
Nanomedicine Strategies for Management of Drug Resistance in Lung Cancer

 ,
,  ,
,  , , , , and
, , , , and 
Abstract
:
1. Introduction
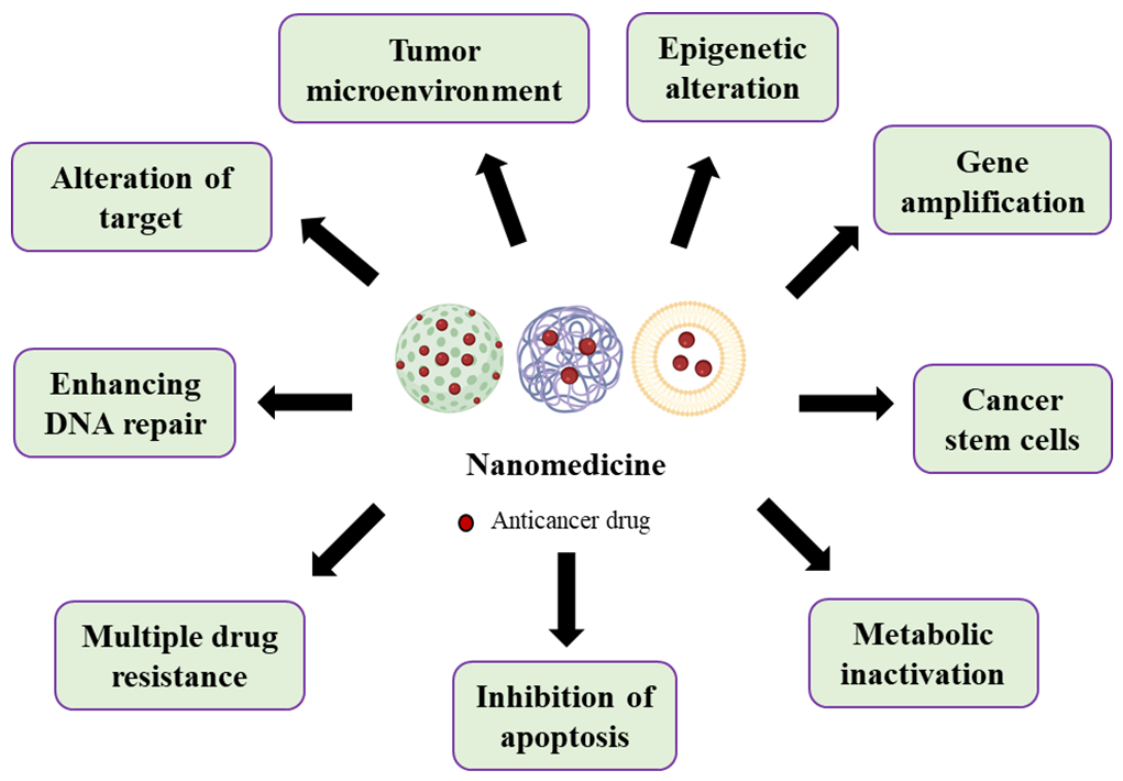
2. Nanomedicine Applications in Management of Lung Cancer Drug Resistance
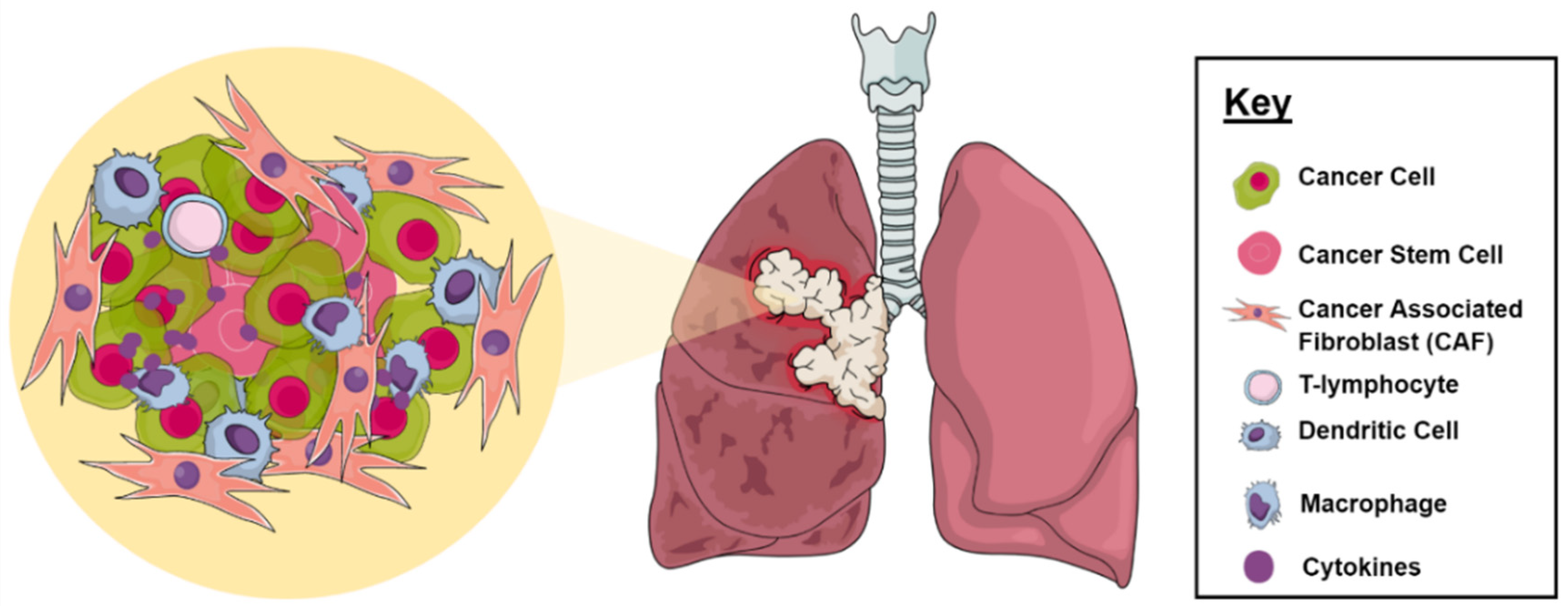
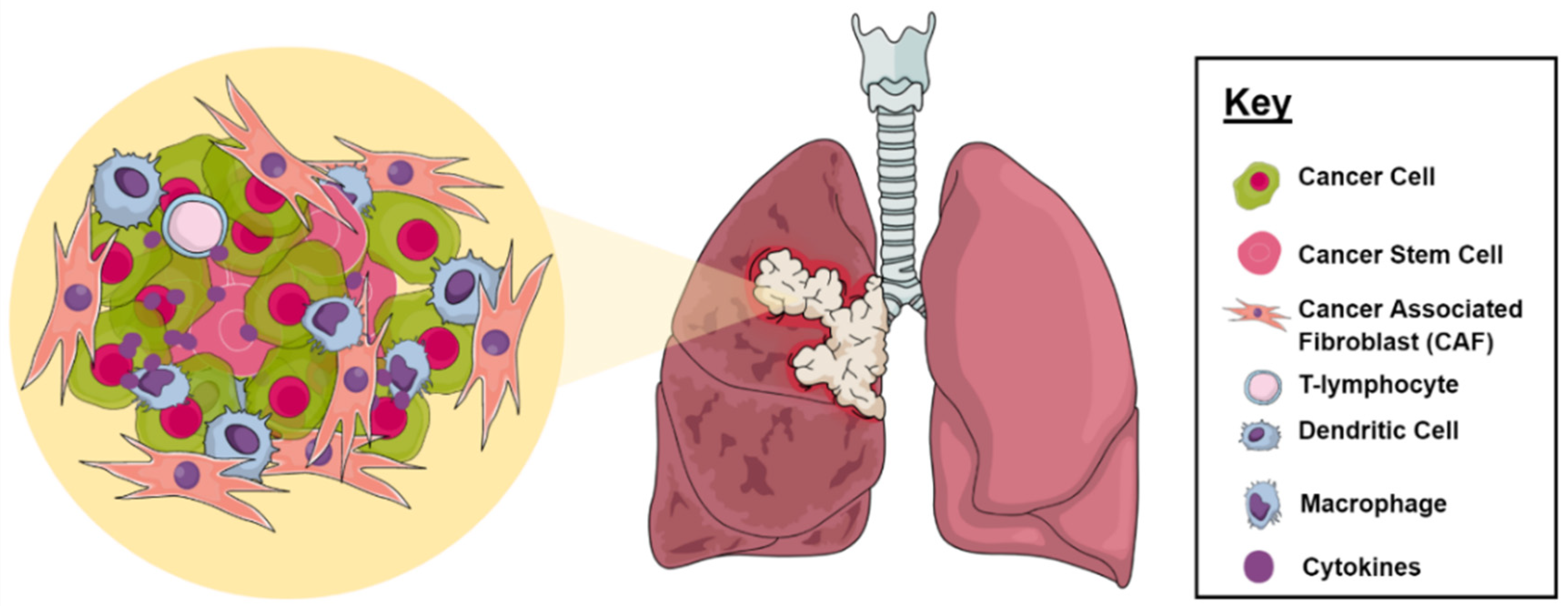
2.1. Tumor Microenvironment
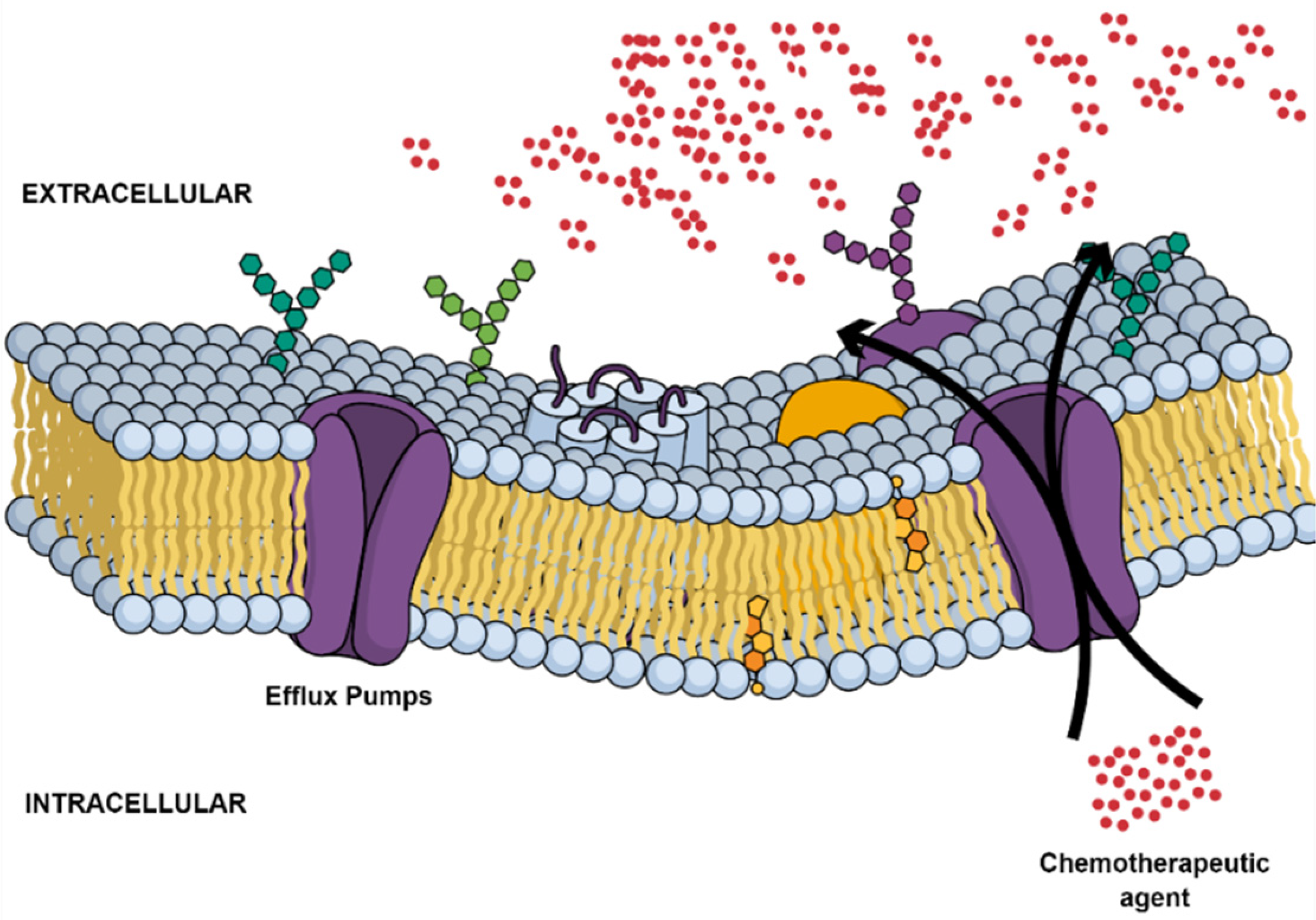
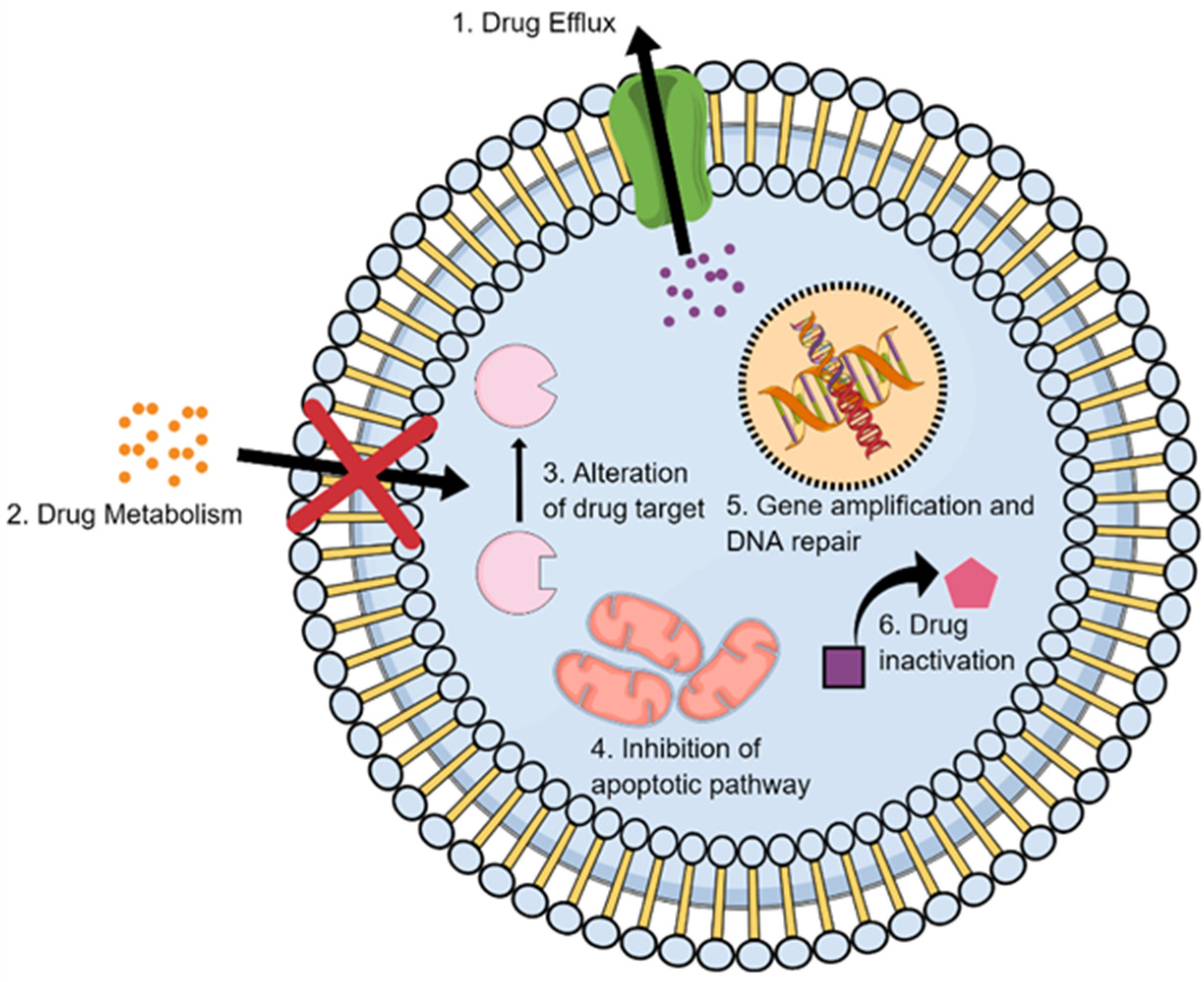
2.2. Multidrug Resistance
2.3. Cancer Stem Cells
2.4. Metabolic Inactivation of the Anticancer Drugs
2.5. Inhibition of the Cell Death
2.6. Alteration of Drug Targets
2.7. Enhancing DNA Repair
2.8. Gene Amplification
2.9. Epigenetic Alteration Caused Drug Resistance
2.10. Clinical Studies Using Nanotechnology for Management of DR in LC
3. Current Limitations and Future Perspectives of Nanomedicine Aimed at Overcoming Drug Resistance
4. Biological Aspects
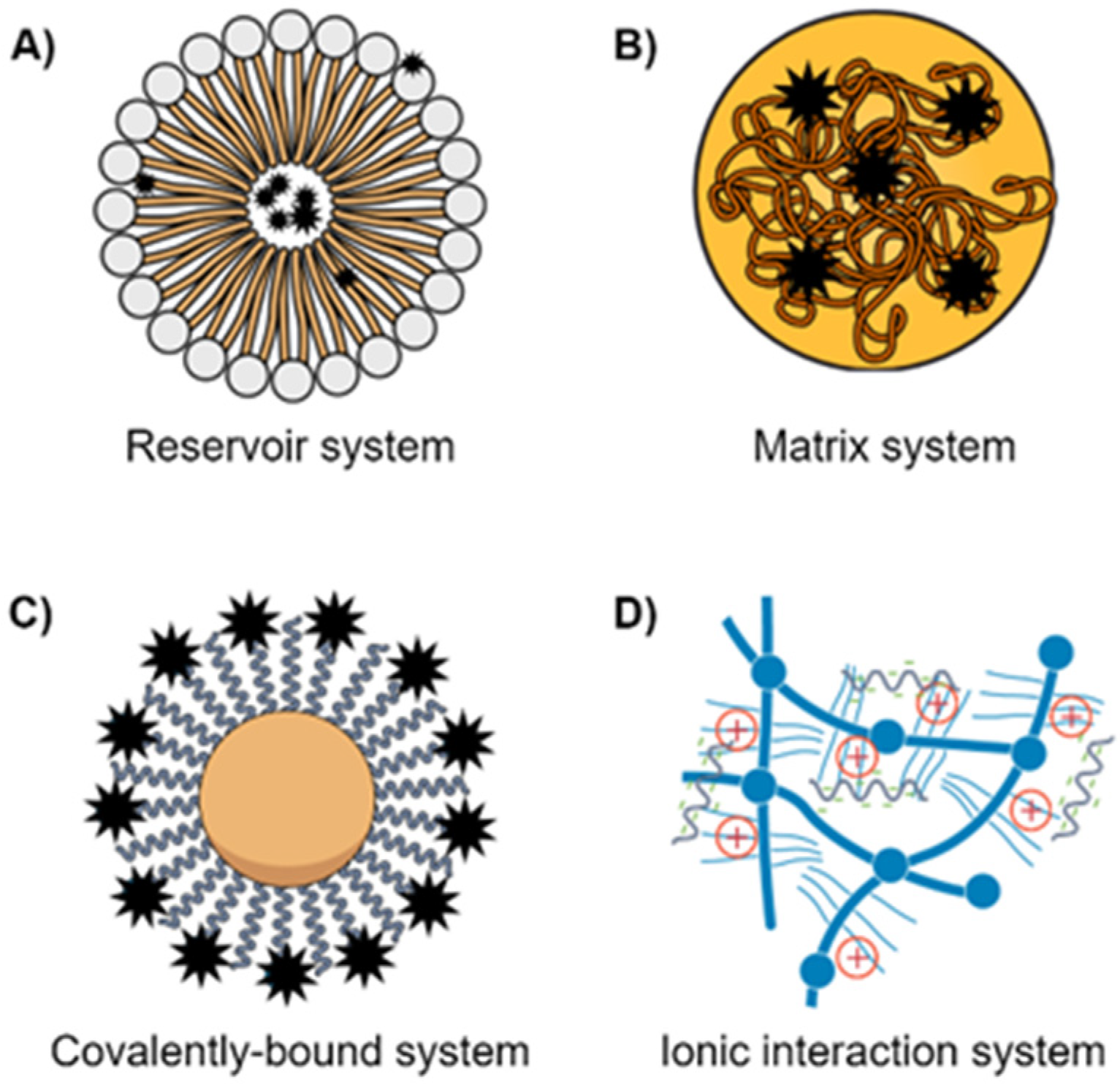
5. Formulation Drawbacks
6. New Approaches for the Use of Nanomedicine in the Treatment of Resistant LC
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dela Cruz, C.S.; Tanoue, L.T.; Matthay, R.A. Lung Cancer: Epidemiology, Etiology, and Prevention. Clin. Chest Med. 2011, 32, 605–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, K.D.; Berns, A. Cell of origin of lung cancer. Mol. Oncol. 2010, 4, 397–403. [Google Scholar] [CrossRef]
- Sutradhar, K.B.; Amin, L. Nanotechnology in Cancer Drug Delivery and Selective Targeting. ISRN Nanotechnol. 2014, 2014, 939378. [Google Scholar] [CrossRef] [Green Version]
- Jabir, N.R.; Tabrez, S.; Ashraf, G.M.; Shakil, S.; Damanhouri, G.A.; Kamal, M.A. Nanotechnology-based approaches in anticancer research. Int. J. Nanomed. 2012, 7, 4391–4408. [Google Scholar] [CrossRef] [Green Version]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Sarkar, S.; Horn, G.; Moulton, K.; Oza, A.; Byler, S.; Kokolus, S.; Longacre, M. Cancer Development, Progression, and Therapy: An Epigenetic Overview. Int. J. Mol. Sci. 2013, 14, 21087–21113. [Google Scholar] [CrossRef]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; del Pilar Rodriguez-Torres, M.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef] [Green Version]
- Palazzolo, S.; Bayda, S.; Hadla, M.; Caligiuri, I.; Corona, G.; Toffoli, G.; Rizzolio, F. The Clinical Translation of Organic Nanomaterials for Cancer Therapy: A Focus on Polymeric Nanoparticles, Micelles, Liposomes and Exosomes. Curr. Med. Chem. 2018, 25, 4224–4268. [Google Scholar] [CrossRef]
- Bahman, F.; Pittalà, V.; Haider, M.; Greish, K. Enhanced Anticancer Activity of Nanoformulation of Dasatinib against Triple-Negative Breast Cancer. J. Pers. Med. 2021, 11, 559. [Google Scholar] [CrossRef]
- Awad, N.S.; Haider, M.; Paul, V.; AlSawaftah, N.M.; Jagal, J.; Pasricha, R.; Husseini, G.A. Ultrasound-Triggered Liposomes Encapsulating Quantum Dots as Safe Fluorescent Markers for Colorectal Cancer. Pharmaceutics 2021, 13, 2073. [Google Scholar] [CrossRef] [PubMed]
- Haider, M.; Zaki, K.Z.; El Hamshary, M.R.; Hussain, Z.; Orive, G.; Ibrahim, H.O. Polymeric nanocarriers: A promising tool for early diagnosis and efficient treatment of colorectal cancer. J. Adv. Res. 2021. [Google Scholar] [CrossRef]
- Ahmed, I.S.; El Hosary, R.; Hassan, M.A.; Haider, M.; Abd-Rabo, M.M. Efficacy and Safety Profiles of Oral Atorvastatin-Loaded Nanoparticles: Effect of Size Modulation on Biodistribution. Mol. Pharm. 2018, 15, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Haider, M.; Elsherbeny, A.; Jagal, J.; Hubatová-Vacková, A.; Ahmed, I.S. Optimization and Evaluation of Poly(lactide-co-glycolide) Nanoparticles for Enhanced Cellular Uptake and Efficacy of Paclitaxel in the Treatment of Head and Neck Cancer. Pharmaceutics 2020, 12, 828. [Google Scholar] [CrossRef]
- Kim, E.-S.; Ahn, E.H.; Chung, E.; Kim, D.-H. Recent advances in nanobiotechnology and high-throughput molecular techniques for systems biomedicine. Mol. Cells 2013, 36, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Haider, M.; Abdin, S.M.; Kamal, L.; Orive, G. Nanostructured Lipid Carriers for Delivery of Chemotherapeutics: A Review. Pharmaceutics 2020, 12, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Kalyane, D.; Raval, N.; Maheshwari, R.; Tambe, V.; Kalia, K.; Tekade, R.K. Employment of enhanced permeability and re-tention effect (EPR): Nanoparticle-based precision tools for targeting of therapeutic and diagnostic agent in cancer. Mater. Sci. Eng. C 2019, 98, 1252–1276. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Cheng, X.; Li, N.; Wang, H.; Chen, H. Nanocarriers and Their Loading Strategies. Adv. Health Mater. 2019, 8, e1801002. [Google Scholar] [CrossRef]
- Rad, H.S.; Monkman, J.; Warkiani, M.E.; Ladwa, R.; O’Byrne, K.; Rezaei, N.; Kulasinghe, A. Understanding the tumor microenvironment for effective immunotherapy. Med. Res. Rev. 2021, 41, 1474–1498. [Google Scholar] [CrossRef]
- Poltavets, V.; Kochetkova, M.; Pitson, S.M.; Samuel, M.S. The Role of the Extracellular Matrix and Its Molecular and Cellular Regulators in Cancer Cell Plasticity. Front. Oncol. 2018, 8, 431. [Google Scholar] [CrossRef] [Green Version]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [Green Version]
- Tan, Z.; Xue, H.; Sun, Y.; Zhang, C.; Song, Y.; Qi, Y. The Role of Tumor Inflammatory Microenvironment in Lung Cancer. Front. Pharmacol. 2021, 12, 688625. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Suares, D.; Yergeri, M.C. Tumor Microenvironment Targeted Nanotherapy. Front. Pharmacol. 2018, 9, 1230. [Google Scholar] [CrossRef] [PubMed]
- Altorki, N.K.; Markowitz, G.J.; Gao, D.; Port, J.L.; Saxena, A.; Stiles, B.; McGraw, T.; Mittal, V. The lung microenvironment: An important regulator of tumour growth and metastasis. Nat. Rev. Cancer 2019, 19, 9–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laflamme, B. Transcriptional cross-talk between tumor and stromal cells. Nat. Genet. 2014, 46, 933. [Google Scholar] [CrossRef]
- Stankovic, B.; Bjørhovde, H.A.K.; Skarshaug, R.; Aamodt, H.; Frafjord, A.; Müller, E.; Hammarström, C.; Beraki, K.; Bækkevold, E.S.; Woldbæk, P.R.; et al. Immune cell composition in human non-small cell lung cancer. Front. Immunol. 2019, 9, 3101. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Hu, Y.; Yao, C. The paradoxical role of tumor-infiltrating immune cells in lung cancer. Intractable Rare Dis. Res. 2017, 6, 234–241. [Google Scholar] [CrossRef] [Green Version]
- Carrasco-Esteban, E.; Domínguez-Rullán, J.A.; Barrionuevo-Castillo, P.; Pelari-Mici, L.; Leaman, O.; Sastre-Gallego, S.; López-Campos, F. Current role of nanoparticles in the treatment of lung cancer. J. Clin. Transl. Res. 2021, 7, 140. [Google Scholar] [CrossRef]
- Mangal, S.; Gao, W.; Li, T.; Zhou, Q. Pulmonary delivery of nanoparticle chemotherapy for the treatment of lung cancers: Challenges and opportunities. Acta Pharmacol. Sin. 2017, 38, 782–797. [Google Scholar] [CrossRef]
- Thakor, A.S.; Gambhir, S.S. Nanooncology: The future of cancer diagnosis and therapy. CA A Cancer J. Clin. 2013, 63, 395–418. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Hu, C.; Tai, Z.; Yao, C.; Tian, J.; Zhang, L.; Xia, Q.; Gong, C.; Gao, Y.; Gao, S. Tumour microenvironment-responsive lipoic acid nanoparticles for targeted delivery of docetaxel to lung cancer. Sci. Rep. 2016, 6, 36281. [Google Scholar] [CrossRef] [Green Version]
- Palanikumar, L.; Al-Hosani, S.; Kalmouni, M.; Nguyen, V.P.; Ali, L.; Pasricha, R.; Barrera, F.N.; Magzoub, M. pH-responsive high stability polymeric nanoparticles for targeted delivery of anticancer therapeutics. Commun. Biol. 2020, 3, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Uthaman, S.; Huh, K.M.; Park, I.-K. Tumor microenvironment-responsive nanoparticles for cancer theragnostic applications. Biomater. Res. 2018, 22, 1–11. [Google Scholar] [CrossRef]
