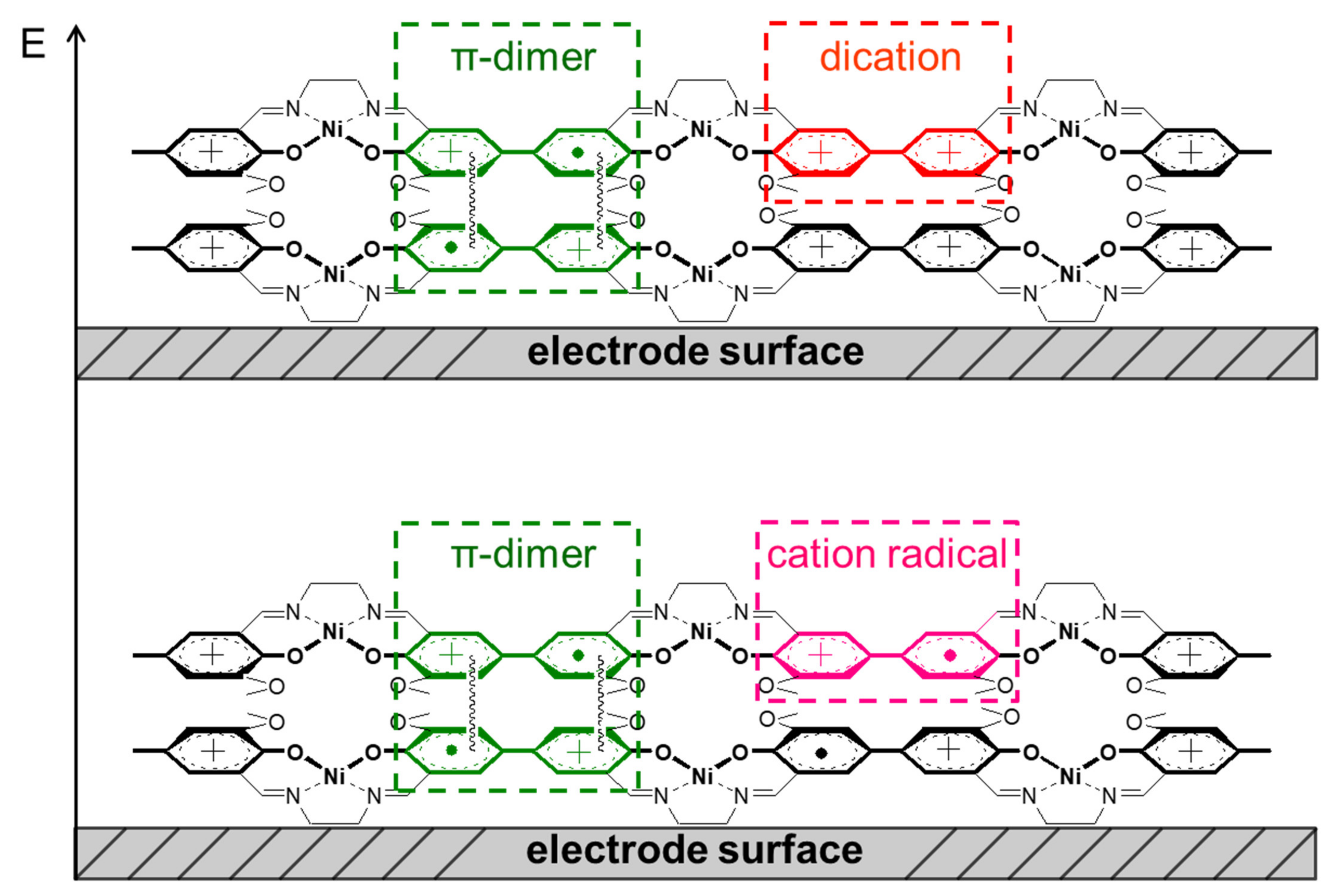
2.6. The Structure and Mechanism of Charge Transfer in the Polymer Films Poly-Ni(3-MeOSalen)
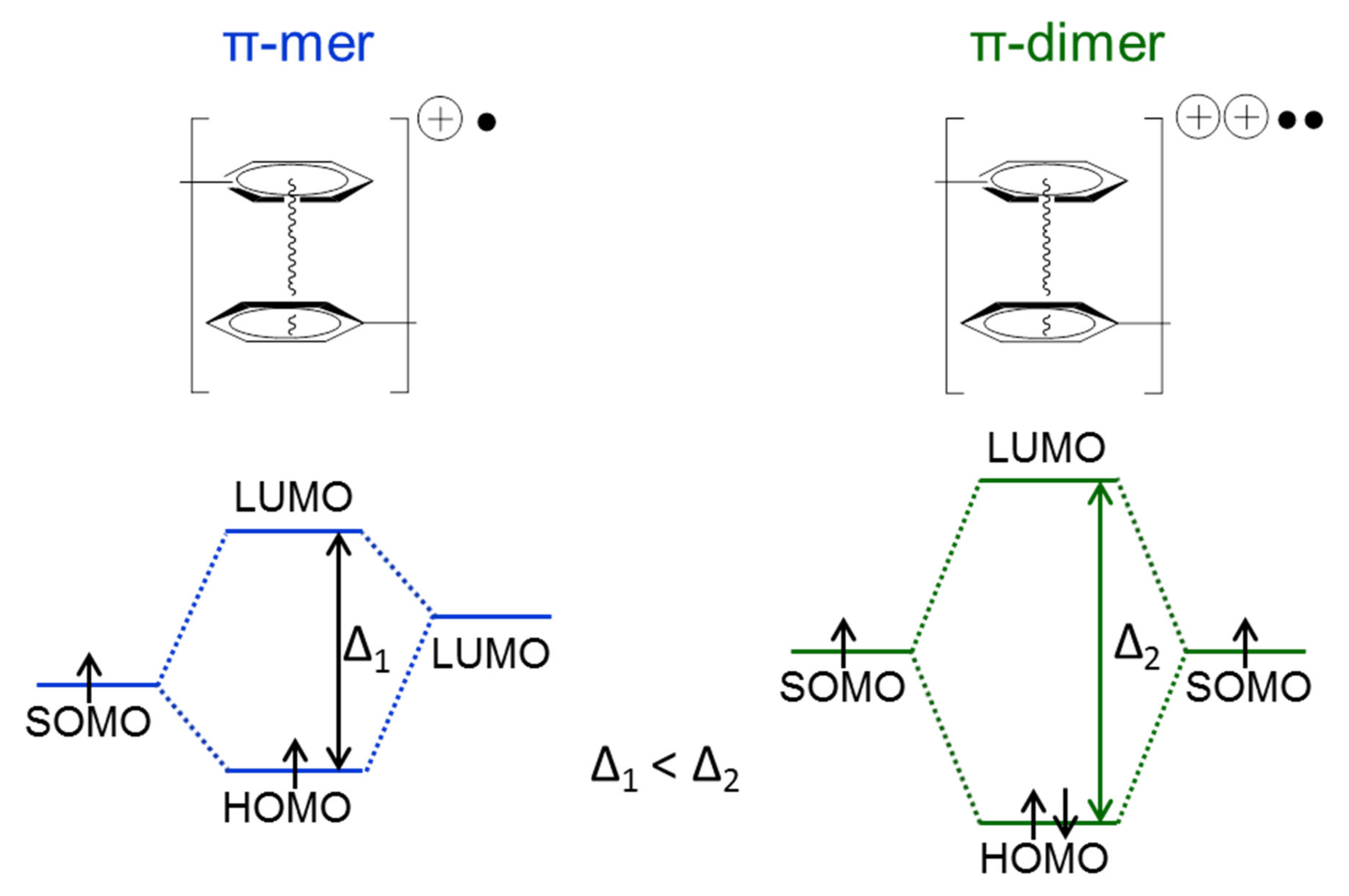
2.7. The Structure and Mechanism of Charge Transfer in the Polymer Films Poly-H2(3-MeOSalen)
3. Materials and Methods
3.1. Chemicals
3.2. Electrochemistry and Electrochemical Quartz Crystal Microbalance (EQCM)
3.3. In Situ UV–VIS Spectroelectrochemistry
3.4. FTIR Spectroscopy
3.5. Scanning Electron Microscopy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhan, C.; Yu, G.; Lu, Y.; Wang, L.; Wujcik, E.; Wei, S. Conductive polymer nanocomposites: A critical review of modern advanced devices. J. Mater. Chem. C 2017, 5, 1569–1585. [Google Scholar] [CrossRef]
- Guo, P.; Hui, T.-W.; Cheung, K.-C.; Wong, K.-Y.; Shiu, K.-K. Charge propagation in nickel 6,6′-bis(2′-hydroxyphenyl)-2,2′-bipyridine polymer film doped with perchlorate anions. J. Electroanal. Chem. 2001, 498, 142–151. [Google Scholar] [CrossRef]
- Muench, S.; Wild, A.; Friebe, C.; Häupler, B.; Janoschka, T.; Schubert, U.S. Polymer-Based Organic Batteries. Chem. Rev. 2016, 116, 9438–9484. [Google Scholar] [CrossRef] [PubMed]
- Gerard, M. Application of conducting polymers to biosensors. Biosens. Bioelectron. 2002, 17, 345–359. [Google Scholar] [CrossRef]
- Rudge, A.; Raistrick, I.; Gottesfeld, S.; Ferraris, J.P. A study of the electrochemical properties of conducting polymers for application in electrochemical capacitors. Electrochimica Acta 1994, 39, 273–287. [Google Scholar] [CrossRef]
- Mortimer, R.J. Organic electrochromic materials. Electrochimica Acta 1999, 44, 2971–2981. [Google Scholar] [CrossRef]
- Polozhentseva, Y.A.; Karushev, M.P.; Rumyantsev, A.M.; Chepurnaya, I.; Timonov, A.M. A Lithium-Ion Supercapacitor with a Positive Electrode Based on a Carbon Material Modified by Polymeric Complexes of Nickel with Schiff Bases. Tech. Phys. Lett. 2020, 46, 196–199. [Google Scholar] [CrossRef]
- Vereshchagin, A.A.; Sizov, V.V.; Verjuzhskij, M.S.; Hrom, S.I.; Volkov, A.I.; Danilova, J.S.; Novozhilova, M.V.; Laaksonen, A.; Levin, O.V. Interaction of amines with electrodes modified by polymeric complexes of Ni with salen-type ligands. Electrochimica Acta 2016, 211, 726–734. [Google Scholar] [CrossRef]
