
Mitochondrial-Targeted Therapy for Doxorubicin-Induced Cardiotoxicity


Abstract
:1. Introduction
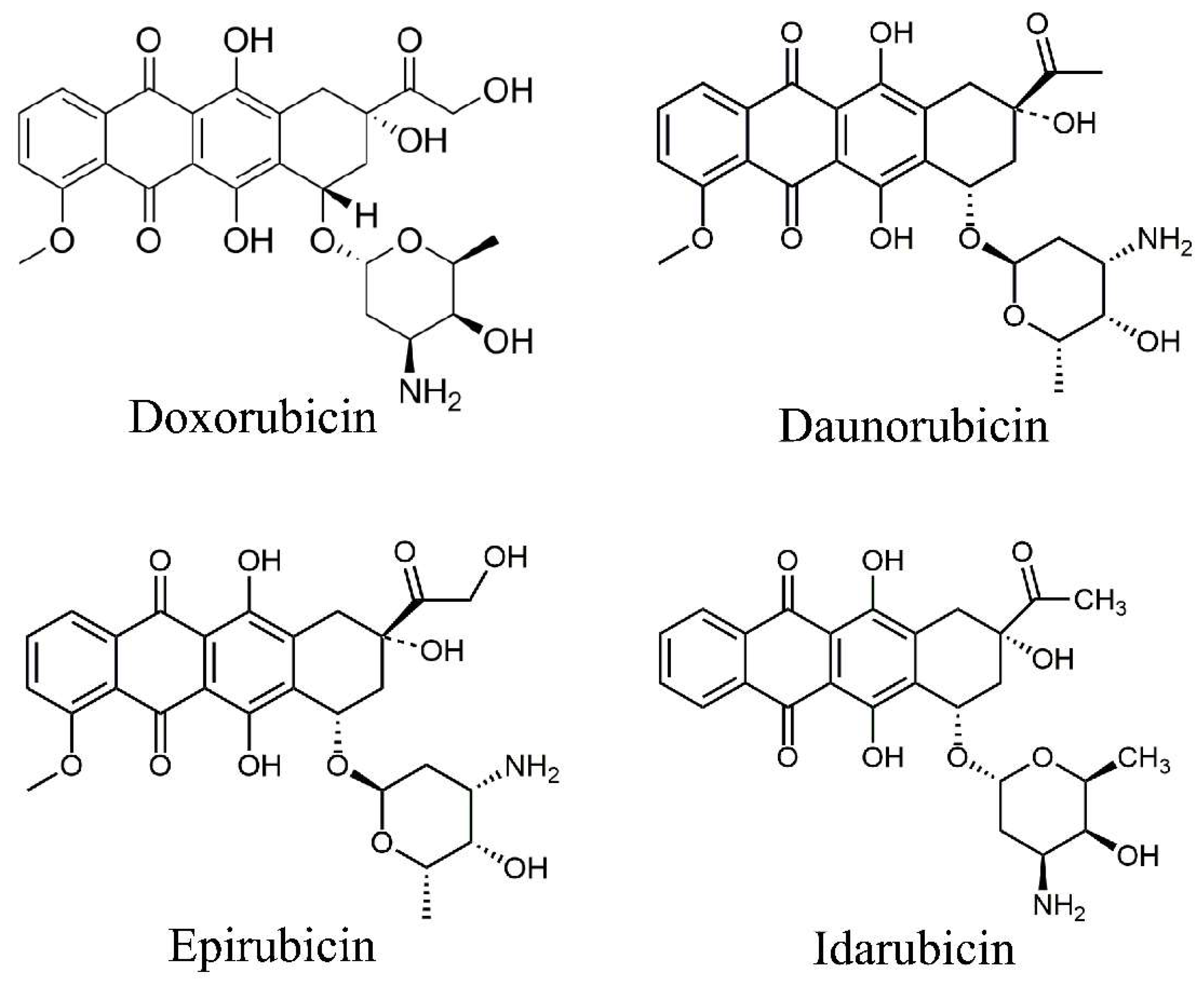
2. Types of Anthracyclines
3. Clinical Features of DOX-Induced Cardiotoxicity
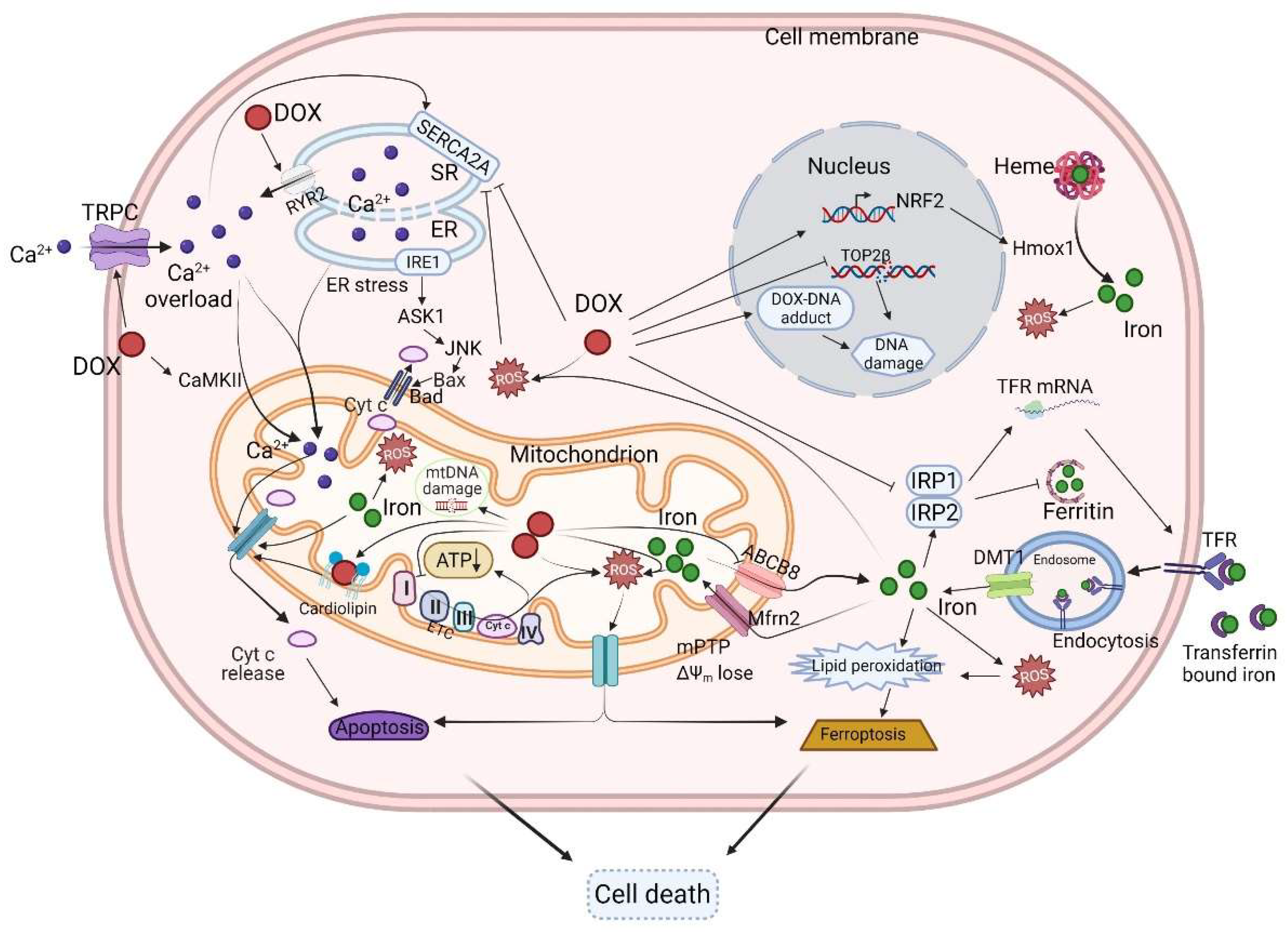
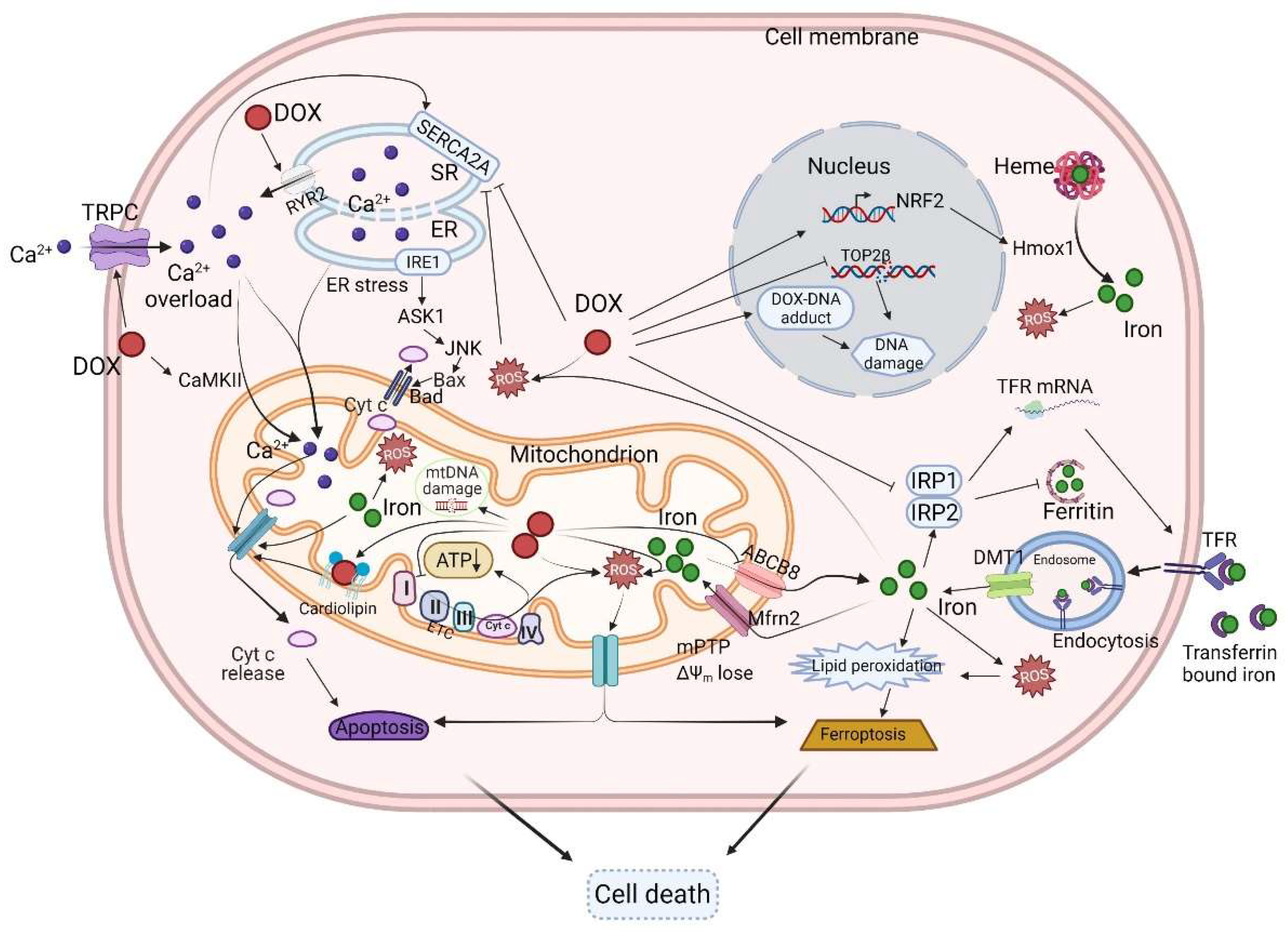
4. The Mechanisms of DOX-Induced Cardiotoxicity
4.1. The Mitochondria
4.2. DOX Accumulates in the Mitochondria and Perturbs Mitochondrial Bioenergetics and Function
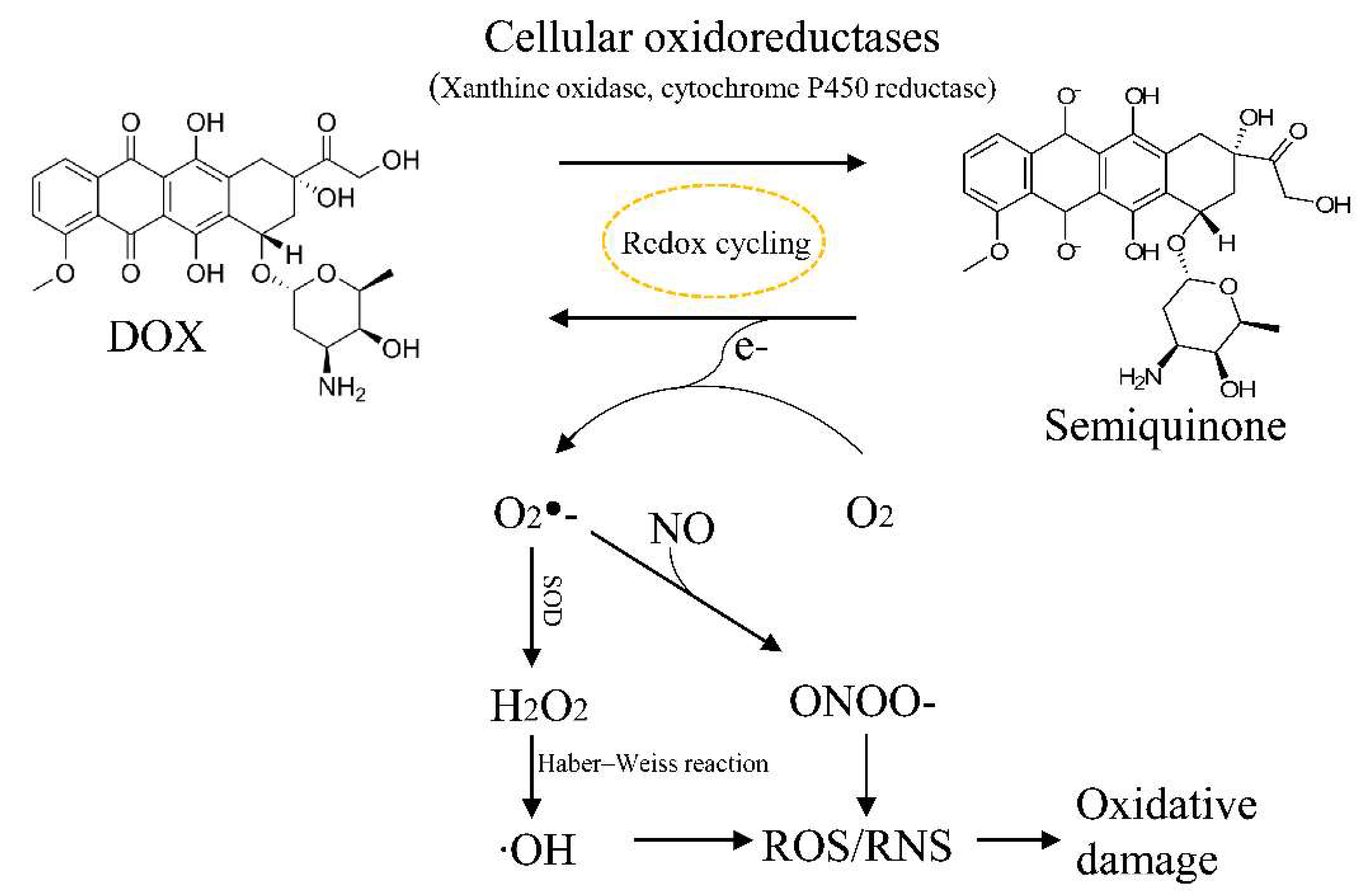
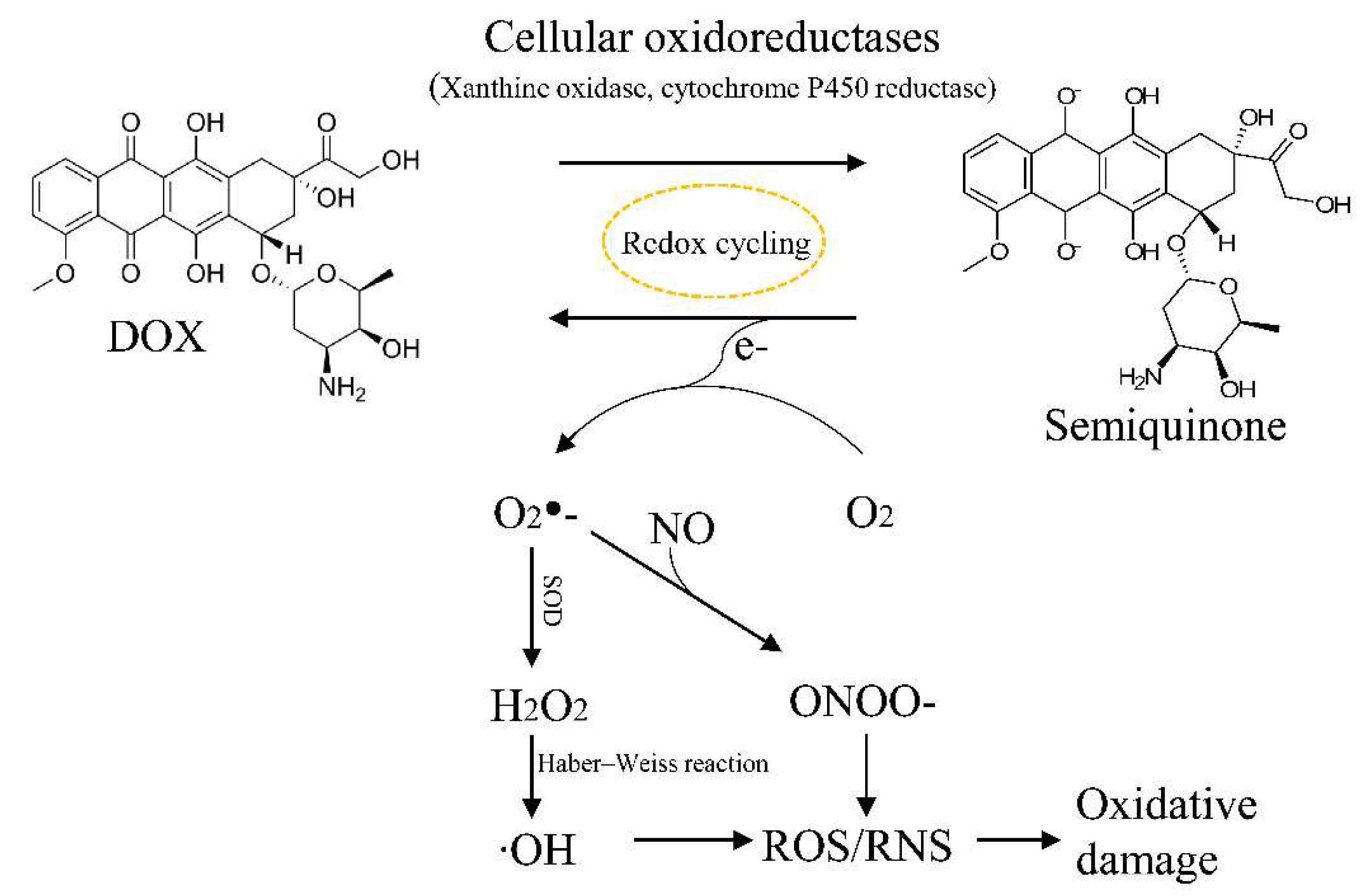
4.3. ROS and DOX-Induced Cardiotoxicity
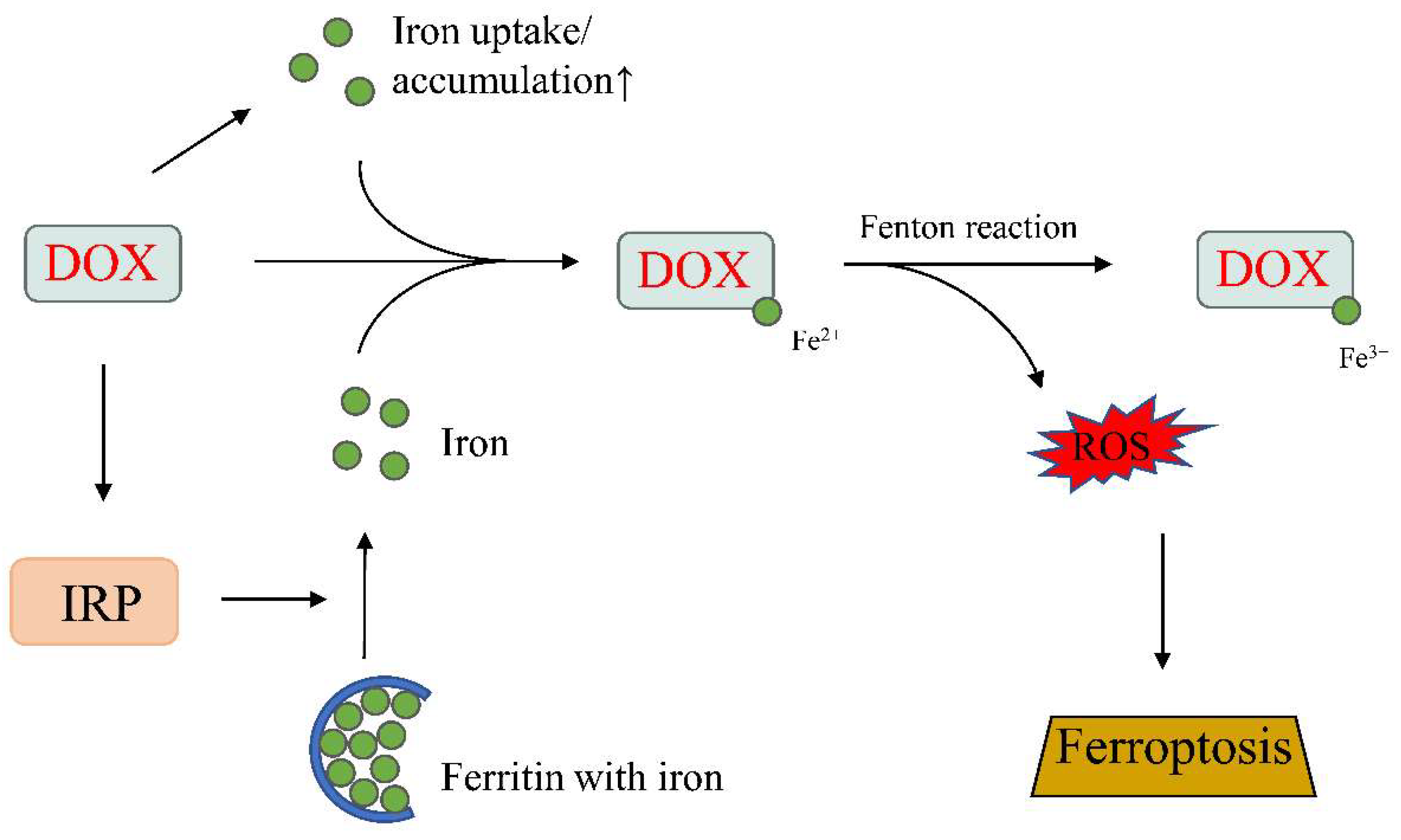
4.4. Iron, Ferroptosis, and DOX-Induced Cardiotoxicity
4.5. Calcium Homeostasis and DOX-Induced Cardiotoxicity
4.6. Topoisomerase and DOX-Induced Cardiotoxicity
4.7. DNA Damage
5. Modulating the Mitochondria to Alleviate DOX-Induced Cardiotoxicity
5.1. Small Molecules
5.2. Antioxidants
5.3. microRNAs (miRs)
5.4. Mitochondrial Transplantation
5.5. Heart Failure Medications
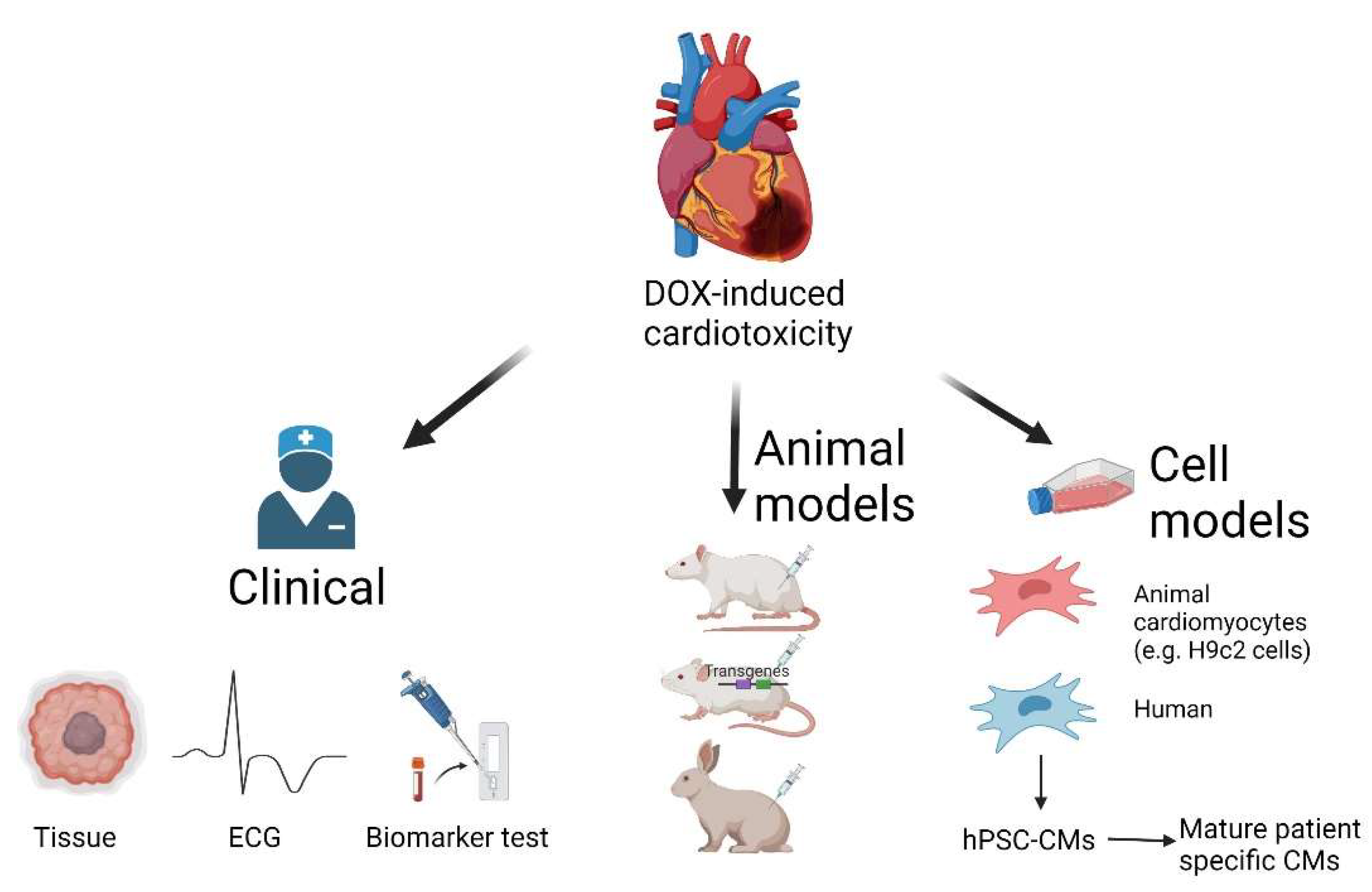
6. Research Models for DOX-Induced Cardiotoxicity
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Harake, D.; Franco, V.I.; Henkel, J.M.; Miller, T.L.; Lipshultz, S.E. Cardiotoxicity in Childhood Cancer Survivors: Strategies for Prevention and Management. Future Cardiol. 2012, 8, 647–670. [Google Scholar] [CrossRef] [Green Version]
- Lipshultz, S.E.; Rifai, N.; Dalton, V.M.; Levy, D.E.; Silverman, L.B.; Lipsitz, S.R.; Colan, S.D.; Asselin, B.L.; Barr, R.D.; Clavell, L.A.; et al. The Effect of Dexrazoxane on Myocardial Injury in Doxorubicin-Treated Children with Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2004, 351, 145–153. [Google Scholar] [CrossRef]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular Advances and Pharmacologie Developments in Antitumor Activity and Cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolaric, K.; Bradamante, V.; Cervek, J.; Cieslinska, A.; Cisarz-Filipcak, E.; Denisov, L.E.; Donat, D.; Drosik, K.; Gershanovic, M.; Hudziec, P.; et al. A Phase II Trial of Cardioprotection with Cardioxane (ICRF-187) in Patients with Advanced Breast Cancer Receiving 5-Fluorouracil, Doxorubicin and Cyclophosphamide. Oncology 1995, 52, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Whaley, F.S.; Gerber, M.C.; Weisberg, S.; York, M.; Spicer, D.; Jones, S.E.; Wadler, S.; Desai, A.; Vogel, C.; et al. Cardioprotection with Dexrazoxane for Doxorubicin-Containing Therapy in Advanced Breast Cancer. J. Clin. Oncol. 1997, 15, 1318–1332. [Google Scholar] [CrossRef]
- Mordente, A.; Meucci, E.; Silvestrini, A.; Martorana, G.E.; Giardina, B. Anthracyclines and Mitochondria. Adv. Exp. Med. Biol. 2012, 942, 385–419. [Google Scholar]
- Seferović, P.M.; Polovina, M.; Bauersachs, J.; Arad, M.; Gal, T.B.; Lund, L.H.; Felix, S.B.; Arbustini, E.; Caforio, A.L.P.; Farmakis, D.; et al. Heart Failure in Cardiomyopathies: A Position Paper from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 553–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govender, J.; Loos, B.; Marais, E.; Engelbrecht, A.M. Mitochondrial Catastrophe during Doxorubicin-Induced Cardiotoxicity: A Review of the Protective Role of Melatonin. J. Pineal Res. 2014, 57, 367–380. [Google Scholar] [CrossRef]
- Mitry, M.A.; Edwards, J.G. Doxorubicin Induced Heart Failure: Phenotype and Molecular Mechanisms. IJC Heart Vasc. 2016, 10, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive Heart Failure in Patients Treated with Doxorubicin: A Retrospective Analysis of Three Trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef]