- Engelberg, S.; Netzer, E.; Assaraf, Y.G.; Livney, Y.D. Selective eradication of human non-small cell lung cancer cells using aptamer-decorated nanoparticles harboring a cytotoxic drug cargo. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Babu, A.; Templeton, A.K.; Munshi, A.; Ramesh, R. Nanoparticle-Based Drug Delivery for Therapy of Lung Cancer: Progress and Challenges. J. Nanomater. 2013, 2013, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Lo, Y.-L.; Huang, X.-S.; Chen, H.-Y.; Huang, Y.-C.; Liao, Z.-X.; Wang, L.-F. ROP and ATRP fabricated redox sensitive micelles based on PCL-SS-PMAA diblock copolymers to co-deliver PTX and CDDP for lung cancer therapy. Colloids Surf. B Biointerfaces 2021, 198, 111443. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.H.; Choi, E.-S.; Kim, S.; Goh, S.-H.; Choi, Y. Redox-Responsive Manganese Dioxide Nanoparticles for Enhanced MR Imaging and Radiotherapy of Lung Cancer. Front. Chem. 2017, 5, 109. [Google Scholar] [CrossRef]
- Yuan, Z.-Q.; Li, J.-Z.; Liu, Y.; Chen, W.-L.; Yang, S.-D.; Zhang, C.-G.; Zhu, W.-J.; Zhou, X.-F.; Liu, C.; Zhang, X.-N. Systemic delivery of micelles loading with paclitaxel using N-succinyl-palmitoyl-chitosan decorated with cRGDyK peptide to inhibit non-small-cell lung cancer. Int. J. Pharm. 2015, 492, 141–151. [Google Scholar] [CrossRef]
- Siriwon, N.; Bejan, D.; Sherif, P.; Pdx, R.; Wang, R.; Pharmaceuticals, P.; Nelson, M.; Zaidan, H.; Hoang, N.; Bindal, A.; et al. Development of novel immunotherapy based on nanoparticle co-delivering PLK1 and PD-L1 inhibitors for lung cancer treatment Moataz Reda PDX Pharmaceuticals Worapol Ngamcherdtrakul PDX Pharmaceuticals. Preprint 2021. [Google Scholar] [CrossRef]
- Fletcher, J.; Williams, R.T.; Henderson, M.J.; Norris, M.D.; Haber, M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist. Updat. 2016, 26, 1–9. [Google Scholar] [CrossRef]
- Bar-Zeev, M.; Livney, Y.D.; Assaraf, Y.G. Targeted nanomedicine for cancer therapeutics: Towards precision medicine overcoming drug resistance. Drug Resist. Updat. 2017, 31, 15–30. [Google Scholar] [CrossRef]
- Gillet, J.-P.; Gottesman, M.M. Mechanisms of Multidrug Resistance in Cancer. Methods Mol. Biol. 2010, 596, 47–76. [Google Scholar] [PubMed]
- Dong, Y.; Feng, S.-S. Poly(d,l-lactide-co-glycolide)/montmorillonite nanoparticles for oral delivery of anticancer drugs. Biomaterials 2005, 26, 6068–6076. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Wang, J.; Lu, C.; Xu, Z.; Chai, J.-J.; Ke, Q.; Deng, X.-Z. Emodin enhances cisplatin sensitivity in non-small cell lung cancer through Pgp downregulation. Oncol. Lett. 2021, 21, 1. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.H.; Wientjes, M.G.; Au, J.L. Kinetics of P-glycoprotein-mediated efflux of paclitaxel. J. Pharmacol. Exp. Ther. 2001, 298, 1236–1242. [Google Scholar] [PubMed]
- Galletti, E.; Magnani, M.; Renzulli, M.L.; Botta, M. Paclitaxel and Docetaxel Resistance: Molecular Mechanisms and Development of New Generation Taxanes. ChemMedChem 2007, 2, 920–942. [Google Scholar] [CrossRef]
- Bergman, A.M.; Pinedo, H.M.; Talianidis, I.; Veerman, G.; Loves, W.J.P.; Van Der Wilt, C.L.; Peters, G.J. Increased sensitivity to gemcitabine of P-glycoprotein and multidrug resistance-associated protein-overexpressing human cancer cell lines. Br. J. Cancer 2003, 88, 1963–1970. [Google Scholar] [CrossRef] [Green Version]
- Berger, W.; Setinek, U.; Hollaus, P.; Zidek, T.; Steiner, E.; Elbling, L.; Cantonati, H.; Attems, J.; Gsur, A.; Micksche, M. Multidrug resistance markers P-glycoprotein, multidrug resistance protein 1, and lung resistance protein in non-small cell lung cancer: Prognostic implications. J. Cancer Res. Clin. Oncol. 2005, 131, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Le, H.; Zhang, Y.; Qian, L.; Sekhar, K.R.; Li, W. Lung Resistance Protein and Multidrug Resistance Protein in Non-Small Cell Lung Cancer and Their Clinical Significance. J. Int. Med. Res. 2011, 39, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Bie, M.; Wang, X.; Fan, M.; Chen, B.; Shi, Q.; Jiang, Y. PGRN exacerbates the progression of non-small cell lung cancer via PI3K/AKT/Bcl-2 antiapoptotic signaling. Genes Dis. 2021. [Google Scholar] [CrossRef]
- Ke, W.; Zhao, X.; Lu, Z. Foeniculum vulgare seed extract induces apoptosis in lung cancer cells partly through the down-regulation of Bcl. Biomed. Pharmacother. 2021, 135, 111213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, N.; Ma, M.; Huang, H.; Handley, M.; Bai, X.; Shan, F. Methionine enkephalin (MENK) suppresses lung cancer by regulating the Bcl-2/Bax/caspase-3 signaling pathway and enhancing natural killer cell-driven tumor immunity. Int. Immunopharmacol. 2021, 98, 107837. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Shi, L.; Banerjee, P.; Liu, X.; Guo, H.-F.; Yu, J.; Bota-Rabassedas, N.; Rodriguez, B.L.; Gibbons, D.L.; Russell, W.K.; et al. A protumorigenic secretory pathway activated by p53 deficiency in lung adenocarcinoma. J. Clin. Investig. 2021, 131, 131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Han, C.Y.; Duan, F.G.; Fan, X.-X.; Yao, X.-J.; Parks, R.J.; Tang, Y.-J.; Wang, M.-F.; Liu, L.; Tsang, B.K.; et al. p53 sensitizes chemoresistant non-small cell lung cancer via elevation of reactive oxygen species and suppression of EGFR/PI3K/AKT signaling. Cancer Cell Int. 2019, 19, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Gibbons, D.L.; Byers, L.A.; Kurie, J.M. Smoking, p53 Mutation, and Lung Cancer. Mol. Cancer Res. 2014, 12, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Murakami, M.; Cabral, H.; Matsumoto, Y.; Wu, S.; Kano, M.R.; Yamori, T.; Nishiyama, N.; Kataoka, K. Improving Drug Potency and Efficacy by Nanocarrier-Mediated Subcellular Targeting. Sci. Transl. Med. 2011, 3, 64ra2. [Google Scholar] [CrossRef]
- Ahmad, J.; Akhter, S.; Khan, M.A.; Wahajuddin, M.; Greig, N.H.; Kamal, M.A.; Midoux, P.; Pichon, C. Engineered Nanoparticles Against MDR in Cancer: The State of the Art and its Prospective. Curr. Pharm. Des. 2016, 22, 4360–4373. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Li, H.; Wang, Y.; Dong, F.; Wang, H.; Zhang, S. Enhanced activity of doxorubicin in drug resistant A549 tumor cells by encapsulation of P-glycoprotein inhibitor in PLGA-based nanovectors. Oncol. Lett. 2013, 7, 387–392. [Google Scholar] [CrossRef]
- Pramual, S.; Lirdprapamongkol, K.; Jouan-Hureaux, V.; Barberi-Heyob, M.; Frochot, C.; Svasti, J.; Niamsiri, N. Overcoming the diverse mechanisms of multidrug resistance in lung cancer cells by photodynamic therapy using pTHPP-loaded PLGA-lipid hybrid nanoparticles. Eur. J. Pharm. Biopharm. 2020, 149, 218–228. [Google Scholar] [CrossRef]
- Zhou, J.; Zhao, W.-Y.; Ma, X.; Ju, R.-J.; Li, X.-Y.; Li, N.; Sun, M.-G.; Shi, J.-F.; Zhang, C.-X.; Lu, W.-L. The anticancer efficacy of paclitaxel liposomes modified with mitochondrial targeting conjugate in resistant lung cancer. Biomaterials 2013, 34, 3626–3638. [Google Scholar] [CrossRef]
- Tang, D.; Zhao, X.; Zhang, L.; Wang, Z.; Wang, C. Identification of hub genes to regulate breast cancer metastasis to brain by bioinformatics analyses. J. Cell. Biochem. 2019, 120, 9522–9531. [Google Scholar] [CrossRef]