- Kakhki, S.; Shams, E. A new bifunctional electrochemical sensor for oxidation of cysteine and reduction of iodate. J. Electroanal. Chem. 2013, 704, 249–254. [Google Scholar] [CrossRef]
- Teixeira, M.; Dadamos, T. An electrochemical sensor for dipyrone determination based on nickel-salen film modified electrode. Procedia Chem. 2009, 1, 297–300. [Google Scholar] [CrossRef] [Green Version]
- Nunes, M.; Araújo, M.; Fonseca, J.; Moura, C.; Hillman, R.; Freire, C. High-Performance Electrochromic Devices Based on Poly[Ni(salen)]-Type Polymer Films. ACS Appl. Mater. Interfaces 2016, 8, 14231–14243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avanesyan, V.T.; Puchkov, M.Y. Photodielectric effect in thin-film structures of the poly[NiSalen] metallopolymer. Phys. Solid State 2009, 51, 2178–2181. [Google Scholar] [CrossRef]
- Ravari, F.B.; Dadgarinezhad, A.; Shekhshoaei, I. Investigation on two salen type schiff base compounds as corrosion inhibition of copper in 0.5 M H2SO4. Gazi Univ. J. Sci. 2009, 22, 175–182. [Google Scholar]
- Ravari, F.B.; Dadgarinezhad, A.; Shekhshoaei, I. STUDIES ON THE EFFECT OF TWO SALEN SCHIFF-BASES ON THE CORROSION OF LOW ALLOY STEEL IN 1M HCL. J. Chil. Chem. Soc. 2010, 55, 328–331. [Google Scholar] [CrossRef] [Green Version]
- Alekseeva, E.V.; Chepurnaya, I.A.; Malev, V.V.; Timonov, A.M.; Levin, O.V. Polymeric nickel complexes with salen-type ligands for modification of supercapacitor electrodes: Impedance studies of charge transfer and storage properties. Electrochimica Acta 2017, 225, 378–391. [Google Scholar] [CrossRef]
- O’Meara, C.; Karushev, M.; Polozhentceva, I.A.; Dharmasena, S.; Cho, H.; Yurkovich, B.J.; Kogan, S.; Kim, J.-H. Nickel–Salen-Type Polymer as Conducting Agent and Binder for Carbon-Free Cathodes in Lithium-Ion Batteries. ACS Appl. Mater. Interfaces 2018, 11, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Chepurnaya, I.A.; Karushev, M.P.; Alekseeva, E.V.; Lukyanov, D.A.; Levin, O.V. Redox-conducting polymers based on metal-salen complexes for energy storage applications. Pure Appl. Chem. 2020, 92, 1239–1258. [Google Scholar] [CrossRef]
- Polozhentseva, Y.A.; Novozhilova, M.V.; Chepurnaya, I.A.; Karushev, M.P. Polymeric Complexes of Nickel with Salen-Type Ligands as Multifunctional Components of Lithium Ion Battery Cathodes. Tech. Phys. Lett. 2021, 47, 83–87. [Google Scholar] [CrossRef]