- Lefrak, E.A.; Piťha, J.; Rosenheim, S.; Gottlieb, J.A. A Clinicopathologic Analysis of Adriamycin Cardiotoxicity. Cancer 1973, 32, 302–314. [Google Scholar] [CrossRef]
- Vejpongsa, P.; Yeh, E.T.H. Topoisomerase 2β: A Promising Molecular Target for Primary Prevention of Anthracycline-Induced Cardiotoxicity. Clin. Pharmacol. Ther. 2014, 95, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Khanna, A.; Pequeno, P.; Gupta, S.; Thavendiranathan, P.; Lee, D.S.; Abdel-Qadir, H.; Nathan, P.C. Increased Risk of All Cardiovascular Disease Subtypes Among Childhood Cancer Survivors. Circulation 2019, 140, 1041–1043. [Google Scholar] [CrossRef] [PubMed]
- Vandecruys, E.; Mondelaers, V.; de Wolf, D.; Benoit, Y.; Suys, B. Late Cardiotoxicity after Low Dose of Anthracycline Therapy for Acute Lymphoblastic Leukemia in Childhood. J. Cancer Surviv. Res. Pract. 2012, 6, 95–101. [Google Scholar] [CrossRef] [Green Version]
- Leger, K.; Slone, T.; Lemler, M.; Leonard, D.; Cochran, C.; Bowman, W.P.; Bashore, L.; Winick, N. Subclinical Cardiotoxicity in Childhood Cancer Survivors Exposed to Very Low Dose Anthracycline Therapy. Pediatr. Blood Cancer 2015, 62, 123–127. [Google Scholar] [CrossRef]
- Meiners, B.; Shenoy, C.; Zordoky, B.N. Clinical and Preclinical Evidence of Sex-Related Differences in Anthracycline-Induced Cardiotoxicity. Biol. Sex Differ. 2018, 9, 38. [Google Scholar] [CrossRef]
- Hequet, O.; Le, Q.H.; Moullet, I.; Pauli, E.; Salles, G.; Espinouse, D.; Dumontet, C.; Thieblemont, C.; Arnaud, P.; Antal, D.; et al. Subclinical Late Cardiomyopathy after Doxorubicin Therapy for Lymphoma in Adults. J. Clin. Oncol. 2004, 22, 1864–1871. [Google Scholar] [CrossRef]
- Zeiss, C.J.; Gatti, D.M.; Toro-Salazar, O.; Davis, C.; Lutz, C.M.; Spinale, F.; Stearns, T.; Furtado, M.B.; Churchill, G.A. Doxorubicin-Induced Cardiotoxicity in Collaborative Cross (CC) Mice Recapitulates Individual Cardiotoxicity in Humans. G3 Genes Genomes Genet. 2019, 9, 2637–2646. [Google Scholar] [CrossRef] [Green Version]
- Eksborg, S. Pharmacokinetics of Anthracyclines. Acta Oncol. 1989, 28, 385–419. [Google Scholar] [CrossRef] [Green Version]
- Casper, E.S.; Gaynor, J.J.; Hajdu, S.I.; Magill, G.B.; Tan, C.; Friedrich, C.; Brennan, M.F. A Prospective Randomized Trial of Adjuvant Chemotherapy with Bolus versus Continuous Infusion of Doxorubicin in Patients with High-grade Extremity Soft Tissue Sarcoma and an Analysis of Prognostic Factors. Cancer 1991, 68, 1221–1229. [Google Scholar] [CrossRef]
- von Hoff, D.D.; Layard, M.W.; Basa, P.; Davis, H.L.; von Hoff, A.L.; Rozencweig, M.; Muggia, F.M. Risk Factors for Doxorubicin-Induced Congestive Heart Failure. Ann. Intern. Med. 1979, 91, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Lipshultz, S.E.; Giantris, A.L.; Lipsitz, S.R.; Dalton, V.K.; Asselin, B.L.; Barr, R.D.; Clavell, L.A.; Hurwitz, C.A.; Moghrabi, A.; Samson, Y.; et al. Doxorubicin Administration by Continuous Infusion Is Not Cardioprotective: The Dana-Farber 91-01 Acute Lymphoblastic Leukemia Protocol. J. Clin. Oncol. 2002, 20, 1677–1682. [Google Scholar] [CrossRef] [PubMed]