- Ma, S.; Li, X.; Ran, M.; Ji, M.; Gou, J.; Yin, T.; He, H.; Wang, Y.; Zhang, Y.; Tang, X. Fabricating nanoparticles co-loaded with survivin siRNA and Pt(IV) prodrug for the treatment of platinum-resistant lung cancer. Int. J. Pharm. 2021, 601, 120577. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.-M.; Jie, J.; Zhang, Y.; Liu, H.; Peng, L.-P. A self-assembled polyjuglanin nanoparticle loaded with doxorubicin and anti-Kras siRNA for attenuating multidrug resistance in human lung cancer. Biochem. Biophys. Res. Commun. 2017, 493, 1430–1437. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, Q.; Mo, J.; Dai, H. Drug-Loaded Polymeric Nanoparticles for Cancer Stem Cell Targeting. Front. Pharmacol. 2017, 8, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabavathy, D.; Swarnalatha, Y.; Ramadoss, N. Lung cancer stem cells—Origin, characteristics and therapy. Stem Cell Investig. 2018, 5, 6. [Google Scholar] [CrossRef] [Green Version]
- Maiuthed, A.; Chantarawong, W.; Chanvorachote, P. Lung Cancer Stem Cells and Cancer Stem Cell-targeting Natural Compounds. Anticancer Res. 2018, 38, 3797–3809. [Google Scholar] [CrossRef] [Green Version]
- Masciale, V.; Grisendi, G.; Banchelli, F.; D’Amico, R.; Maiorana, A.; Sighinolfi, P.; Stefani, A.; Morandi, U.; Dominici, M.; Aramini, B. Isolation and Identification of Cancer Stem-Like Cells in Adenocarcinoma and Squamous Cell Carcinoma of the Lung: A Pilot Study. Front. Oncol. 2019, 9, 1394. [Google Scholar] [CrossRef] [Green Version]
- Herreros-Pomares, A.; De-Maya-Girones, J.D.; Calabuig-Fariñas, S.; Lucas, R.; Martínez, A.; Pardo-Sánchez, J.M.; Alonso, S.; Blasco, A.; Guijarro, R.; Martorell, M.; et al. Lung tumorspheres reveal cancer stem cell-like properties and a score with prognostic impact in resected non-small-cell lung cancer. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Li, Y.; Shi, S.; Ming, Y.; Wang, L.; Li, C.; Luo, M.; Li, Z.; Li, B.; Chen, J. Specific cancer stem cell-therapy by albumin nanoparticles functionalized with CD44-mediated targeting. J. Nanobiotechnol. 2018, 16, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Hong, Y.; Li, Y.; Hu, C.; Yip, T.-C.; Yu, W.K.; Zhu, Y.; Fong, C.-C.; Wang, W.; Au, S.-K.; et al. Targeted destruction of cancer stem cells using multifunctional magnetic nanoparticles that enable combined hyperthermia and chemotherapy. Theranostics 2020, 10, 1181. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.-L.; Wang, S.-S.; Jiang, J.; Liang, X.-H. Links between cancer stem cells and epithelial–mesenchymal transition. OncoTargets Ther. 2015, 8, 2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, H.; Liu, Y.; Gao, Z.; Huang, W. Recent advances in drug delivery systems for targeting cancer stem cells. Acta Pharm. Sin. B 2021, 11, 55–70. [Google Scholar] [CrossRef]
- Chiou, G.-Y.; Cherng, J.-Y.; Hsu, H.-S.; Wang, M.-L.; Tsai, C.-M.; Lu, K.-H.; Chien, Y.; Hung, S.-C.; Chen, Y.-W.; Wong, C.-I.; et al. Cationic polyurethanes-short branch PEI-mediated delivery of Mir145 inhibited epithelial–mesenchymal transdifferentiation and cancer stem-like properties and in lung adenocarcinoma. J. Control. Release 2012, 159, 240–250. [Google Scholar] [CrossRef]
- Karachaliou, N.; Rosell, R.; Viteri, S. The role of SOX2 in small cell lung cancer, lung adenocarcinoma and squamous cell carcinoma of the lung. Transl. Lung Cancer Res. 2013, 2, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Andey, T.; Bora-Singhal, N.; Chellappan, S.P.; Singh, M. Cationic lipoplexes for treatment of cancer stem cell-derived murine lung tumors. Nanomed. Nanotechnol. Biol. Med. 2019, 18, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Di Nicolantonio, F.; Mercer, S.J.; Knight, L.; Gabriel, F.G.; Whitehouse, P.; Sharma, S.; Fernando, A.; Glaysher, S.; Di Palma, S.; Johnson, P.; et al. Cancer cell adaptation to chemotherapy. BMC Cancer 2005, 5, 78. [Google Scholar] [CrossRef] [Green Version]
- Satta, T.; Isobe, K.-I.; Yamauchi, M.; Nakashima, I.; Takagi, H. Expression of MDR1 and glutatione S transferase-π genes and chemosensitivities in human gastrointestinal cancer. Cancer 1992, 69, 941–946. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochrome P450 and Chemical Toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef]
- Jancova, P.; Anzenbacher, P.; Anzenbacherova, E. Phase II Drug Metabolizing Enzymes. Biomed. Pap. 2010, 154, 103–116. [Google Scholar] [CrossRef] [Green Version]
- Iyanagi, T. Molecular Mechanism of Phase I and Phase II Drug-Metabolizing Enzymes: Implications for Detoxification. Int. Rev. Cytol. 2007, 260, 35–112. [Google Scholar] [CrossRef]
- Patel, M.; Taskar, K.S.; Zamek-Gliszczynski, M.J. Importance of Hepatic Transporters in Clinical Disposition of Drugs and Their Metabolites. J. Clin. Pharmacol. 2016, 56, S23–S39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sau, A.; Tregno, F.P.; Valentino, F.; Federici, G.; Caccuri, A.M. Glutathione transferases and development of new principles to overcome drug resistance. Arch. Biochem. Biophys. 2010, 500, 116–122. [Google Scholar] [CrossRef]
- Tew, K.D.; Gaté, L. Glutathione S-transferases as emerging therapeutic targets. Expert Opin. Ther. Targets 2001, 5, 477–489. [Google Scholar] [CrossRef]
- Chen, C.-S.; Lin, J.T.; Goss, K.A.; He, Y.-A.; Halpert, J.R.; Waxman, D.J. Activation of the Anticancer Prodrugs Cyclophosphamide and Ifosfamide: Identification of Cytochrome P450 2B Enzymes and Site-Specific Mutants with Improved Enzyme Kinetics. Mol. Pharmacol. 2004, 65, 1278–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopeček, J.; Kopečková, P. HPMA copolymers: Origins, early developments, present, and future. Adv. Drug Deliv. Rev. 2010, 62, 122–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brozovic, A.; Ambriović-Ristov, A.; Osmak, M. The relationship between cisplatin-induced reactive oxygen species, glutathione, and BCL-2 and resistance to cisplatin. Crit. Rev. Toxicol. 2010, 40, 347–359. [Google Scholar] [CrossRef]
- Allocati, N.; Masulli, M.; Di Ilio, C.; Federici, L. Glutathione transferases: Substrates, inihibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogene 2018, 7, 1–15. [Google Scholar] [CrossRef]
- Ma, L.; Xu, Y.; Su, J.; Yu, H.; Kang, J.; Li, H.; Li, X.; Xie, Q.; Yu, C.; Sun, L.; et al. Autophagic flux promotes cisplatin resistance in human ovarian carcinoma cells through ATP-mediated lysosomal function. Int. J. Oncol. 2015, 47, 1890–1900. [Google Scholar] [CrossRef] [Green Version]
- Aoyama, K.; Nakaki, T. Impaired Glutathione Synthesis in Neurodegeneration. Int. J. Mol. Sci. 2013, 14, 21021–21044. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, Y.C.; Singh, S.V.; Ahmad, H.; Moller, P.C. Immunocytochemical evidence for the expression of GST1, GST2, and GST3 gene loci for glutathione S-transferase in human lung. Lung 1987, 165, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Di Illio, C.; Del Boccio, G.; Aceto, A.; Casaccia, R.; Mucilli, F.; Federici, G. Elevation of glutathione transferase activity in human lung tumor. Carcinogenesis 1988, 9, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.W.; Kuo, M.T. Role of glutathione in the regulation of Cisplatin resistance in cancer chemotherapy. Met.-Based Drugs 2010, 2010, 430939. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Heiden, M.G.V.; Kroemer, G. Metabolic targets for cancer therapy. Nat. Rev. Drug Discov. 2013, 12, 829–846. [Google Scholar] [CrossRef] [PubMed]
- Yi, T.; Cho, S.-G.; Yi, Z.; Pang, X.; Rodriguez, M.; Wang, Y.; Sethi, G.; Aggarwal, B.B.; Liu, M. Thymoquinone inhibits tumor angiogenesis and tumor growth through suppressing AKT and extracellular signal-regulated kinase signaling pathways. Mol. Cancer Ther. 2008, 7, 1789–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2011, 31, 1869–1883. [Google Scholar] [CrossRef] [Green Version]