- Polozhentseva, Y.A.; Novozhilova, M.V.; Bykov, V.A.; Karushev, M.P. Modification of Porous Carbon Material with Polymeric Cobalt Complex with a Schiff Base of Salen-Type for Electrodes of Electrochemical Supercapacitors. Tech. Phys. Lett. 2020, 46, 913–915. [Google Scholar] [CrossRef]
- Smirnova, E.A.; Timonov, A.M. A novel functional material for the electrochemical reduction of chlorinated organic compounds. Bull. Acad. Sci. USSR Div. Chem. Sci. 2021, 70, 1618–1621. [Google Scholar] [CrossRef]
- Besedina, M.A.; Smirnova, E.A.; Poturai, D.O.; Karushev, M.P. The activity of monomeric and polymeric nickel complexes with Salen-type ligands as photosensitive materials for electrochemical solar cells. Bull. Acad. Sci. USSR Div. Chem. Sci. 2021, 70, 107–112. [Google Scholar] [CrossRef]
- Tomczyk, D.; Bukowski, W.; Bester, K. Redox processes in the solution of Ni(II) complex with salen type ligand and in the polymer films. Electrochimica Acta 2018, 267, 181–194. [Google Scholar] [CrossRef]
- Łępicka, K.; Pieta, P.; Shkurenko, A.; Borowicz, P.; Majewska, M.; Rosenkranz, M.; Avdoshenko, S.; Popov, A.A.; Kutner, W. Spectroelectrochemical Approaches to Mechanistic Aspects of Charge Transport in meso-Nickel(II) Schiff Base Electrochromic Polymer. J. Phys. Chem. C 2017, 121, 16710–16720. [Google Scholar] [CrossRef]
- Dmitrieva, E.; Rosenkranz, M.; Danilova, J.S.; Smirnova, E.; Karushev, M.; Chepurnaya, I.; Timonov, A.M. Radical formation in polymeric nickel complexes with N2O2 Schiff base ligands: An in situ ESR and UV–vis–NIR spectroelectrochemical study. Electrochimica Acta 2018, 283, 1742–1752. [Google Scholar] [CrossRef]
- Clarke, R.M.; Herasymchuk, K.; Storr, T. Electronic structure elucidation in oxidized metal–salen complexes. Coord. Chem. Rev. 2017, 352, 67–82. [Google Scholar] [CrossRef]
- Dunn, T.J.; Webb, M.I.; Hazin, K.; Verma, P.; Wasinger, E.C.; Shimazaki, Y.; Storr, T. Double oxidation localizes spin in a Ni bis-phenoxyl radical complex. Dalton Trans. 2013, 42, 3950–3956. [Google Scholar] [CrossRef]
- Audebert, P.; Capdevielle, P.; Maumy, M. Redox and conducting polymers based on salen-type metal units; electrochemical study and some characteristics. New J. Chem. 1992, 16, 697–703. [Google Scholar]