- Lipshultz, S.E.; Miller, T.L.; Lipsitz, S.R.; Neuberg, D.S.; Dahlberg, S.E.; Colan, S.D.; Silverman, L.B.; Henkel, J.M.; Franco, V.I.; Cushman, L.L.; et al. Continuous versus Bolus Infusion of Doxorubicin in Children with ALL: Long-Term Cardiac Outcomes. Pediatrics 2012, 130, 1003–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipshultz, S.E.; Lipsitz, S.R.; Sallan, S.E.; Dalton, V.M.; Mone, S.M.; Gelber, R.D.; Colan, S.D. Chronic Progressive Cardiac Dysfunction Years after Doxorubicin Therapy for Childhood Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2005, 23, 2629–2636. [Google Scholar] [CrossRef]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Energy Metabolism in Heart Failure. J. Physiol. 2004, 555, 1–13. [Google Scholar] [CrossRef]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial Protein Organization: From Biogenesis to Networks and Function. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS Signaling in Organismal Homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [Green Version]
- Tokarska-Schlattner, M.; Zaugg, M.; Zuppinger, C.; Wallimann, T.; Schlattner, U. New Insights into Doxorubicin-Induced Cardiotoxicity: The Critical Role of Cellular Energetics. J. Mol. Cell. Cardiol. 2006, 41, 389–405. [Google Scholar] [CrossRef]
- Goormaghtigh, E.; Huart, P.; Brasseur, R.; Ruysschaert, J.M. Mechanism of Inhibition of Mitochondrial Enzymatic Complex I-III by Adriamycin Derivatives. Biochim. Biophys. Acta-Biomembr. 1986, 861, 83–94. [Google Scholar] [CrossRef]
- Marcillat, O.; Zhang, Y.; Davies, K.J.A. Oxidative and Non-Oxidative Mechanisms in the Inactivation of Cardiac Mitochondrial Electron Transport Chain Components by Doxorubicin. Biochem. J. 1989, 259, 181–189. [Google Scholar] [CrossRef] [Green Version]
- Davies, K.J.A.; Doroshow, J.H. Redox Cycling of Anthracyclines by Cardiac Mitochondria. I. Anthracycline Radical Formation by NADH Dehydrogenase. J. Biol. Chem. 1986, 261, 3060–3067. [Google Scholar] [CrossRef]
- Basit, F.; van Oppen, L.M.P.E.; Schöckel, L.; Bossenbroek, H.M.; van Emst-De Vries, S.E.; Hermeling, J.C.W.; Grefte, S.; Kopitz, C.; Heroult, M.; Willems, P.H.G.M.; et al. Mitochondrial Complex i Inhibition Triggers a Mitophagy-Dependent ROS Increase Leading to Necroptosis and Ferroptosis in Melanoma Cells. Cell Death Dis. 2017, 8, e2716. [Google Scholar] [CrossRef]
- Papadopoulou, L.C.; Theophilidis, G.; Thomopoulos, G.N.; Tsiftsoglou, A.S. Structural and Functional Impairmemt of Mitochondria in Adriamycin-Induced Cardiomyopathy in Mice: Suppression of Cytochrome c Oxidase II Gene Expression. Biochem. Pharmacol. 1999, 57, 481–489. [Google Scholar] [CrossRef]
- Lebrecht, D.; Setzer, B.; Ketelsen, U.P.; Haberstroh, J.; Walker, U.A. Time-Dependent and Tissue-Specific Accumulation of MtDNA and Respiratory Chain Defects in Chronic Doxorubicin Cardiomyopathy. Circulation 2003, 108, 2423–2429. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.W.; Klein, J.B.; Kang, Y.J. Metallothionein Inhibits Doxorubicin-Induced Mitochondrial Cytochrome c Release and Caspase-3 Activation in Cardiomyocytes. J. Pharmacol. Exp. Ther. 2001, 298, 461–468. [Google Scholar]
- Zhang, P.; Chen, Z.; Lu, D.; Wu, Y.; Fan, M.; Qian, J.; Ge, J. Overexpression of COX5A Protects H9c2 Cells against Doxorubicin-Induced Cardiotoxicity. Biochem. Biophys. Res. Commun. 2020, 524, 43–49. [Google Scholar] [CrossRef]
- Zhang, J.; Xiang, H.; Liu, J.; Chen, Y.; He, R.R.; Liu, B. Mitochondrial Sirtuin 3: New Emerging Biological Function and Therapeutic Target. Theranostics 2020, 10, 8315–8342. [Google Scholar] [CrossRef]
- He, L.; Liu, F.; Li, J. Mitochondrial Sirtuins and Doxorubicin-Induced Cardiotoxicity. Cardiovasc. Toxicol. 2021, 21, 179–191. [Google Scholar] [CrossRef]