- Borst, P.; Evers, R.; Kool, M.; Wijnholds, J. A Family of Drug Transporters: The Multidrug Resistance-Associated Proteins. J. Natl. Cancer Inst. 2000, 92, 1295–1302. [Google Scholar] [CrossRef]
- Wangpaichitr, M.; Wu, C.; Li, Y.Y.; Nguyen, D.J.M.; Kandemir, H.; Shah, S.; Chen, S.; Feun, L.G.; Prince, J.S.; Kuo, M.T.; et al. Exploiting ROS and metabolic differences to kill cisplatin resistant lung cancer. Oncotarget 2017, 8, 49275–49292. [Google Scholar] [CrossRef] [Green Version]
- Damia, G.; D’Incalci, M. Mechanisms of resistance to alkylating agents. Mult. Drug Resist. Cancer 1998, 27, 165–173. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, Q.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, H.; Huang, C.; Lei, Y. Cancer drug resistance: Redox resetting renders a way. Oncotarget 2016, 7, 42740–42761. [Google Scholar] [CrossRef] [Green Version]
- Ramesh, R.; Shanker, M.; Willcutts, D.; Roth, J. Drug resistance in lung cancer. Lung Cancer Targets Ther. 2010, 1, 23–36. [Google Scholar] [CrossRef] [Green Version]
- Moreira, A.F.; Dias, D.R.; Correia, I.J. Stimuli-responsive mesoporous silica nanoparticles for cancer therapy: A review. Microporous Mesoporous Mater. 2016, 236, 141–157. [Google Scholar] [CrossRef]
- Wang, J.; Sun, X.; Mao, W.; Sun, W.; Tang, J.; Sui, M.; Shen, Y.; Gu, Z. Tumor Redox Heterogeneity-Responsive Prodrug Nanocapsules for Cancer Chemotherapy. Adv. Mater. 2013, 25, 3670–3676. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Hu, Q.; Zhu, W.; Zhao, M.; Ping, Y.; Tang, G. Structure-Invertible Nanoparticles for Triggered Co-Delivery of Nucleic Acids and Hydrophobic Drugs for Combination Cancer Therapy. Adv. Funct. Mater. 2015, 25, 3380–3392. [Google Scholar] [CrossRef]
- Hu, Y.-W.; Du, Y.-Z.; Liu, N.; Liu, X.; Meng, T.-T.; Cheng, B.-L.; He, J.-B.; You, J.; Yuan, H.; Hu, F.-Q. Selective redox-responsive drug release in tumor cells mediated by chitosan based glycolipid-like nanocarrier. J. Control. Release 2015, 206, 91–100. [Google Scholar] [CrossRef]
- Zhang, W.; Lin, W.; Pei, Q.; Hu, X.; Xie, Z.; Jing, X. Redox-Hypersensitive Organic Nanoparticles for Selective Treatment of Cancer Cells. Chem. Mater. 2016, 28, 4440–4446. [Google Scholar] [CrossRef]
- Stephen, Z.; Kievit, F.; Veiseh, O.; Chiarelli, P.A.; Fang, C.; Wang, K.; Hatzinger, S.J.; Ellenbogen, R.G.; Silber, J.R.; Zhang, M. Redox-Responsive Magnetic Nanoparticle for Targeted Convection-Enhanced Delivery of O6-Benzylguanine to Brain Tumors. ACS Nano 2014, 8, 10383–10395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Q.; Wang, Y.; Zhang, L.; Zhang, X.; Zhou, S. A Nanoplatform with Precise Control over Release of Cargo for Enhanced Cancer Therapy. Small 2016, 12, 1378–1390. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Yu, H.; Zhang, Z.; Meng, Q.; Sun, H.; Chen, X.; Yin, Q.; Li, Y. Hydrogen-bonded and reduction-responsive micelles loading atorvastatin for therapy of breast cancer metastasis. Biomaterials 2014, 35, 7574–7587. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.T.; Tran, T.H.; Amiji, M.; Lu, X.; Kasi, R.M. Redox-sensitive nanoparticles from amphiphilic cholesterol-based block copolymers for enhanced tumor intracellular release of doxorubicin. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 2071–2082. [Google Scholar] [CrossRef] [Green Version]
- Hu, K.; Zhou, H.; Liu, Y.; Liu, Z.; Liu, J.; Tang, J.; Li, J.; Zhang, J.; Sheng, W.; Zhao, Y.; et al. Hyaluronic acid functional amphipathic and redox-responsive polymer particles for the co-delivery of doxorubicin and cyclopamine to eradicate breast cancer cells and cancer stem cells. Nanoscale 2015, 7, 8607–8618. [Google Scholar] [CrossRef]
- Park, H.-K.; Lee, S.J.; Oh, J.-S.; Lee, S.-G.; Jeong, Y.-I.; Lee, H.C. Smart Nanoparticles Based on Hyaluronic Acid for Redox-Responsive and CD44 Receptor-Mediated Targeting of Tumor. Nanoscale Res. Lett. 2015, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.; Hu, Y.; Cai, K.; Ding, X.; Zhang, Q.; Li, M.; Ma, X.; Zhang, B.; Zeng, Y.; Li, P.; et al. Intracellular redox-activated anticancer drug delivery by functionalized hollow mesoporous silica nanoreservoirs with tumor specificity. Biomaterials 2014, 35, 7951–7962. [Google Scholar] [CrossRef]
- Zhang, B.; Luo, Z.; Liu, J.; Ding, X.; Li, J.; Cai, K. Cytochrome c end-capped mesoporous silica nanoparticles as redox-responsive drug delivery vehicles for liver tumor-targeted triplex therapy in vitro and in vivo. J. Control. Release 2014, 192, 192–201. [Google Scholar] [CrossRef]
- Wang, H.; Li, Y.; Zhang, M.; Wu, D.; Shen, Y.; Tang, G.; Ping, Y. Redox-Activatable ATP-Depleting Micelles with Dual Modulation Characteristics for Multidrug-Resistant Cancer Therapy. Adv. Health Mater. 2017, 6, 1601293. [Google Scholar] [CrossRef]
- Wang, X.; Cai, X.; Hu, J.; Shao, N.; Wang, F.; Zhang, Q.; Xiao, J.; Cheng, Y. Glutathione-Triggered “Off–On” Release of Anticancer Drugs from Dendrimer-Encapsulated Gold Nanoparticles. J. Am. Chem. Soc. 2013, 135, 9805–9810. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Julien, O.; Wells, J.A. Caspases and their substrates. Cell Death Differ. 2017, 24, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, R.M.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.-Y.; Lin, L.-T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 2015, 35, S78–S103. [Google Scholar] [CrossRef]
- Balaji, S.; Terrero, D.; Tiwari, A.K.; Ashby, C.R.; Raman, D. Alternative approaches to overcome chemoresistance to apoptosis in cancer. Adv. Protein Chem. Struct. Biol. 2021, 126, 91–122. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lee, E.; Thompson, G.V.; Wardak, A.Z.; Fairlie, W.; Colman, P.M. Mutation to Bax beyond the BH3 Domain Disrupts Interactions with Pro-survival Proteins and Promotes Apoptosis. J. Biol. Chem. 2011, 286, 7123–7131. [Google Scholar] [CrossRef] [Green Version]
- Le Gallo, M.; Poissonnier, A.; Blanco, P.; Legembre, P. CD95/Fas, Non-Apoptotic Signaling Pathways, and Kinases. Front. Immunol. 2017, 8, 1216. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.-J.; Whang, Y. PTEN sensitizes prostate cancer cells to death receptor-mediated and drug-induced apoptosis through a FADD-dependent pathway. Oncogene 2002, 21, 319–327. [Google Scholar] [CrossRef]
- Fulda, S.; Vucic, D. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 2012, 11, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Rathore, R.; McCallum, J.E.; Varghese, E.; Florea, A.-M.; Büsselberg, D. Overcoming chemotherapy drug resistance by targeting inhibitors of apoptosis proteins (IAPs). Apoptosis 2017, 22, 898–919. [Google Scholar] [CrossRef]
- Yang, D.; Chen, M.-B.; Wang, L.-Q.; Yang, L.; Liu, C.-Y.; Lu, P.-H. Bcl-2 expression predicts sensitivity to chemotherapy in breast cancer: A systematic review and meta-analysis. J. Exp. Clin. Cancer Res. 2013, 32, 105. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, L.; Wang, R.; Chen, S.; Wang, S.; Chen, H.; Pan, B.; Sun, Y. Prognostic value of Bcl-2 expression in patients with non-small-cell lung cancer: A meta-analysis and systemic review. OncoTargets Ther. 2015, 8, 3361–3369. [Google Scholar] [CrossRef] [Green Version]
- Sjöström, J.; Von Boguslawski, K.; Saksela, E.; Blomqvist, C.; Bengtsson, N.O.; Malmström, P.; Mjaaland, I.; Ostenstadt, B.; Wist, E.; Valvere, V.; et al. The predictive value of bcl-2, bax, bcl-xL, bag-1, fas, and fasL for chemotherapy response in advanced breast cancer. Clin. Cancer Res. 2002, 8, 811–816. [Google Scholar]
- Krug, L. Bcl-2 and bax expression in advanced non-small cell lung cancer: Lack of correlation with chemotherapy response or survival in patients treated with docetaxel plus vinorelbine. Lung Cancer 2003, 39, 139–143. [Google Scholar] [CrossRef]
