
Influenza A Virus NS1 Protein Structural Flexibility Analysis According to Its Structural Polymorphism Using Computational Approaches


Abstract
:1. Introduction
2. Results
2.1. NS1 Protein Properties Depending on Three Strains
2.1.1. Strain Specific Properties of NS1
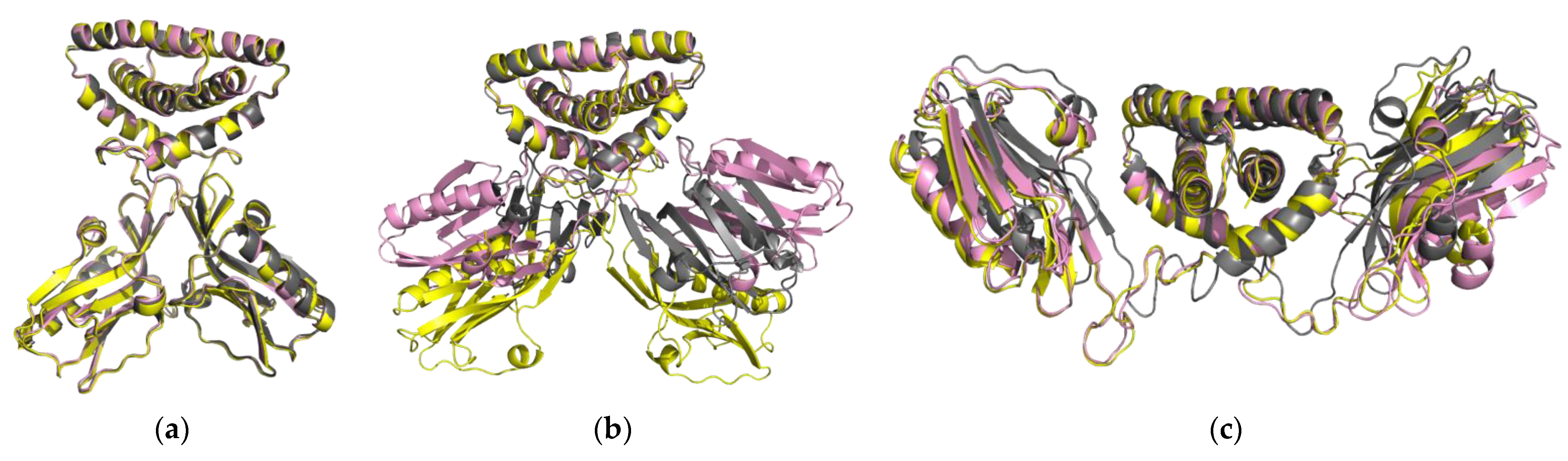
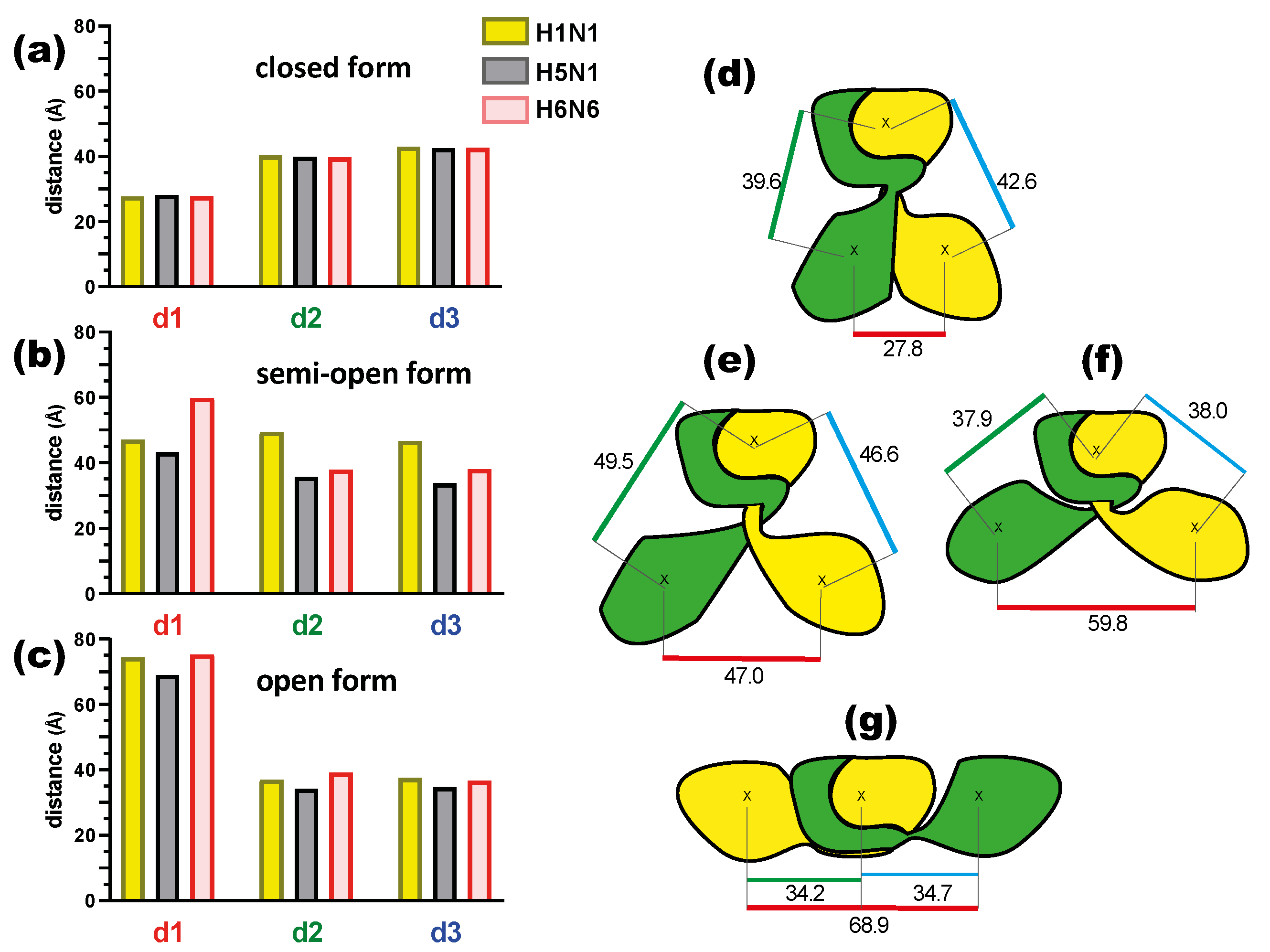
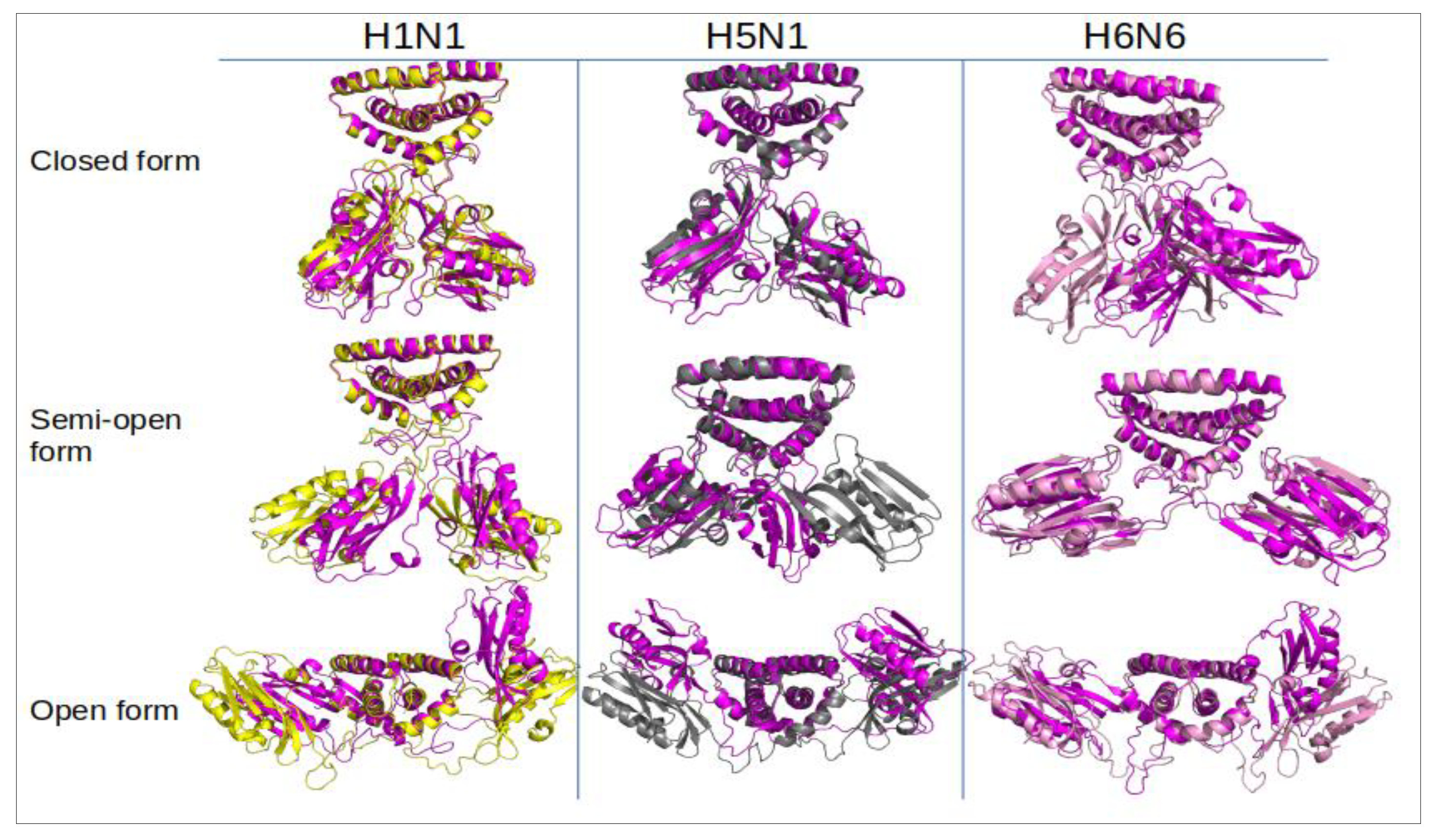
2.1.2. NS1 Structural Properties According to the Three Forms
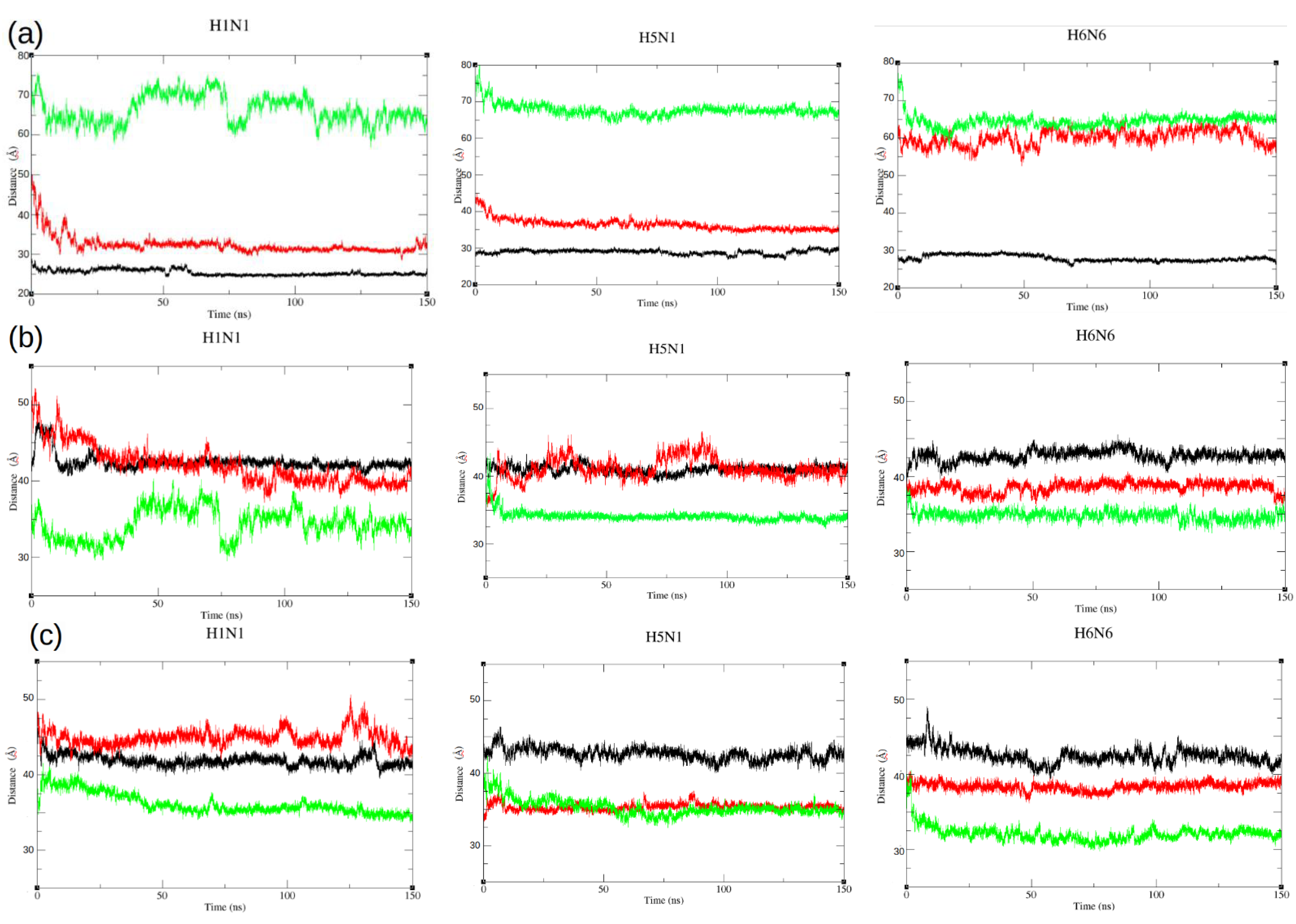