- Cheung, K.G.; Cole, L.K.; Xiang, B.; Chen, K.; Ma, X.; Myal, Y.; Hatch, G.M.; Tong, Q.; Dolinsky, V.W. Sirtuin-3 (SIRT3) Protein Attenuates Doxorubicin-Induced Oxidative Stress and Improves Mitochondrial Respiration in H9c2 Cardiomyocytes. J. Biol. Chem. 2015, 290, 10981–10993. [Google Scholar] [CrossRef] [Green Version]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 Blocks the Cardiac Hypertrophic Response by Augmenting Foxo3a-Dependent Antioxidant Defense Mechanisms in Mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [CrossRef] [Green Version]
- Samant, S.A.; Zhang, H.J.; Hong, Z.; Pillai, V.B.; Sundaresan, N.R.; Wolfgeher, D.; Archer, S.L.; Chan, D.C.; Gupta, M.P. SIRT3 Deacetylates and Activates OPA1 To Regulate Mitochondrial Dynamics during Stress. Mol. Cell. Biol. 2014, 34, 807–819. [Google Scholar] [CrossRef] [Green Version]
- Coelho, A.R.; Martins, T.R.; Couto, R.; Deus, C.; Pereira, C.V.; Simões, R.F.; Rizvanov, A.A.; Silva, F.; Cunha-Oliveira, T.; Oliveira, P.J.; et al. Berberine-Induced Cardioprotection and Sirt3 Modulation in Doxorubicin-Treated H9c2 Cardiomyoblasts. Biochim. Biophys. Acta-Mol. Basis Dis. 2017, 1863, 2904–2923. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Ma, H.; Jiang, Z.; Niu, L.; Zhang, X.; Liu, Y.; Wang, Y.; Cheng, S.; Deng, Y.; Qi, H.; et al. Dosing Depending on SIRT3 Activity Attenuates Doxorubicin-Induced Cardiotoxicity via Elevated Tolerance against Mitochondrial Dysfunction and Oxidative Stress. Biochem. Biophys. Res. Commun. 2019, 517, 111–117. [Google Scholar] [CrossRef]
- Pillai, V.B.; Bindu, S.; Sharp, W.; Fang, Y.H.; Kim, G.; Gupta, M.; Samant, S.; Gupta, M.P. Sirt3 Protects Mitochondrial DNA Damage and Blocks the Development of Doxorubicin-Induced Cardiomyopathy in Mice. Am. J. Physiol.-Heart Circul. Physiol. 2016, 310, H962–H972. [Google Scholar] [CrossRef] [Green Version]
- Tanno, M.; Sakamoto, J.; Miura, T.; Shimamoto, K.; Horio, Y. Nucleocytoplasmic Shuttling of the NAD+-Dependent Histone Deacetylase SIRT1. J. Biol. Chem. 2007, 282, 6823–6832. [Google Scholar] [CrossRef] [Green Version]
- Soni, S.K.; Basu, P.; Singaravel, M.; Sharma, R.; Pandi-Perumal, S.R.; Cardinali, D.P.; Reiter, R.J. Sirtuins and the Circadian Clock Interplay in Cardioprotection: Focus on Sirtuin 1. Cell. Mol. Life Sci. 2021, 78, 2503–2515. [Google Scholar] [CrossRef]
- Vikram, A.; Lewarchik, C.M.; Yoon, J.Y.; Naqvi, A.; Kumar, S.; Morgan, G.M.; Jacobs, J.S.; Li, Q.; Kim, Y.R.; Kassan, M.; et al. Sirtuin 1 Regulates Cardiac Electrical Activity by Deacetylating the Cardiac Sodium Channel. Nat. Med. 2017, 23, 361–367. [Google Scholar] [CrossRef]
- Hsu, C.P.; Zhai, P.; Yamamoto, T.; Maejima, Y.; Matsushima, S.; Hariharan, N.; Shao, D.; Takagi, H.; Oka, S.; Sadoshima, J. Silent Information Regulator 1 Protects the Heart from Ischemia/Reperfusion. Circulation 2010, 122, 2170–2182. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Guo, J.; Zhang, Q.; Yin, J.; Li, J.; Zhou, W.; Zhang, T.; Yuan, H.; Zhao, J.; Zhang, L.; et al. Erythropoietin Activates SIRT1 to Protect Human Cardiomyocytes against Doxorubicin-Induced Mitochondrial Dysfunction and Toxicity. Toxicol. Lett. 2017, 275, 28–38. [Google Scholar] [CrossRef]
- Wu, Y.Z.; Zhang, L.; Wu, Z.X.; Shan, T.T.; Xiong, C. Berberine Ameliorates Doxorubicin-Induced Cardiotoxicity via a SIRT1/P66Shc-Mediated Pathway. Oxid. Med. Cell. Longev. 2019, 2019, 2150394. [Google Scholar] [CrossRef]