- Hoferkamp, L.A.; Goldsby, K.A. Surface-modified electrodes based on nickel(II) and copper(II) bis(salicylaldimine) complexes. Chem. Mater. 1989, 1, 348–352. [Google Scholar] [CrossRef]
- Vilas-Boas, M.; Freire, C.; de Castro, B.; Hillman, A.R. Electrochemical Characterization of a Novel Salen-Type Modified Electrode. J. Phys. Chem. B 1998, 102, 8533–8540. [Google Scholar] [CrossRef] [Green Version]
- Rodyagina, T.Y.; Gaman’Kov, P.V.; Dmitrieva, E.A.; Chepurnaya, I.; Vasil’Eva, S.V.; Timonov, A.M. Structuring Redox Polymers Poly[M(Schiff)] (M = Ni, Pd; Schiff = Schiff Bases) on a Molecular Level: Methods and Results of an Investigation. Russ. J. Electrochem. 2005, 41, 1101–1110. [Google Scholar] [CrossRef]
- Vereschagin, A.A.; Sizov, V.V.; Vlasov, P.S.; Alekseeva, E.V.; Konev, A.S.; Levin, O.V. Water-stable [Ni(salen)]-type electrode material based on phenylazosubstituted salicylic aldehyde imine ligand. New J. Chem. 2017, 41, 13918–13928. [Google Scholar] [CrossRef] [Green Version]
- Pereira, C.F.; Olean-Oliveira, A.; David-Parra, D.N.; Teixeira, M.F. A chemiresistor sensor based on a cobalt(salen) metallopolymer for dissolved molecular oxygen. Talanta 2018, 190, 119–125. [Google Scholar] [CrossRef]
- Chiang, L.; Kochem, A.; Jarjayes, O.; Dunn, T.J.; Vezin, H.; Sakaguchi, M.; Ogura, T.; Orio, M.; Shimazaki, Y.; Thomas, F.; et al. Radical Localization in a Series of Symmetric NiIIComplexes with Oxidized Salen Ligands. Chem. A Eur. J. 2012, 18, 14117–14127. [Google Scholar] [CrossRef] [PubMed]
- Yankin, A.N.; Lukyanov, D.A.; Beletskii, E.V.; Bakulina, O.Y.; Vlasov, P.S.; Levin, O.V. Aryl-Aryl Coupling of Salicylic Aldehydes through Oxidative CH-activation in Nickel Salen Derivatives. ChemistrySelect 2019, 4, 8886–8890. [Google Scholar] [CrossRef]
- Swager, T.M. 50th Anniversary Perspective: Conducting/Semiconducting Conjugated Polymers. A Personal Perspective on the Past and the Future. Macromolecules 2017, 50, 4867–4886. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Jones, R.A.; Holliday, B.J. Understanding the Effect of Metal Centers on Charge Transport and Delocalization in Conducting Metallopolymers. Macromolecules 2017, 50, 872–883. [Google Scholar] [CrossRef]