- Shim, M.K.; Moon, Y.; Yang, S.; Kim, J.; Cho, H.; Lim, S.; Yoon, H.Y.; Seong, J.-K.; Kim, K. Cancer-specific drug-drug nanoparticles of pro-apoptotic and cathepsin B-cleavable peptide-conjugated doxorubicin for drug-resistant cancer therapy. Biomaterials 2020, 261, 120347. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, F.; Wen, H.; Shi, W.; Huang, Q.; Huang, Y.; Xie, J.; Li, P.; Chen, J.; Qin, L.; et al. Tumor- and mitochondria-targeted nanoparticles eradicate drug resistant lung cancer through mitochondrial pathway of apoptosis. J. Nanobiotechnol. 2020, 18, 8. [Google Scholar] [CrossRef]
- Marrache, S.; Pathak, R.K.; Dhar, S. Detouring of cisplatin to access mitochondrial genome for overcoming resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 10444–10449. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Zhou, Y.; Liu, L.; Xu, Y.; Chen, Q.; Wang, Y.; Wu, S.; Deng, Y.; Zhang, J.; Shao, A. Nanoparticle-Based Drug Delivery in Cancer Therapy and Its Role in Overcoming Drug Resistance. Front. Mol. Biosci. 2020, 7, 193. [Google Scholar] [CrossRef]
- Choi, K.Y.; Correa, S.; Min, J.; Li, J.; Roy, S.; Laccetti, K.H.; Dreaden, E.; Kong, S.; Heo, R.; Roh, Y.H.; et al. Binary Targeting of siRNA to Hematologic Cancer Cells In Vivo Using Layer-by-Layer Nanoparticles. Adv. Funct. Mater. 2019, 29, 1900018. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H. Novel cationic solid lipid nanoparticles enhanced p53 gene transfer to lung cancer cells. Eur. J. Pharm. Biopharm. 2008, 68, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Turajlic, S.; Furney, S.; Stamp, G.; Rana, S.; Ricken, G.; Oduko, Y.; Saturno, G.; Springer, C.; Hayes, A.; Gore, M.; et al. Whole-genome sequencing reveals complex mechanisms of intrinsic resistance to BRAF inhibition. Ann. Oncol. 2014, 25, 959–967. [Google Scholar] [CrossRef]
- Dearden, S.; Stevens, J.; Wu, Y.-L.; Blowers, D. Mutation incidence and coincidence in non small-cell lung cancer: Meta-analyses by ethnicity and histology (mutMap). Ann. Oncol. 2013, 24, 2371–2376. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, T.M.; Kim, D.-W.; Go, H.; Keam, B.; Lee, S.-H.; Ku, J.-L.; Chung, D.H.; Heo, D.S. Heterogeneity of Genetic Changes Associated with Acquired Crizotinib Resistance in ALK-Rearranged Lung Cancer. J. Thorac. Oncol. 2013, 8, 415–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitiushkina, N.V.; Iyevleva, A.G.; Poltoratskiy, A.N.; Ivantsov, A.O.; Togo, A.V.; Polyakov, I.S.; Orlov, S.V.; Matsko, D.E.; Novik, V.I.; Imyanitov, E.N. Detection ofEGFRmutations andEML4-ALKrearrangements in lung adenocarcinomas using archived cytological slides. Cancer Cytopathol. 2013, 121, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leischler, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Eur. J. Cancer 2016, 69, S138. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Shaurova, T.; Shoemaker, S.; Petkovich, M.; Hershberger, P.A.; Wu, Y. Tumor-Targeted Nanoparticles Deliver a Vitamin D-Based Drug Payload for the Treatment of EGFR Tyrosine Kinase Inhibitor-Resistant Lung Cancer. Mol. Pharm. 2018, 15, 3216–3226. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, S.; Gopinath, S.C.B.; Arshad, M.K.M.; Poopalan, P.; Anbu, P. A DNA based visual and colorimetric aggregation assay for the early growth factor receptor (EGFR) mutation by using unmodified gold nanoparticles. Mikrochim. Acta 2019, 186, 546. [Google Scholar] [CrossRef]
- Milella, M.; Falcone, I.; Conciatori, F.; Cesta Incani, U.; Del Curatolo, A.; Inzerilli, N.; Nuzzo, C.M.; Vaccaro, V.; Vari, S.; Cognetti, F.; et al. PTEN: Multiple Functions in Human Malignant Tumors. Front. Oncol. 2015, 5, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantley, L.C.; Neel, B.G. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 4240–4245. [Google Scholar] [CrossRef] [Green Version]
- Sos, M.L.; Koker, M.; Weir, B.A.; Heynck, S.; Rabinovsky, R.; Zander, T.; Seeger, J.M.; Weiss, J.; Fischer, F.; Frommolt, P.; et al. PTEN Loss Contributes to Erlotinib Resistance in EGFR-Mutant Lung Cancer by Activation of Akt and EGFR. Cancer Res. 2009, 69, 3256–3261. [Google Scholar] [CrossRef] [Green Version]
- Niu, G.; Chen, X. Vascular endothelial growth factor as an anti-angiogenic target for cancer therapy. Curr. Drug Targets 2010, 11, 1000–1017. [Google Scholar] [CrossRef] [PubMed]
- Loges, S.; Schmidt, T.; Carmeliet, P. Mechanisms of Resistance to Anti-Angiogenic Therapy and Development of Third-Generation Anti-Angiogenic Drug Candidates. Genes Cancer 2010, 1, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Tokunaga, T.; Hatanaka, H.; Kijima, H.; Yamazaki, H.; Abe, Y.; Osamura, Y.; Inoue, H.; Ueyama, Y.; Nakamura, M. Neuropilin 1 and neuropilin 2 co-expression is significantly correlated with increased vascularity and poor prognosis in nonsmall cell lung carcinoma. Cancer 2002, 95, 2196–2201. [Google Scholar] [CrossRef]
- Gridelli, C.; de Marinis, F.; Ardizzoni, A.; Novello, S.; Fontanini, G.; Cappuzzo, F.; Grossi, F.; Santo, A.; Cortinovis, D.; Favaretto, A.; et al. Advanced non-small cell lung cancer management in patients progressing after first-line treatment: Results of the cross-sectional phase of the Italian LIFE observational study. J. Cancer Res. Clin. Oncol. 2014, 140, 1783–1793. [Google Scholar] [CrossRef] [PubMed]
- Gridelli, C.; De Marinis, F.; Di Maio, M.; Cortinovis, D.; Cappuzzo, F.; Mok, T. Gefitinib as first-line treatment for patients with advanced non-small-cell lung cancer with activating Epidermal Growth Factor Receptor mutation: Implications for clinical practice and open issues. Lung Cancer 2011, 72, 3–8. [Google Scholar] [CrossRef]
- Bell, D.W.; Gore, I.; Okimoto, R.A.; Godin-Heymann, N.; Sordella, R.; Mulloy, R.; Sharma, S.V.; Brannigan, B.W.; Mohapatra, G.; Settleman, J.; et al. Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat. Genet. 2005, 37, 1315–1316. [Google Scholar] [CrossRef]
- Tang, J.; Salama, R.; Gadgeel, S.M.; Sarkar, F.H.; Ahmad, A. Erlotinib Resistance in Lung Cancer: Current Progress and Future Perspectives. Front. Pharmacol. 2013, 4, 15. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Wei, S.; Song, Y. T790M and acquired resistance of EGFR TKI: A literature review of clinical reports. J. Thorac. Dis. 2011, 3, 10–18. [Google Scholar] [CrossRef]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR Mutations in Lung Cancer: Correlation with Clinical Response to Gefitinib Therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [Green Version]
- Suda, K.; Onozato, R.; Yatabe, Y.; Mitsudomi, T. EGFR T790M Mutation: A Double Role in Lung Cancer Cell Survival? J. Thorac. Oncol. 2009, 4, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, H.; Park, E.; Yun, C.H.; Sng, N.J.; Lucena-Araujo, A.R.; Yeo, W.L.; Huberman, M.S.; Cohen, D.W.; Nakayama, S.; Ishioka, K.; et al. Structural, biochemical, and clinical characterization of Epidermal Growth Factor Receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci. Transl. Med. 2014, 6, 216ra177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, H.; Chen, B.; Huang, W.; Tang, Y.; Jiang, Y.; Zhang, W.; Huang, Y. Reprogramming Tumor-Associated Macrophages to Reverse EGFRT790M Resistance by Dual-Targeting Codelivery of Gefitinib/Vorinostat. Nano Lett. 2017, 17, 7684–7690. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Yu, X.; Kang, X.; Zhao, Y.; Zhao, P.; Jin, H.; Fu, X.; Wan, Y.; Peng, C.; Huang, Y. Remodeling Tumor-Associated Macrophages and Neovascularization Overcomes EGFRT790M-Associated Drug Resistance by PD-L1 Nanobody-Mediated Codelivery. Small 2018, 14, 1802372. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Ulldemolins, A.; Seras-Franzoso, J.; Andrade, F.; Rafael, D.; Abasolo, I.; Gener, P.; Jr, S.S. Perspectives of nano-carrier drug delivery systems to overcome cancer drug resistance in the clinics. Cancer Drug Resist 2021, 4, 44–68. [Google Scholar] [CrossRef]