2.2. Dynamic Properties of NS1 Structures
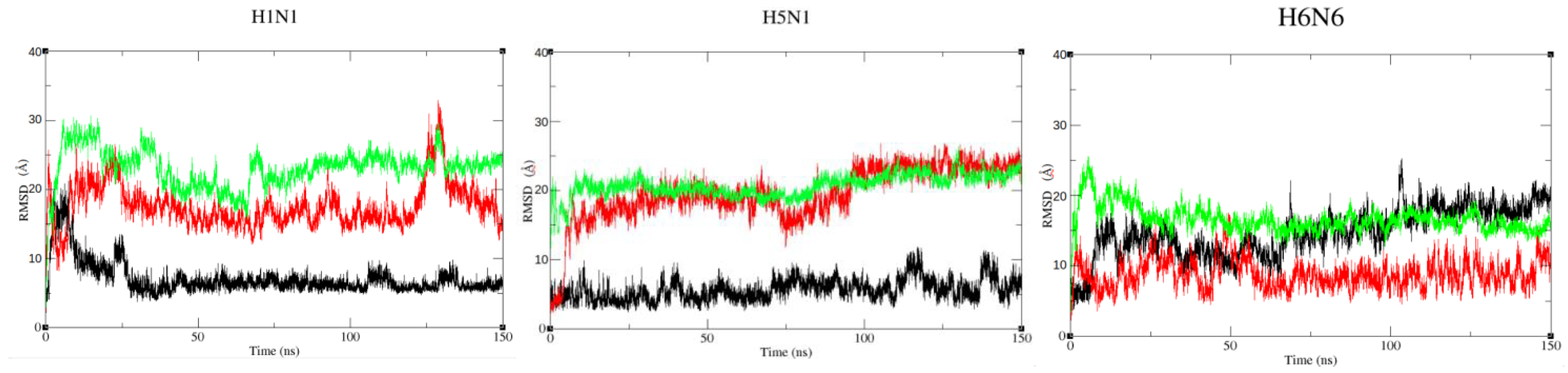
2.2.1. Stability of NS1 Structures from the Three Strains in Different Forms
2.2.2. Identification of Flexible Regions in the NS1 Structures