- Audebert, P.; Hapiot, P.; Capdevielle, P.; Maumy, M. Electrochemical polymerization of several salen-type complexes. Kinetic studies in the microsecond time range. J. Electroanal. Chem. 1992, 338, 269–278. [Google Scholar] [CrossRef]
- Vilas-Boas, M.; Freire, C.; de Castro, B.; Christensen, A.P.A.; Hillman§, A.R. New Insights into the Structure and Properties of Electroactive Polymer Films Derived from [Ni(salen)]. Inorg. Chem. 1997, 36, 4919–4929. [Google Scholar] [CrossRef]
- Chiang, L.; Herasymchuk, K.; Thomas, F.; Storr, T. Influence of Electron-Withdrawing Substituents on the Electronic Structure of Oxidized Ni and Cu Salen Complexes. Inorg. Chem. 2015, 54, 5970–5980. [Google Scholar] [CrossRef]
- Shimazaki, Y.; Stack, T.D.P.; Storr, T. Detailed Evaluation of the Geometric and Electronic Structures of One-Electron Oxidized Group 10 (Ni, Pd, and Pt) Metal(II)-(Disalicylidene)diamine Complexes. Inorg. Chem. 2009, 48, 8383–8392. [Google Scholar] [CrossRef] [Green Version]
- Kiss, L.; Bősz, D.; Kovács, F.; Li, H.; Nagy, G.; Kunsági-Máté, S. Investigation of phenol electrooxidation in aprotic non-aqueous solvents by using cyclic and normal pulse voltammetry. Polym. Bull. 2019, 76, 5849–5864. [Google Scholar] [CrossRef] [Green Version]
- Tessensohn, M.E.; Hirao, H.; Webster, R.D. Electrochemical Properties of Phenols and Quinones in Organic Solvents are Strongly Influenced by Hydrogen-Bonding with Water. J. Phys. Chem. C 2013, 117, 1081–1090. [Google Scholar] [CrossRef]
- Richards, J.A.; Whitson, P.E.; Evans, D.H. Electrochemical oxidation of 2,4,6-tri-tert-butylphenol. J. Electroanal. Chem. Interfacial Electrochem. 1975, 63, 311–327. [Google Scholar] [CrossRef]
- Coetzee, J.F.; Padmanabhan, G.R. Dissociation and Homoconjugation of Certain Phenols in Acetonitrile. J. Phys. Chem. 1965, 69, 3193–3196. [Google Scholar] [CrossRef]
- Thomas, F.; Jarjayes, O.; Duboc, C.; Philouze, C.; Saint-Aman, E.; Pierre, J.-L. Intramolecularly hydrogen-bonded versus copper(ii) coordinated mono- and bis-phenoxyl radicals. Dalton Trans. 2004, 2662–2669. [Google Scholar] [CrossRef]
- Benisvy, L.; Bill, E.; Blake, A.J.; Collison, D.; Davies, E.S.; Garner, C.D.; McArdle, G.; McInnes, E.J.L.; McMaster, J.; Ross, S.H.K.; et al. Phenoxyl radicals: H-bonded and coordinated to Cu(ii) and Zn(ii). Dalton Trans. 2005, 258–267. [Google Scholar] [CrossRef]