- Desai, A.; Yan, Y.; Gerson, S.L. Advances in therapeutic targeting of the DNA damage response in cancer. DNA Repair 2018, 66-67, 24–29. [Google Scholar] [CrossRef]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73, e478s. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.K.; Wang, Z.; Fong, C.-C.; Liu, D.; Yip, T.-C.; Au, S.-K.; Zhu, G.; Yang, M. Chemoresistant lung cancer stem cells display high DNA repair capability to remove cisplatin-induced DNA damage. Br. J. Pharmacol. 2017, 174, 302–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olaussen, K.A.; Dunant, A.; Fouret, P.; Brambilla, E.; Andre, F.; Haddad, V.; Taranchon, E.; Filipits, M.; Pirker, R.; Popper, H.H.; et al. DNA Repair by ERCC1 in Non–Small-Cell Lung Cancer and Cisplatin-Based Adjuvant Chemotherapy. N. Engl. J. Med. 2006, 355, 983–991. [Google Scholar] [CrossRef]
- Tell, G.; Damante, G.; Caldwell, D.; Kelley, M.R. The Intracellular Localization of APE1/Ref-1: More than a Passive Phenomenon? Antioxid. Redox Signal. 2005, 7, 367–384. [Google Scholar] [CrossRef]
- Huang, R.; Zhou, P.-K. DNA damage repair: Historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 254. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Li, J.; Jiang, H.-G.; Lan, T.; Chen, Y.-C. Curcumin reverses cisplatin resistance in cisplatin-resistant lung caner cells by inhibiting FA/BRCA pathway. Tumor Biol. 2014, 36, 3591–3599. [Google Scholar] [CrossRef]
- Hong, Y.; Che, S.; Hui, B.; Wang, X.; Zhang, X.; Ma, H. Combination Therapy of Lung Cancer Using Layer-by-Layer Cisplatin Prodrug and Curcumin Co-Encapsulated Nanomedicine. Drug Des. Dev. Ther. 2020, 14, 2263–2274. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.W.; Dreaden, E.C.; Morandell, S.; Zhou, W.; Dhara, S.S.; Sriram, G.; Lam, F.C.; Patterson, J.C.; Quadir, M.; Dinh, A.; et al. Enhancing chemotherapy response through augmented synthetic lethality by co-targeting nucleotide excision repair and cell-cycle checkpoints. Nat. Commun. 2020, 11, 4124. [Google Scholar] [CrossRef]
- Chen, Y.-Y.; Lin, Y.-J.; Huang, W.-T.; Hung, C.-C.; Lin, H.-Y.; Tu, Y.-C.; Liu, D.-M.; Lan, S.-J.; Sheu, M.-J. Demethoxycurcumin-Loaded Chitosan Nanoparticle Downregulates DNA Repair Pathway to Improve Cisplatin-Induced Apoptosis in Non-Small Cell Lung Cancer. Molecules 2018, 23, 3217. [Google Scholar] [CrossRef] [Green Version]
- Godin-Heymann, N.; Ulkus, L.; Brannigan, B.W.; McDermott, U.; Lamb, J.; Maheswaran, S.; Settleman, J.; Haber, D.A. The T790M “gatekeeper” mutation in EGFR mediates resistance to low concentrations of an irreversible EGFR inhibitor. Mol. Cancer Ther. 2008, 7, 874–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.-M.; Zhao, X.; Christensen, J.; et al. MET Amplification Leads to Gefitinib Resistance in Lung Cancer by Activating ERBB3 Signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.-L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and Clonal Selection of MET Amplification in EGFR Mutant NSCLC. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Sattler, M.; Hasina, R.; Reddy, M.M.; Gangadhar, T.; Salgia, R. The role of the c-Met pathway in lung cancer and the potential for targeted therapy. Ther. Adv. Med Oncol. 2011, 3, 171–184. [Google Scholar] [CrossRef] [Green Version]
- Shi, P.; Oh, Y.-T.; Zhang, G.; Yao, W.; Yue, P.; Li, Y.; Kanteti, R.; Riehm, J.; Salgia, R.; Owonikoko, T.K.; et al. Met gene amplification and protein hyperactivation is a mechanism of resistance to both first and third generation EGFR inhibitors in lung cancer treatment. Cancer Lett. 2016, 380, 494–504. [Google Scholar] [CrossRef]
- Le, X.; Puri, S.; Negrao, M.V.; Nilsson, M.B.; Robichaux, J.; Boyle, T.; Hicks, J.K.; Lovinger, K.L.; Roarty, E.; Rinsurongkawong, W.; et al. Landscape of EGFR-Dependent and -Independent Resistance Mechanisms to Osimertinib and Continuation Therapy Beyond Progression in EGFR-Mutant NSCLC. Clin. Cancer Res. 2018, 24, 6195–6203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yabuki, N.; Sakata, K.; Yamasaki, T.; Terashima, H.; Mio, T.; Miyazaki, Y.; Fujii, T.; Kitada, K. Gene amplification and expression in lung cancer cells with acquired paclitaxel resistance. Cancer Genet. Cytogenet. 2007, 173, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-J.; Chin, J.E.; Ueda, K.; Clark, D.P.; Pastan, I.; Gottesman, M.M.; Roninson, I.B. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell 1986, 47, 381–389. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP–dependent transporters. Nat. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Saad, M.; Garbuzenko, O.B.; Minko, T. Co-delivery of siRNA and an anticancer drug for treatment of multidrug-resistant cancer. Nanomedicine 2008, 3, 761–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Reinberg, D. Epigenetic inheritance: Uncontested? Cell Res. 2011, 21, 435–441. [Google Scholar] [CrossRef] [Green Version]
- Roberti, A.; Valdes, A.F.; Torrecillas, R.; Fraga, M.F.; Fernandez, A.F. Epigenetics in cancer therapy and nanomedicine. Clin. Epigenet. 2019, 11, 81. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [Green Version]
- Wilting, R.H.; Dannenberg, J.-H. Epigenetic mechanisms in tumorigenesis, tumor cell heterogeneity and drug resistance. Drug Resist. Updat. 2012, 15, 21–38. [Google Scholar] [CrossRef] [Green Version]
- Glozak, M.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamdani, H.; Jalal, S.I. Histone Deacetylase Inhibition in Non-small Cell Lung Cancer: Hype or Hope? Front. Cell Dev. Biol. 2020, 8, 582370. [Google Scholar] [CrossRef]
- Wang, L.; Li, H.; Ren, Y.; Zou, S.; Fang, W.; Jiang, X.; Jia, L.; Li, M.; Liu, X.; Yuan, X.; et al. Targeting HDAC with a novel inhibitor effectively reverses paclitaxel resistance in non-small cell lung cancer via multiple mechanisms. Cell Death Dis. 2016, 7, e2063. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.C.; Min, Y.; Palm, R.C.; Fiordalisi, J.J.; Wagner, K.T.; Hyder, N.; Cox, A.D.; Caster, J.; Tian, X.; Wang, A.Z. Nanoparticle formulations of histone deacetylase inhibitors for effective chemoradiotherapy in solid tumors. Biomaterials 2015, 51, 208–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, T.H.; Ramasamy, T.; Truong, D.H.; Shin, B.S.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Development of Vorinostat-Loaded Solid Lipid Nanoparticles to Enhance Pharmacokinetics and Efficacy against Multidrug-Resistant Cancer Cells. Pharm. Res. 2014, 31, 1978–1988. [Google Scholar] [CrossRef]
- Tu, B.; Zhang, M.; Liu, T.; Huang, Y. Nanotechnology-Based Histone Deacetylase Inhibitors for Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 8. [Google Scholar] [CrossRef]
- Jiang, T.; Sun, W.; Zhu, Q.; Burns, N.A.; Khan, S.A.; Mo, R.; Gu, Z. Furin-Mediated Sequential Delivery of Anticancer Cytokine and Small-Molecule Drug Shuttled by Graphene. Adv. Mater. 2015, 27, 1021–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, A.T.N.; Yoon, J.; Ganbold, E.-O.; Singh, D.; Kim, D.; Cho, K.-H.; Lee, S.Y.; Choo, J.; Lee, K.; Joo, S.-W. Colloidal gold nanoparticle conjugates of gefitinib. Colloids Surf. B Biointerfaces 2014, 123, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.T.N.; Yoon, J.; Ganbold, E.-O.; Singh, D.K.; Kim, D.; Cho, K.-H.; Son, S.J.; Choo, J.; Lee, S.Y.; Kim, S.; et al. Adsorption and desorption of tyrosine kinase inhibitor erlotinib on gold nanoparticles. J. Colloid Interface Sci. 2014, 425, 96–101. [Google Scholar] [CrossRef]