- Brookins Danz, E.D.; Skramsted, J.; Henry, N.; Bennett, J.A.; Keller, R.S. Resveratrol Prevents Doxorubicin Cardiotoxicity through Mitochondrial Stabilization and the Sirt1 Pathway. Free Radic. Biol. Med. 2009, 46, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.; Dong, C.; Patel, J.; Duan, C.; Wang, X.; Wu, X.; Cao, Y.; Pu, L.; Lu, D.; Shen, T.; et al. SIRT1 Suppresses Doxorubicin-Induced Cardiotoxicity by Regulating the Oxidative Stress and P38MAPK Pathways. Cell. Physiol. Biochem. 2015, 35, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Wang(a), J.; Zhang, J.; Xiao, M.; Wang, S.; Wang(b), J.; Guo, Y.; Tang, Y.; Gu, J. Molecular Mechanisms of Doxorubicin-Induced Cardiotoxicity: Novel Roles of Sirtuin 1-Mediated Signaling Pathways. Cell. Mol. Life Sci. 2021, 78, 3105–3125. [Google Scholar] [CrossRef]
- Cui, M.; Atmanli, A.; Morales, M.G.; Tan, W.; Chen, K.; Xiao, X.; Xu, L.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Nrf1 Promotes Heart Regeneration and Repair by Regulating Proteostasis and Redox Balance. Nat. Commun. 2021, 12, 5270. [Google Scholar] [CrossRef]
- Nicolay, K.; de Kruijff, B. Effects of Adriamycin on Respiratory Chain Activities in Mitochondria from Rat Liver, Rat Heart and Bovine Heart. Evidence for a Preferential Inhibition of Complex III and IV. Biochim. Biophys. Acta-Bioenerg. 1987, 892, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Goormaghtigh, E.; Huart, P.; Praet, M.; Brasseur, R.; Ruysschaert, J.M. Structure of the Adriamycin-Cardiolipin Complex. Role in Mitochondrial Toxicity. Biophys. Chem. 1990, 35, 247–257. [Google Scholar] [CrossRef]
- Sokolove, P.M. Interactions of Adriamycin Aglycones with Mitochondria May Mediate Adriamycin Cardiotoxicity. Int. J. Biochem. 1994, 26, 1341–1350. [Google Scholar] [CrossRef]
- Sinibaldi, F.; Howes, B.D.; Droghetti, E.; Polticelli, F.; Piro, M.C.; di Pierro, D.; Fiorucci, L.; Coletta, M.; Smulevich, G.; Santucci, R. Role of Lysines in Cytochrome C-Cardiolipin Interaction. Biochemistry 2013, 52, 4578–4588. [Google Scholar] [CrossRef] [Green Version]
- Wallace, K.B.; Sardão, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ.Res. 2020, 126, 926–941. [Google Scholar] [CrossRef]
- Gebert, N.; Ryan, M.T.; Pfanner, N.; Wiedemann, N.; Stojanovski, D. Mitochondrial Protein Import Machineries and Lipids: A Functional Connection. Biochim. Biophys. Acta-Biomembr. 2011, 1808, 1002–1011. [Google Scholar] [CrossRef] [Green Version]
- Eilers, M.; Endo, T.; Schatz, G. Adriamycin, a Drug Interacting with Acidic Phospholipids, Blocks Import of Precursor Proteins by Isolated Yeast Mitochondria. J. Biol. Chem. 1989, 264, 2945–2950. [Google Scholar] [CrossRef]
- Guven, C.; Sevgiler, Y.; Taskin, E. Mitochondrial Dysfunction Associated with Doxorubicin. In Mitochondrial Diseases; BoD: Norderstedt, Germany, 2018. [Google Scholar]
- Wallace, K.B. Doxorubicin-Induced Cardiac Mitochondrionopathy. Pharmacol. Toxicol. 2003, 93, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Bellance, N.; Furt, F.; Melser, S.; Lalou, C.; Thoraval, D.; Maneta-Peyret, L.; Lacombe, D.; Moreau, P.; Rossignol, R. Doxorubicin Inhibits Phosphatidylserine Decarboxylase and Modifies Mitochondrial Membrane Composition in Hela Cells. Int. J. Mol. Sci. 2020, 21, 1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voelker, D.R. Adriamycin Disrupts Phosphatidylserine Import into the Mitochondria of Permeabilized CHO-K1 Cells. J. Biol. Chem. 1991, 266, 12185–12188. [Google Scholar] [CrossRef]
- Songbo, M.; Lang, H.; Xinyong, C.; Bin, X.; Ping, Z.; Liang, S. Oxidative Stress Injury in Doxorubicin-Induced Cardiotoxicity. Toxicol. Lett. 2019, 307, 41–48. [Google Scholar] [CrossRef]