- Ahmad, J.U.; Nieger, M.; Sundberg, M.R.; Leskelä, M.; Repo, T. Solid and solution structures of bulky tert-butyl substituted salicylaldimines. J. Mol. Struct. 2011, 995, 9–19. [Google Scholar] [CrossRef]
- Kingsborough, R.P.; Swager, T.M. Polythiophene Hybrids of Transition-Metal Bis(salicylidenimine)s: Correlation between Structure and Electronic Properties. J. Am. Chem. Soc. 1999, 121, 8825–8834. [Google Scholar] [CrossRef]
- Tomczyk, D.; Bukowski, W.; Bester, K.; Urbaniak, P.; Seliger, P.; Andrijewski, G.; Skrzypek, S. The mechanism of electropolymerization of nickel(ii) salen type complexes. New J. Chem. 2017, 41, 2112–2123. [Google Scholar] [CrossRef] [Green Version]
- Signorini, O.; Dockal, E.; Castellano, G.; Oliva, G. Synthesis and characterization of aquo[N,N′-ethylenebis(3-ethoxysalicylideneaminato)]dioxouranium(VI). Polyhedron 1996, 15, 245–255. [Google Scholar] [CrossRef]
- Dockal, E.R. Tetradentate Schiff base oxovanadium(IV) complexes. Transit. Met. Chem. 1996, 21, 370–376. [Google Scholar] [CrossRef]
- Danilova, J.S.; Avdoshenko, S.M.; Karushev, M.P.; Timonov, A.M.; Dmitrieva, E. Infrared spectroscopic study of nickel complexes with salen-type ligands and their polymers. J. Mol. Struct. 2021, 1241, 130668. [Google Scholar] [CrossRef]
- Vráblová, A.; Tomás, M.; Titiš, J.; Černák, J.; Falvello, L.R. On new solvatomorphs of the metalloligand [Ni(o-van-en)]. Inorganica Chim. Acta 2020, 512, 119874. [Google Scholar] [CrossRef]
- Sauerbrey, G. Verwendung von Schwingquarzen zur Wägung danner Schichten und zur Mikrowägung. Z. Phys. 2005, 155, 206–222. [Google Scholar] [CrossRef]
- Ohmori, H.; Matsumoto, A.; Masui, M.; Sayo, H. Anodic Oxidation of Schiff Bases Derived from 2-Hydroxy-3-Methoxybenzaldehyde. J. Electrochem. Soc. 1977, 124, 1849–1854. [Google Scholar] [CrossRef]
- Tanaka, M.; Yoshida, H.; Ogasawara, M. Ionic processes in pulse-irradiated fluid solutions of poly(4-vinylbiphenyl). Int. J. Radiat. Appl. Instrum. Part C Radiat. Phys. Chem. 1989, 34, 591–595. [Google Scholar] [CrossRef]
- Talipov, M.R.; Navale, T.S.; Hossain, M.M.; Shukla, R.; Ivanov, M.V.; Rathore, R. Dihedral-Angle-Controlled Crossover from Static Hole Delocalization to Dynamic Hopping in Biaryl Cation Radicals. Angew. Chem. Int. Ed. 2016, 56, 266–269. [Google Scholar] [CrossRef] [Green Version]