- Morton, S.W.; Lee, M.J.; Deng, Z.J.; Dreaden, E.C.; Siouve, E.; Shopsowitz, K.E.; Shah, N.J.; Yaffe, M.B.; Hammond, P.T. A Nanoparticle-Based Combination Chemotherapy Delivery System for Enhanced Tumor Killing by Dynamic Rewiring of Signaling Pathways. Sci. Signal. 2014, 7, ra44. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Zhang, C.-X.; Wang, X.-X.; Zhang, L.; Ma, X.; Zhou, J.; Ju, R.-J.; Li, X.-Y.; Zhao, W.-Y.; Lu, W.-L. Development of targeting lonidamine liposomes that circumvent drug-resistant cancer by acting on mitochondrial signaling pathways. Biomaterials 2013, 34, 3366–3380. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Fan, L.; Pang, Z.; Ren, J.; Ren, Y.; Li, J.; Chen, J.; Wen, Z.; Jiang, X. TRAIL and doxorubicin combination enhances anti-glioblastoma effect based on passive tumor targeting of liposomes. J. Control. Release 2011, 154, 93–102. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, H.; Gu, K.; Chen, J.; Rui, M.; Jiang, G.-L. In vitro and in vivo study of a nanoliposomal cisplatin as a radiosensitizer. Int. J. Nanomed. 2011, 6, 437–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Peng, L.; Mumper, R.J.; Huang, L. Combinational delivery of c-myc siRNA and nucleoside analogs in a single, synthetic nanocarrier for targeted cancer therapy. Biomaterials 2013, 34, 8459–8468. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Schwerbrock, N.M.; Rogers, A.B.; Kim, W.Y.; Huang, L. Codelivery of VEGF siRNA and Gemcitabine Monophosphate in a Single Nanoparticle Formulation for Effective Treatment of NSCLC. Mol. Ther. 2013, 21, 1559–1569. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Luo, M.; Wang, B.-L.; Li, Y.-L.; Zhang, Q.; Qian, Z.-Y.; Shi, H.-S.; Fan, M.; Liu, Z. Codelivery of curcumin and doxorubicin by MPEG-PCL results in improved efficacy of systemically administered chemotherapy in mice with lung cancer. Int. J. Nanomed. 2013, 8, 3521–3531. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Yin, Q.; Chen, L.; Zhang, Z.; Li, Y. Co-delivery of paclitaxel and survivin shRNA by pluronic P85-PEI/TPGS complex nanoparticles to overcome drug resistance in lung cancer. Biomaterials 2012, 33, 8613–8624. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Shi, L.; Ren, L.; Zhou, L.; Li, T.; Qiao, Y.; Wang, H. A nanomedicine approach enables co-delivery of cyclosporin A and gefitinib to potentiate the therapeutic efficacy in drug-resistant lung cancer. Signal Transduct. Target. Ther. 2018, 3, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Tang, J.; Wang, Y.; Ramishetti, S.; Fu, Q.; Racette, K.; Liu, F. Multifunctional Nanoparticles Based on a Single-Molecule Modification for the Treatment of Drug-Resistant Cancer. Mol. Pharm. 2013, 10, 1465–1469. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.-Y.; Wang, L.; Sun, X.; Tang, M.; Quan, H.-T.; Zhang, L.-S.; Lou, L.-G.; Gou, S.-H. SHR-A1403, a novel c-Met antibody-drug conjugate, exerts encouraging anti-tumor activity in c-Met-overexpressing models. Acta Pharmacol. Sin. 2019, 40, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Manikhas, G.M.; Orlov, S.; Afanasyev, B.; Makhson, A.M.; Bhar, P.; Hawkins, M.J. Abraxane®, a novel Cremophor®-free, albumin-bound particle form of paclitaxel for the treatment of advanced non-small-cell lung cancer. Ann. Oncol. 2006, 17, 1263–1268. [Google Scholar] [CrossRef]
- Arms, L.; Smith, D.W.; Flynn, J.; Palmer, W.; Martin, A.; Woldu, A.; Hua, S. Advantages and Limitations of Current Techniques for Analyzing the Biodistribution of Nanoparticles. Front. Pharmacol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic. Bioeng. Transl. Med. 2016, 1, 10–29. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Lallana, E.; Sousa-Herves, A.; Fernandez-Trillo, P.; Riguera, R.; Fernandez-Megia, E. Click Chemistry for Drug Delivery Nanosystems. Pharm. Res. 2011, 29, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Shoba, G.; Joy, D.; Joseph, T.; Majeed, M.; Rajendran, R.; Srinivas, P.S. Influence of Piperine on the Pharmacokinetics of Curcumin in Animals and Human Volunteers. Planta Med. 1998, 64, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.; Al Zaki, A.; Hui, J.Z.; Muzykantov, V.R.; Tsourkas, A. Multifunctional Nanoparticles: Cost Versus Benefit of Adding Targeting and Imaging Capabilities. Science 2012, 338, 903–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleck, L.M. The Costs of Caring: Who Pays? Who Profits? Who Panders? Hast. Cent. Rep. 2006, 36, 13–17. [Google Scholar] [CrossRef]
- Desai, N. Challenges in Development of Nanoparticle-Based Therapeutics. AAPS J. 2012, 14, 282–295. [Google Scholar] [CrossRef] [Green Version]
- Alexiou, C.; Schmid, R.J.; Jurgons, R.; Kremer, M.; Wanner, G.; Bergemann, C.; Huenges, E.; Nawroth, T.; Arnold, W.; Parak, F.G. Targeting cancer cells: Magnetic nanoparticles as drug carriers. Eur. Biophys. J. 2006, 35, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, D.K.; Fong, L.S.; Zhang, Y. Nanoparticles in photodynamic therapy: An emerging paradigm. Adv. Drug Deliv. Rev. 2008, 60, 1627–1637. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Li, H.; Han, D.; Yang, K.; Kang, L. Inhibition of autophagy enhances apoptosis induced by Ce6-photodynamic therapy in human colon cancer cells. Photodiagn. Photodyn. Ther. 2021, 36, 102605. [Google Scholar] [CrossRef]
- Deng, X.; Song, Q.; Zhang, Y.; Liu, W.; Hu, H.; Zhang, Y. Tumour microenvironment-responsive nanoplatform based on biodegradable liposome-coated hollow MnO2 for synergistically enhanced chemotherapy and photodynamic therapy. J. Drug Target. 2021, 14, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Crous, A.; Abrahamse, H. Aluminium (III) phthalocyanine chloride tetrasulphonate is an effective photosensitizer for the eradication of lung cancer stem cells. R. Soc. Open Sci. 2021, 8, 210148. [Google Scholar] [CrossRef]
- Yu, T.-T.; Sang, X.-Y.; Han, N.; Peng, X.-C.; Li, Q.-R.; Xu, X.; Xiao, R.-C.; Xu, H.-Z.; Chen, X.; Wang, M.-F.; et al. Macrophages mediated delivery of chlorin e6 and treatment of lung cancer by photodynamic reprogramming. Int. Immunopharmacol. 2021, 100, 108164. [Google Scholar] [CrossRef]
- Shi, C.; Huang, H.; Zhou, X.; Zhang, Z.; Ma, H.; Yao, Q.; Shao, K.; Sun, W.; Du, J.; Fan, J.; et al. Reversing Multidrug Resistance by Inducing Mitochondrial Dysfunction for Enhanced Chemo-Photodynamic Therapy in Tumor. ACS Appl. Mater. Interfaces 2021, 13, 45259–45268. [Google Scholar] [CrossRef]
- Reshma, P.; Unnikrishnan, B.; Preethi, G.; Syama, H.; Archana, M.; Remya, K.; Shiji, R.; Sreekutty, J.; Sreelekha, T. Overcoming drug-resistance in lung cancer cells by paclitaxel loaded galactoxyloglucan nanoparticles. Int. J. Biol. Macromol. 2019, 136, 266–274. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Nanomedicine | Drug Load | Targets | Cells/Cancer | References |
|---|---|---|---|---|
| PCL-SS-PMAA micelles | CIS/PTX | pH/redox responsive | In vitro NCI-H358 LC cells | [38] |
| Magnetic NPs | Manganese dioxide | Redox responsive/radiosensitive | In vitro hypoxic-induced gefitinib-resistant PC9 human LC cells (PC9GR) | [39] |
| cRGDyK-SPCS micelles | PTX | pH responsive/protein targeting | In vitro Luc-A549 LC cells and in vivo Luc-A549 cells implanted subcutaneously into the right upper flanks of a female nude mouse | [40] |
| Mesoporous silica nanoparticle decorated with PD-L1 antibody (ARAC) | Volasertib | Immune cells modulation | In vivo LLC-JSP murine LC cells (200K) inoculated in right flank of C57BL/6 female mice | [41] |