- Ide, T.; Tsutsui, H.; Kinugawa, S.; Suematsu, N.; Hayashidani, S.; Ichikawa, K.; Utsumi, H.; Machida, Y.; Egashira, K.; Takeshita, A. Direct Evidence for Increased Hydroxyl Radicals Originating from Superoxide in the Failing Myocardium. Circ.Res. 2000, 86, 52–57. [Google Scholar] [CrossRef]
- Pecoraro, M.; Pala, B.; di Marcantonio, M.C.; Muraro, R.; Marzocco, S.; Pinto, A.; Mincione, G.; Popolo, A. Doxorubicin-Induced Oxidative and Nitrosative Stress: Mitochondrial Connexin 43 Is at the Crossroads. Int. J. Mol. Med. 2020, 46, 1197–1209. [Google Scholar] [CrossRef]
- Angsutararux, P.; Luanpitpong, S.; Issaragrisil, S. Chemotherapy-Induced Cardiotoxicity: Overview of the Roles of Oxidative Stress. Oxid. Med. Cell. Longev. 2015, 2015, 795602. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Kim, H.S.; Seo, Y.R. Understanding of ROS-Inducing Strategy in Anticancer Therapy. Oxid. Med. Cell. Longev. 2019, 2019, 5381692. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Rajesh, M.; Bátkai, S.; Kashiwaya, Y.; Haskó, G.; Liaudet, L.; Szabó, C.; Pacher, P. Role of Superoxide, Nitric Oxide, and Peroxynitrite in Doxorubicin-Induced Cell Death in Vivo and in Vitro. Am. J. Physiol.-Heart Circul. Physiol. 2009, 296, H1466–H1483. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Nagoshi, T.; Yoshii, A.; Oi, Y.; Takahashi, H.; Kimura, H.; Ito, K.; Kashiwagi, Y.; Tanaka, T.D.; Yoshimura, M. Xanthine Oxidase Inhibition Attenuates Doxorubicin-Induced Cardiotoxicity in Mice. Free Radic. Biol. Med. 2021, 162, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, T.; Ikeda, M.; Ide, T.; Deguchi, H.; Ikeda, S.; Okabe, K.; Ishikita, A.; Matsushima, S.; Koumura, T.; Yamada, K.I.; et al. Mitochondria-Dependent Ferroptosis Plays a Pivotal Role in Doxorubicin Cardiotoxicity. JCI Insight 2020, 5, e132747. [Google Scholar] [CrossRef] [PubMed]
- James Kang, Y.; Chen, Y.; Epstein, P.N. Suppression of Doxorubicin Cardiotoxicity by Overexpression of Catalase in the Heart of Transgenic Mice. J. Biol. Chem. 1996, 271, 12610–12616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yen, H.C.; Oberley, T.D.; Vichitbandha, S.; Ho, Y.S.; St. Clair, D.K. The Protective Role of Manganese Superoxide Dismutase against Adriamycin-Induced Acute Cardiac Toxicity in Transgenic Mice. J. Clin. Investig. 1996, 98, 1253–1260. [Google Scholar] [CrossRef] [Green Version]
- Shioji, K.; Kishimoto, C.; Nakamura, H.; Masutani, H.; Yuan, Z.; Oka, S.-I.; Yodoi, J. Overexpression of Thioredoxin-1 in Transgenic Mice Attenuates Adriamycin-Induced Cardiotoxicity. Circulation 2002, 106, 1403–1409. [Google Scholar] [CrossRef]
- Zhou, Z.; Kang, Y.J. Immunocytochemical Localization of Metallothionein and Its Relation to Doxorubicin Toxicity in Transgenic Mouse Heart. Am. J. Pathol. 2000, 156, 1653–1662. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Xiong, Y.; Ho, Y.S.; Liu, X.; Chua, C.C.; Xu, X.; Wang, H.; Hamdy, R.; Chua, B.H.L. Glutathione Peroxidase 1-Deficient Mice Are More Susceptible to Doxorubicin-Induced Cardiotoxicity. Biochim. Biophys. Acta-Mol. Cell Res. 2008, 1783, 2020–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludke, A.; Akolkar, G.; Ayyappan, P.; Sharma, A.K.; Singal, P.K. Time Course of Changes in Oxidative Stress and Stress-Induced Proteins in Cardiomyocytes Exposed to Doxorubicin and Prevention by Vitamin C. PLoS ONE 2017, 12, e0179452. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, S.; Tatsumi, T.; Shiraishi, J.; Mano, A.; Keira, N.; Matoba, S.; Asayama, J.; Fushiki, S.; Fliss, H.; Nakagawa, M. Amlodipine Inhibits Doxorubicin-Induced Apoptosis in Neonatal Rat Cardiac Myocytes. J. Am. Coll. Cardiol. 2003, 41, 870–878. [Google Scholar] [CrossRef] [Green Version]