- Lü, J.-M.; Rosokha, A.S.V.; Kochi, J.K. Stable (Long-Bonded) Dimers via the Quantitative Self-Association of Different Cationic, Anionic, and Uncharged π-Radicals: Structures, Energetics, and Optical Transitions. J. Am. Chem. Soc. 2003, 125, 12161–12171. [Google Scholar] [CrossRef]
- Ganesan, V.; Rosokha, S.V.; Kochi, J.K. Isolation of the Latent Precursor Complex in Electron-Transfer Dynamics. Intermolecular Association and Self-Exchange with Acceptor Anion Radicals. J. Am. Chem. Soc. 2003, 125, 2559–2571. [Google Scholar] [CrossRef]
- Baeuerle, P.; Segelbacher, U.; Maier, A.; Mehring, M. Electronic structure of mono- and dimeric cation radicals in end-capped oligothiophenes. J. Am. Chem. Soc. 1993, 115, 10217–10223. [Google Scholar] [CrossRef]
- Hasegawa, M.; Daigoku, K.; Hashimoto, K.; Nishikawa, H.; Iyoda, M. Face-to-Face Dimeric Tetrathiafulvalenes and Their Cation Radical and Dication Species as Models of Mixed Valence and π-Dimer States. Bull. Chem. Soc. Jpn. 2012, 85, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Punyain, K.; Kelterer, A.-M.; Grampp, G. Theoretical studies on the dimerization of substituted paraphenylenediamine radical cations. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2011, 83, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Swager, T.M. π-Dimer Formation as the Driving Force for Calix[4]arene-Based Molecular Actuators. Org. Lett. 2008, 10, 3575–3578. [Google Scholar] [CrossRef] [PubMed]
- Nishinaga, T.; Sotome, Y. Stable Radical Cations and Their π-Dimers Prepared from Ethylene- and Propylene-3,4-dioxythiophene Co-oligomers: Combined Experimental and Theoretical Investigations. J. Org. Chem. 2017, 82, 7245–7253. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Endo, T.; Takase, M.; Iyoda, M.; Nishinaga, T. Structural, Optical, and Electronic Properties of a Series of 3,4-Propylenedioxythiophene Oligomers in Neutral and Various Oxidation States. J. Am. Chem. Soc. 2011, 133, 11339–11350. [Google Scholar] [CrossRef]
- Losada, J.; del Peso, I.; Beyer, L. Redox and electrocatalytic properties of electrodes modified by films of polypyrrole nickel(II) Schiff-base complexes. J. Electroanal. Chem. 1998, 447, 147–154. [Google Scholar] [CrossRef]