2.3. Dynamic Motions of the ED Domains during Simulations
3. Discussion
4. Materials and Methods
4.1. Preparation of the Crystal Structures
4.2. Building Structural Models by Homology
4.3. Molecular Dynamics (MD) Simulation
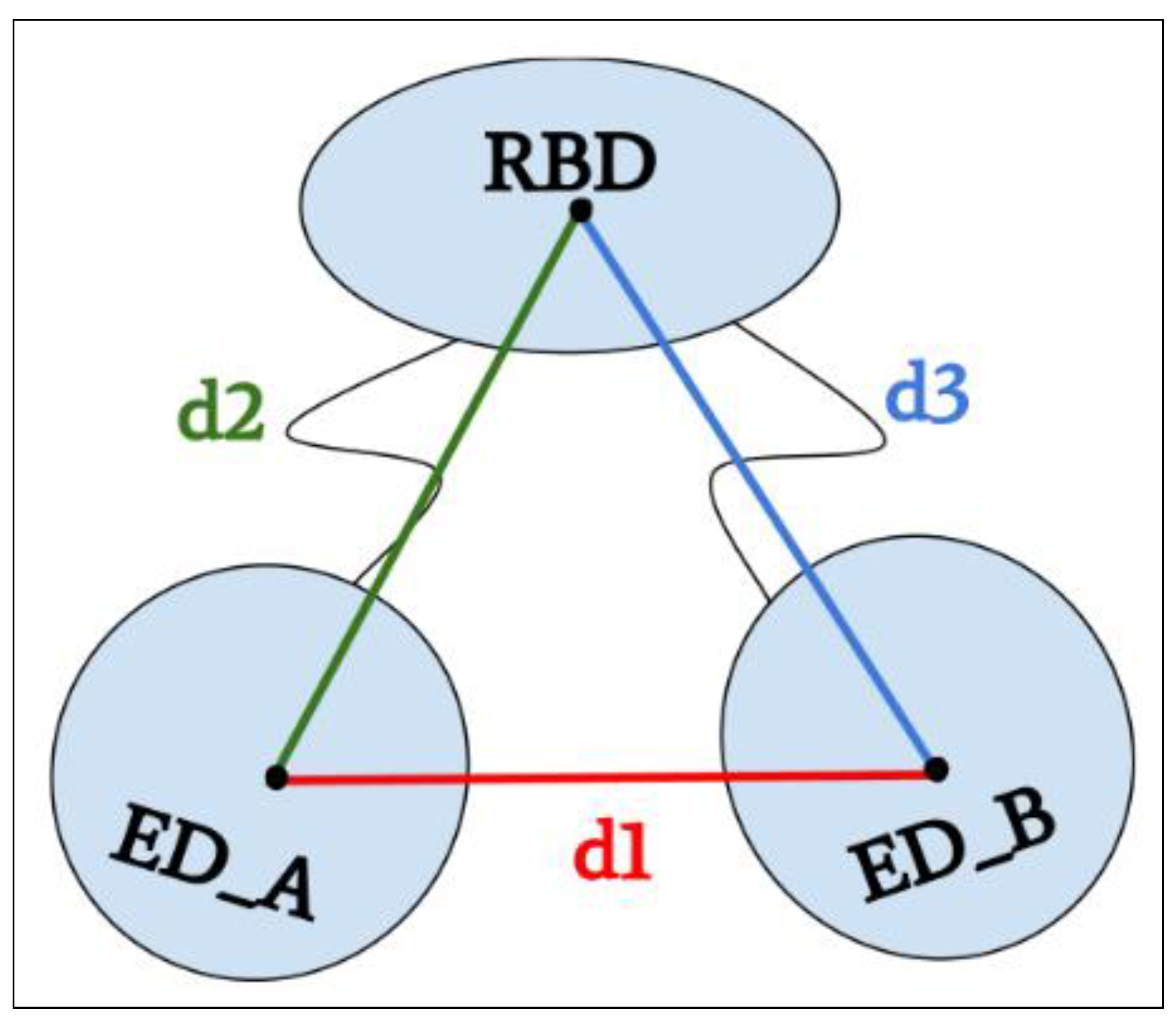
4.4. Distance Calculations between Domains
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Influenza (Seasonal). Available online: https://www.who.int/en/news-room/fact-sheets/detail/influenza-(seasonal) (accessed on 20 December 2021).