- Gulati, V.; Harikrishnan, P.; Palaniswamy, C.; Aronow, W.S.; Jain, D.; Frishman, W.H. Cardiac Involvement in Hemochromatosis. Cardiol. Rev. 2014, 22, 56–68. [Google Scholar] [CrossRef]
- Miranda, C.J.; Makui, H.; Soares, R.J.; Bilodeau, M.; Mui, J.; Vali, H.; Bertrand, R.; Andrews, N.C.; Santos, M.M. Hfe Deficiency Increases Susceptibility to Cardiotoxicity and Exacerbates Changes in Iron Metabolism Induced by Doxorubicin. Blood 2003, 102, 2574–2580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vela, D. Keeping Heart Homeostasis in Check through the Balance of Iron Metabolism. Acta Physiol. 2020, 228, e13324. [Google Scholar] [CrossRef] [PubMed]
- Minotti, G.; Salvatorelli, E.; Menna, P.; Ronchi, R.; Cairo, G. Doxorubicin Irreversibly Inactivates Iron Regulatory Proteins 1 and 2 in Cardiomyocytes: Evidence for Distinct Metabolic Pathways and Implications for Iron-Mediated Cardiotoxicity of Antitumor Therapy. Cancer Res. 2001, 61, 8422–8428. [Google Scholar] [PubMed]
- Shi, Y.; Moon, M.; Dawood, S.; McManus, B.; Liu, P.P. Mechanisms and Management of Doxorubicin Cardiotoxicity. Herz 2011, 36, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Christidi, E.; Brunham, L.R. Regulated Cell Death Pathways in Doxorubicin-Induced Cardiotoxicity. Cell Death Dis. 2021, 12, 339. [Google Scholar] [CrossRef]
- Gordan, R.; Wongjaikam, S.; Gwathmey, J.K.; Chattipakorn, N.; Chattipakorn, S.C.; Xie, L.H. Involvement of Cytosolic and Mitochondrial Iron in Iron Overload Cardiomyopathy: An Update. Heart Fail. Rev. 2018, 23, 801–816. [Google Scholar] [CrossRef]
- Cavadini, P.; Biasiotto, G.; Poli, M.; Levi, S.; Verardi, R.; Zanella, I.; Derosas, M.; Ingrassia, R.; Corrado, M.; Arosio, P. RNA Silencing of the Mitochondrial ABCB7 Transporter in HeLa Cells Causes an Iron-Deficient Phenotype with Mitochondrial Iron Overload. Blood 2007, 109, 3552–3559. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Naga Prasad, S.V.; Mutharasan, R.K.; Jairaj Naik, T.; Ardehali, H. Cardiotoxicity of Doxorubicin Is Mediated through Mitochondrial Iron Accumulation. J. Clin. Investig. 2014, 124, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Kumfu, S.; Chattipakorn, S.; Fucharoen, S.; Chattipakorn, N. Mitochondrial Calcium Uniporter Blocker Prevents Cardiac Mitochondrial Dysfunction Induced by Iron Overload in Thalassemic Mice. BioMetals 2012, 25, 1167–1175. [Google Scholar] [CrossRef]
- Sripetchwandee, J.; Kenknight, S.B.; Sanit, J.; Chattipakorn, S.; Chattipakorn, N. Blockade of Mitochondrial Calcium Uniporter Prevents Cardiac Mitochondrial Dysfunction Caused by Iron Overload. Acta Physiol. 2014, 210, 330–341. [Google Scholar] [CrossRef]
- Qin, Y.; Guo, T.; Wang, Z.; Zhao, Y. The Role of Iron in Doxorubicin-Induced Cardiotoxicity: Recent Advances and Implication for Drug Delivery. J. Mater. Chem. B 2021, 9, 4793–4803. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the Ferroptosis Regulator Gpx4 Triggers Acute Renal Failure in Mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a Target for Protection against Cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasinoff, B.B.; Schnabl, K.L.; Marusak, R.A.; Patel, D.; Huebner, E. Dexrazoxane (ICRF-187) Protects Cardiac Myocytes against Doxorubicin by Preventing Damage to Mitochondria. Cardiovasc. Toxicol. 2003, 3, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.L.; Kerrigan, J.E.; Lin, C.P.; Azarova, A.M.; Tsai, Y.C.; Ban, Y.; Liu, L.F. Topoisomerase IIβ-Mediated DNA Double-Strand Breaks: Implications in Doxorubicin Cardiotoxicity and Prevention by Dexrazoxane. Cancer Res. 2007, 67, 8839–8846. [Google Scholar] [CrossRef] [Green Version]