- Pahor, N.B.; Calligaris, M.; Nardin, G.; Randaccio, L. N,N’-Ethylenebis(salicylideneimine). Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1978, 34, 1360–1363. [Google Scholar] [CrossRef]
- Pfeiffer, P.; Breith, E.; Lübbe, E.; Tsumaki, T. Tricyclische orthokondensierte Nebenvalenzringe. Eur. J. Org. Chem. 1933, 503, 84–130. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ν(C=N), cm−1 | ν(C-O), cm−1 | ν(C-N), cm−1 | ν(O-H), cm−1 |
|---|---|---|---|---|
| H2(3-MeOSalen) | 1633(s) | 1250(s) | 1409(w) | 2600 |
| Ni(3-MeOSalen) | 1622(s) | 1315(s) | 1411(w) | - |
| poly-H2(3-MeOSalen) | 1628(s) | 1250(s) | 1409(w) | 2600 |
| poly-Ni(3-MeOSalen) | 1617(s) | 1315(s) | 1405(w) | - |
| Poly-Ni(3-MeOSalen) | Poly-H2(3-MeOSalen) | ||
|---|---|---|---|
| Potential range, V | 0 ÷ 0.77 | 0.77 ÷ 1.30 | 0 ÷ 1.30 |
| Molar mass of charge-transferring species, g mol−1 | 45 ± 5 | 147 ± 12 | 46 ± 4 |
| Number of electrons per a monomer unit | 1.4 ± 0.1 | 1.1 ± 0.1 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polozhentseva, J.; Novozhilova, M.; Karushev, M. Reversible Redox Processes in Polymer of Unmetalated Salen-Type Ligand: Combined Electrochemical in Situ Studies and Direct Comparison with Corresponding Nickel Metallopolymer. Int. J. Mol. Sci. 2022, 23, 1795. https://doi.org/10.3390/ijms23031795
Polozhentseva J, Novozhilova M, Karushev M. Reversible Redox Processes in Polymer of Unmetalated Salen-Type Ligand: Combined Electrochemical in Situ Studies and Direct Comparison with Corresponding Nickel Metallopolymer. International Journal of Molecular Sciences. 2022; 23(3):1795. https://doi.org/10.3390/ijms23031795
Chicago/Turabian StylePolozhentseva, Julia, Maria Novozhilova, and Mikhail Karushev. 2022. "Reversible Redox Processes in Polymer of Unmetalated Salen-Type Ligand: Combined Electrochemical in Situ Studies and Direct Comparison with Corresponding Nickel Metallopolymer" International Journal of Molecular Sciences 23, no. 3: 1795. https://doi.org/10.3390/ijms23031795
APA StylePolozhentseva, J., Novozhilova, M., & Karushev, M. (2022). Reversible Redox Processes in Polymer of Unmetalated Salen-Type Ligand: Combined Electrochemical in Situ Studies and Direct Comparison with Corresponding Nickel Metallopolymer. International Journal of Molecular Sciences, 23(3), 1795. https://doi.org/10.3390/ijms23031795