| PLGA NPs | DOX/Cyclosporin | P-gp | In vitro PTX-resistant LC A549 cell line (A549-Taxol) and in vivo A549-Taxol cells implanted subcutaneously into female BALB/c mice | [60] |
| TPGS1000-TPP | Paclitaxel | P-gp and mitochondrial targeting | In vitro CIS-resistant human LC cells, A549/cDDP cells and in vivo A549/cDDP xenografts subcutaneously injected into female BALB/c nude | [62] |
| Graphene | TRAIL + DOX | FADD | Human LC | [198] |
| Gold | Gefitinib | EGFR | LC (in vitro) | [199] |
| Gold | Erlotinib | EGFR | Human adenocarcinoma and NSCLC (in vitro) | [200] |
| Liposomal | Erlotinib DOX | EGFR | Human breast and LC | [201] |
| Liposomal | siRNA (MRP1/BCL2) DOX | MRP1/BCL2 | Human LC | [187] |
| Liposomal | Lonidamine + epirubicin (in a separate liposomal formulation) | Mitochondrial hexokinase | Human LC | [202] |
| Liposomal | TRAIL + DOX (in separate NPs) | FADD | Human LC | [203] |
| Nanoliposomes in combination with radiation therapy | CIS (CDDP), radiation therapy | CIS alkylating and crosslinking DNA, sensation to radiation lesions | Human Lewis lung carcinoma A549 cells subcutaneously inoculated into C57BL/6N mice, n ivivo model | [204] |
| LCP NPs | siRNA (c-Myc) Gemcitabine monophosphate | c-Myc | Human LC | [205] |
| LCP NPs | SiRNA (VEGF) Gemcitabine monophosphate | VEGF | Human LC | [206] |
| (MPEG-PCL) micelles | CUR + DOX | ABC pumps/NF-κB | Murine LC | [207] |
| Polymeric micelles | Paclitaxel and survivin shRNA, which down-regulate survivin gene expression by RNA interference | Co-delivery of drug and gene-enhanced antitumor effect | LC | [208] |
| PLGA NPs | Cyclosporin A + DOX | P-gp | Human LC | [60] |
| PEG-PLA NPs | Gefitinib, cyclosporin A | EGFR | LC | [209] |
| PEG 1000 succinate-containing micellar NPs | PTX, fluorouracil (5-FU) | Inhibition of P-gp, inhibition of cell division by PTX, irreversible inhibition of thymidylate synthase, synergism of PTX/5-FU | H460/TaxR human NSCLC overexpressing P-gp in vitro mode | [210] |
| SHR-A1403 Polymeric NPs | Anti-c-Met monoclonal antibody (c-Met mAb) conjugated to a micro-tubule inhibitor | c-Met | Non-small cell LC cells | [211] |
| PCL-SS-PMAA: Poly(ε-caprolactone)-SS-poly(methacrylic acid), cRGDyK-SPCS: micelles N-succinyl-palmitoyl-chitosan decorated with cRGDyK peptide, TPGS1000-TPP: liposomes decorated with d-α-tocopheryl polyethylene glycol 1000 succinate-triphenylphosphine, PI3K: phosphoinositide 3-kinase, EGFR: epidermal growth factor receptor, VEGFR: vascular endothelial growth factor receptor, BCL2: B-cell lymphoma 2, LCP: lipid/calcium/phosphate, MPEG-PCL: methoxy poly(ethylene glycol)-poly(caprolactone), PEG-PLA: polyethylene glycol-block-poly(D, L-lactic acid), MDR1: multidrug resistance 1, MRP1: multidrug resistance-associated protein, FADD: Fas-associated protein with death domain, TRAIL: tumor necrosis factor-related apoptosis-inducing ligand, BCL2: B-cell lymphoma 2; siRNA: small interferin RNA | ||||
| Type of NPs | Cargo/Therapy | Status | Patient/Ccondition | Stage | ClinicalTrials.gov Identifier: |
|---|---|---|---|---|---|
| Liposomes | Drug: LY01610 (Irinotecan hydrochloride liposome injection) | Recruiting | SCLC | Phase 2 | NCT04381910 |
| Liposomes | Device: Liposomal DOX combined with ifosfamide | Unknown | SCLC | Phase 2 | NCT01872416 |
| Liposomes | Drug: PLM60 | Recruiting | SCLC | Phase 2 | NCT04352413 |
| Liposomes | Drug: MRX34 | Terminated | SCLC | Phase 1 | NCT01829971 |
| Polymeric-PEG | Drug: ADI-PEG 20 (Arginine deiminase pegylated) | Terminated | SCLC | Phase 2 | NCT01266018 |
| Polymeric-PEG | Drug: LCL161 Drug: Topotecan Drug: Pegylated GCSF (PEG-GCSF) | Terminated | LC | Phase 1, Phase 2 | NCT02649673 |
| Polymeric-PEG | Drug: Pegylated irinotecan | Completed | SCLC | Phase 2 | NCT01876446 |
| Polymeric-PEG | Drug: Pegylated irinotecan | Recurrent Small Cell | LC | Phase 2 | NCT01876446 |
| Polymeric-PEG | Drug: PEG-rhG-CSF | Unknown | SCLC | Not Applicable | NCT03776604 |
| Polymeric-PEG | Drug: ADI-PEG 20 | Completed | Solid tumors NSCLC | Phase 1 | NCT01497925 |
| Polymeric-PEG | Drug: Pegylated recombinant human endostatin (PEG-ENDO) | Recruiting | Solid tumors NSCLC | Phase 1 | NCT04413227 |
| Polymeric-PEG | Drug: PEG-rhG-CSF | Completed | Malignant Solid Tumor LC | Phase 4 | NCT02805166 |
| Polymeric-PEG | Drug: YPEG-rhG-CSF, 20 μg/kg, single s.c. at 48 h after chemotherapy for each experimental cycle Drug: YPEG-rhG-CSF, 30 μg/kg, single s.c. at 48 h after chemotherapy for each experimental cycle Drug: YPEG-rhG-CSF, 45 μg/kg, single s.c. at 48 h after chemotherapy for each experimental cycle Drug: PEG-rhG-CSF, 100 μg/kg, single s.c. at 48 h after chemotherapy for each experimental cycle | Completed | Phase 2 | NCT02005458 | |
| Polymeric-PEG | Drug: ADI-PEG 20 | Terminated | Non-squamous NSCLC | Phase 1 | NCT02029690 |
| NPs | Drug: EP0057 Drug: Olaparib | Recruiting | Lung neoplasms | Phase 1 Phase 2 | NCT02769962 |
| NPs | Drug: BIND-014 | Completed | NSCLC | Phase 2 | NCT01792479 |
| NPs | Drug: BIND-014 (Docetaxel NPs for injectable suspension) | Completed | KRAS-positive patients with NSCLC Squamous cell NSCLC | Phase 2 | NCT02283320 |
| NPs | Drug: AGuIX Radiation: Radiotherapy | Recruiting | NSCLC | Phase 1, Phase 2 | NCT04789486 |
| Micelles | Drug: PTX (Genexol) Drug: PTX-loaded polymeric micelle (Genexol-PM) | Completed | NSCLC | Phase 2 | NCT01023347 |
| Micelles | Drug: PTX micelles for injection Drug: PTX injection Drug: CIS | Active, not recruiting | NSCLC | Phase 3 | NCT02667743 |
| Albumin | Drug: Nanoparticle albumin-bound PTX/carboplatin | Unknown | NSCLC | Phase 2 | NCT01872403 |
| Albumin | Drug: Carboplatin Drug: Erlotinib hydrochloride Drug: PTX albumin-stabilized nanoparticle formulation Radiation: Radiation therapy | Completed | LC | Phase 2 | NCT00553462 |
| Albumin | Drug: HLX10 Drug: Carboplatin and nab paclitaxel Drug: Placebo | Recruiting | NSCLC | Phase 3 | NCT04033354 |
| Albumin | Drug: Nanoparticle albumin-bound PTX | Unknown | NSCLC | Phase 2 | NCT02016209 |
| Albumin | Drug: Albumin paclitaxel Drug: Simvastatin | Recruiting | SCLC | Phase 2 | NCT04698941 |
| Albumin | Drug: PTX/Albumin-bound PTX Drug: IBI308 | Recruiting | SCLC | Phase 2 | NCT04056949 |
| Radioactive 18F-Fluoropaclitaxel (FPAC) | Drug: FPAC | Terminated | LC | Phase 1 | NCT01086696 |
| NPs | Drug: TargomiRs | Completed | NSCLC | Phase 1 | NCT02369198 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haider, M.; Elsherbeny, A.; Pittalà, V.; Consoli, V.; Alghamdi, M.A.; Hussain, Z.; Khoder, G.; Greish, K. Nanomedicine Strategies for Management of Drug Resistance in Lung Cancer. Int. J. Mol. Sci. 2022, 23, 1853. https://doi.org/10.3390/ijms23031853
Haider M, Elsherbeny A, Pittalà V, Consoli V, Alghamdi MA, Hussain Z, Khoder G, Greish K. Nanomedicine Strategies for Management of Drug Resistance in Lung Cancer. International Journal of Molecular Sciences. 2022; 23(3):1853. https://doi.org/10.3390/ijms23031853
Chicago/Turabian StyleHaider, Mohamed, Amr Elsherbeny, Valeria Pittalà, Valeria Consoli, Maha Ali Alghamdi, Zahid Hussain, Ghalia Khoder, and Khaled Greish. 2022. "Nanomedicine Strategies for Management of Drug Resistance in Lung Cancer" International Journal of Molecular Sciences 23, no. 3: 1853. https://doi.org/10.3390/ijms23031853
APA StyleHaider, M., Elsherbeny, A., Pittalà, V., Consoli, V., Alghamdi, M. A., Hussain, Z., Khoder, G., & Greish, K. (2022). Nanomedicine Strategies for Management of Drug Resistance in Lung Cancer. International Journal of Molecular Sciences, 23(3), 1853. https://doi.org/10.3390/ijms23031853