- Iuliano, A.D.; Roguski, K.M.; Chang, H.H.; Muscatello, D.J.; Palekar, R.; Tempia, S.; Cohen, C.; Gran, J.M.; Schanzer, D.; Cowling, B.J.; et al. Estimates of global seasonal influenza-associated respiratory mortality: A modelling study. Lancet 2018, 391, 1285–1300. [Google Scholar] [CrossRef]
- Hale, B.G.; Randall, R.E.; Ortín, J.; Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef] [PubMed]
- Zhirnov, O.P.; Konakova, T.E.; Wolff, T.; Klenk, H.D. NS1 Protein of Influenza A Virus Down-Regulates Apoptosis. J. Virol. 2002, 76, 1617–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, D.A. The influenza virus NS1 protein as a therapeutic target. Antivir. Res. 2013, 99, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Rosário-Ferreira, N.; Preto, A.J.; Melo, R.; Moreira, I.S.; Brito, R.M.M. The Central Role of Non-Structural Protein 1 (NS1) in Influenza Biology and Infection. Int. J. Mol. Sci. 2020, 21, 1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marc, D.; Barbachou, S.; Soubieux, D. The RNA-binding domain of influenza virus non-structural protein-1 cooperatively binds to virus-specific RNA sequences in a structure-dependent manner. Nucleic Acids Res. 2012, 41, 434–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, S.; Robertus, J.D. X-ray structures of NS1 effector domain mutants. Arch. Biochem. Biophys. 2010, 494, 198–204. [Google Scholar] [CrossRef] [Green Version]
- Krug, R.M. Functions of the influenza A virus NS1 protein in antiviral defense. Curr. Opin. Virol. 2015, 12, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.; Wong, S.M.; Yuan, Y.A. Structural basis for dsRNA recognition by NS1 protein of influenza A virus. Cell Res. 2008, 19, 187–195. [Google Scholar] [CrossRef]
- Trapp, S.; Soubieux, D.; Lidove, A.; Esnault, E.; Lion, A.; Guillory, V.; Wacquiez, A.; Kut, E.; Quéré, P.; Larcher, T.; et al. Major contribution of the RNA-binding domain of NS1 in the pathogenicity and replication potential of an avian H7N1 influenza virus in chickens. Virol. J. 2018, 15, 55. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marc, D. NS1 des Virus Influenza: Une Protéine Très « Influente ». Virologie (Montrouge) 2012, 16, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Kerry, P.S.; Ayllon, J.; Taylor, M.A.; Hass, C.; Lewis, A.; García-Sastre, A.; Randall, R.E.; Hale, B.G.; Russell, R.J. A Transient Homotypic Interaction Model for the Influenza A Virus NS1 Protein Effector Domain. PLoS ONE 2011, 6, e17946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrillo, B.; Choi, J.M.; Bornholdt, Z.A.; Sankaran, B.; Rice, A.P.; Prasad, B.V.V. The Influenza A Virus Protein NS1 Displays Structural Polymorphism. J. Virol. 2014, 88, 4113–4122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hale, B.G. Conformational plasticity of the influenza A virus NS1 protein. J. Gen. Virol. 2014, 95, 2099–2105. [Google Scholar] [CrossRef] [PubMed]
- Aramini, J.M.; Ma, L.C.; Zhou, L.; Schauder, C.M.; Hamilton, K.; Amer, B.R.; Mack, T.R.; Lee, H.W.; Ciccosanti, C.T.; Zhao, L.; et al. Dimer Interface of the Effector Domain of Non-structural Protein 1 from Influenza A Virus. J. Biol. Chem. 2011, 286, 26050–26060. [Google Scholar] [CrossRef] [Green Version]
- Shen, Q.; Cho, J.H. The structure and conformational plasticity of the nonstructural protein 1 of the 1918 influenza A virus. Biochem. Biophys. Res. Commun. 2019, 518, 178–182. [Google Scholar] [CrossRef]
- Jureka, A.; Kleinpeter, A.; Cornilescu, G.; Cornilescu, C.; Petit, C. Structural Basis for a Novel Interaction between the NS1 Protein Derived from the 1918 Influenza Virus and RIG-I. Structure 2015, 23, 2001–2010. [Google Scholar] [CrossRef] [Green Version]
- Bornholdt, Z.A.; Prasad, B.V.V. X-ray structure of NS1 from a highly pathogenic H5N1 influenza virus. Nature 2008, 456, 985–988. [Google Scholar] [CrossRef]
- Koliopoulos, M.G.; Lethier, M.; van der Veen, A.G.; Haubrich, K.; Hennig, J.; Kowalinski, E.; Stevens, R.V.; Martin, S.R.; Reis, E.; Sousa, C.; et al. Molecular mechanism of influenza A NS1-mediated TRIM25 recognition and inhibition. Nat. Commun. 2018, 9, 1820. [Google Scholar] [CrossRef]
- Mitra, S.; Kumar, D.; Hu, L.; Sankaran, B.; Moosa, M.M.; Rice, A.P.; Ferreon, J.C.; Ferreon, A.C.M.; Prasad, B.V.V. Influenza A Virus Protein NS1 Exhibits Strain-Independent Conformational Plasticity. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.S.; Huang, I.C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; García-Sastre, A. Influenza A Virus NS1 Targets the Ubiquitin Ligase TRIM25 to Evade Recognition by the Host Viral RNA Sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinpeter, A.B.; Jureka, A.S.; Falahat, S.M.; Green, T.J.; Petit, C.M. Structural analyses reveal the mechanism of inhibition of influenza virus NS1 by two antiviral compounds. J. Biol. Chem. 2018, 293, 14659–14668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abi Hussein, H.; Geneix, C.; Cauvin, C.; Marc, D.; Flatters, D.; Camproux, A.C. Molecular Dynamics Simulations of Influenza A Virus NS1 Reveal a Remarkably Stable RNA-Binding Domain Harboring Promising Druggable Pockets. Viruses 2020, 12, 537. [Google Scholar] [CrossRef]
- Cho, J.H.; Zhao, B.; Shi, J.; Savage, N.; Shen, Q.; Byrnes, J.; Yang, L.; Hwang, W.; Li, P. Molecular recognition of a host protein by NS1 of pandemic and seasonal influenza A viruses. Proc. Natl. Acad. Sci. USA 2020, 117, 6550–6558. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using Modeller. Curr. Protoc. Bioinform. 2016, 54. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- De Lano, W.L. The PyMOL Molecular Graphics System. 2002. Available online: www.pymol.org (accessed on 15 December 2021).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains/Forms | Closed Form | Semi-Open Form | Open Form | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Strains | H1N1HM | H5N1HM | H6N6HM | H1N1XR | H5N1HM | H6N6XR | H1N1HM | H5N1XR | H6N6HM |
| RMSD Values (in Å) | |||||||||
| RMSD All | 3.5 ± 0.5 | 2.3 ± 0.4 | 5.0 ± 1.1 | 10.2 ± 1.0 | 8.6 ± 1.4 | 4.2 ± 0.7 | 10.3 ± 1.6 | 11.9 ± 2.1 | 8.9 ± 0.7.9 |
| RMSD RBD | 1.9 ± 0.1 | 1.9 ± 0.1 | 2.4 ± 0.2 | 3.3 ± 0.1 | 2.3 ± 0.1 | 2.2 ± 0.2 | 3.1 ± 0.01 | 3.0 ± 0.1 | 2.7 ± 0.1 |
| RMSD ED_A | 1.5 ± 0.1 | 1.2 ± 0.1 | 1.1 ± 0.1 | 1.2 ± 1 | 1.6 ± 0.2 | 1.3 ± 0.2 | 1.8 ± 0.2 | 2.5 ± 0.2 | 2.1 ± 0.1 |
| RMSD ED_B | 1.6 ± 0.1 | 1.2 ± 0.2 | 1.3 ± 0.1 | 1.2 ± 0.1 | 1.1 ± 0.1 | 1.1 ± 0.1 | 1.4 ± 0.1 | 1.9 ± 0.2 | 2.1 ± 0.1 |
| RMSD ED fit to RBD | 7.1 ± 2.4 | 5.6 ± 1.5 | 14.7 ± 3.4 | 17.6 ± 3.1 | 19.5 ± 4 | 8.8 ± 2.0 | 23.1 ± 2.7 | 20.4 ± 2.9 | 16.6 ± 1.8 |
| Distances values | |||||||||
| Distance d1 | 25.4 ± 0.7 | 28.8 ± 0.6 | 27.8 ± 0.8 | 32.4 ± 2.3 | 36.4 ± 1.5 | 60.0 ± 2.0 | 66.3 ± 3.4 | 66.5 ± 1.7 | 64.5 ± 1.7 |
| Distance d2 | 42.4 ± 1.0 | 40.9 ± 0.5 | 42.7 ± 0.7 | 42.0 ± 2.0 | 41.1 ± 1.6 | 38.5 ± 0.6 | 34.3 ± 1.9 | 36.3 ± 1.0 | 34.7 ± 0.6 |
| Distance d3 | 41.9 ± 0.7 | 42.6 ± 0.8 | 42.4 ± 1.0 | 44.9 ± 1.0 | 35.3 ± 0.4 | 38.3 ± 0.5 | 36.1 ± 1.3 | 34.5 ± 1.3 | 32.0 ± 1.0 |
| Strains/Forms | Closed Form | Semi-Open Form | Open Form |
|---|---|---|---|
| H1N1 | H1N1HM | H1N1XR | H1N1HM |
| Uniprot identifier: P03496 | Uniprot identifier: P03496 | Uniprot identifier: P03496 | |
| Xray structure template: 4OPA | Xray structure: 5NT2 | Xray structure template: 6OQE | |
| Reverse mutations: R38A/K41A/W187A | Reverse mutations: R38A/K41A/W187A | Reverse mutations: R38A/K41A/W187A | |
| H5N1 | H5N1HM | H5N1HM | H5N1XR |
| Uniprot identifier: A5A5U1 | Uniprot identifier: A5A5U1 | Uniprot identifier: A5A5U1 | |
| Xray structure template: 4OPA | Xray structure template: 5NT2 | Xray structure: 3F5T | |
| Reverse mutations: R38A/K41A | Reverse mutations: R38A/K41A | Reverse mutations: R38A/K41A | |
| H6N6 | H6N6HM | H6N6XR | H6N6HM |
| Uniprot identifier: Q20NS3 | Uniprot identifier: Q20NS3 | Uniprot identifier: Q20NS3 | |
| Xray structure template: 4OPA | Xray structure: 4OPH | Xray structure template: 6OQE | |
| Reverse mutations: R38A/K41A | Reverse mutations: R38A/K41A | Reverse mutations: R38A/K41A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naceri, S.; Marc, D.; Camproux, A.-C.; Flatters, D. Influenza A Virus NS1 Protein Structural Flexibility Analysis According to Its Structural Polymorphism Using Computational Approaches. Int. J. Mol. Sci. 2022, 23, 1805. https://doi.org/10.3390/ijms23031805
Naceri S, Marc D, Camproux A-C, Flatters D. Influenza A Virus NS1 Protein Structural Flexibility Analysis According to Its Structural Polymorphism Using Computational Approaches. International Journal of Molecular Sciences. 2022; 23(3):1805. https://doi.org/10.3390/ijms23031805
Chicago/Turabian StyleNaceri, Sarah, Daniel Marc, Anne-Claude Camproux, and Delphine Flatters. 2022. "Influenza A Virus NS1 Protein Structural Flexibility Analysis According to Its Structural Polymorphism Using Computational Approaches" International Journal of Molecular Sciences 23, no. 3: 1805. https://doi.org/10.3390/ijms23031805