- Hasinoff, B.B.; Patel, D.; Wu, X. The Oral Iron Chelator ICL670A (Deferasirox) Does Not Protect Myocytes against Doxorubicin. Free Radic. Biol. Med. 2003, 35, 1469–1479. [Google Scholar] [CrossRef]
- Hasinoff, B.B.; Patel, D. The Iron Chelator Dp44mT Does Not Protect Myocytes against Doxorubicin. J. Inorg. Biochem. 2009, 103, 1093–1101. [Google Scholar] [CrossRef]
- Wang, H.; Cheng, X.; Tian, J.; Xiao, Y.; Tian, T.; Xu, F.; Hong, X.; Zhu, M.X. TRPC Channels: Structure, Function, Regulation and Recent Advances in Small Molecular Probes. Pharmacol. Ther. 2020, 209, 107497. [Google Scholar] [CrossRef]
- Peng, W.; Shen, H.; Wu, J.; Guo, W.; Pan, X.; Wang, R.; Chen, S.R.W.; Yan, N. Structural Basis for the Gating Mechanism of the Type 2 Ryanodine Receptor RyR2. Science 2016, 354, aah5324. [Google Scholar] [CrossRef]
- Kho, C.; Lee, A.; Hajjar, R.J. Altered Sarcoplasmic Reticulum Calcium Cycling—Targets for Heart Failure Therapy. Nat. Rev. Cardiol. 2012, 9, 717–733. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.-C.; Sun, G.-B.; Ye, J.-X.; Wang, J.; Zhang, M.-D.; Sun, X.-B. Salvianolic Acid B Attenuates Doxorubicin-Induced ER Stress by Inhibiting TRPC3 and TRPC6 Mediated Ca2+ Overload in Rat Cardiomyocytes. Toxicol. Lett. 2017, 276, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Hanna, A.D.; Lam, A.; Tham, S.; Dulhunty, A.F.; Beard, N.A. Adverse Effects of Doxorubicin and Its Metabolic Product on Cardiac RyR2 and SERCA2A. Mol. Pharmacol. 2014, 86, 438–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sag, C.M.; Köhler, A.C.; Anderson, M.E.; Backs, J.; Maier, L.S. CaMKII-Dependent SR Ca Leak Contributes to Doxorubicin-Induced Impaired Ca Handling in Isolated Cardiac Myocytes. J. Mol. Cell. Cardiol. 2011, 51, 749–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial Calcium Overload Is a Key Determinant in Heart Failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalivendi, S.V.; Konorev, E.A.; Cunningham, S.; Vanamala, S.K.; Kaji, E.H.; Joseph, J.; Kalyanaraman, B. Doxorubicin Activates Nuclear Factor of Activated T-Lymphocytes and Fas Ligand Transcription: Role of Mitochondrial Reactive Oxygen Species and Calcium. Biochem. J. 2005, 389, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Sruthi Sritharan, N.S. A Comprehensive Review on Time-Tested Anticancer Drug Doxorubicin. Life Sci. 2021, 278, 119527. [Google Scholar] [CrossRef]
- Zhang, Y.; Xia, Z.; la Cour, K.H.; Ren, J. Activation of Akt Rescues Endoplasmic Reticulum Stress-Impaired Murine Cardiac Contractile Function via Glycogen Synthase Kinase-3β-Mediated Suppression of Mitochondrial Permeation Pore Opening. Antioxid. Redox Signal. 2011, 15, 2407–2424. [Google Scholar] [CrossRef] [Green Version]
- Deniaud, A.; Sharaf El Dein, O.; Maillier, E.; Poncet, D.; Kroemer, G.; Lemaire, C.; Brenner, C. Endoplasmic Reticulum Stress Induces Calcium-Dependent Permeability Transition, Mitochondrial Outer Membrane Permeabilization and Apoptosis. Oncogene 2008, 27, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Montalvo, R.N.; Doerr, V.; Min, K.; Szeto, H.H.; Smuder, A.J. Doxorubicin-Induced Oxidative Stress Differentially Regulates Proteolytic Signaling in Cardiac and Skeletal Muscle. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2020, 318, R227–R233. [Google Scholar] [CrossRef]
- Yarmohammadi, F.; Rezaee, R.; Haye, A.W.; Karimi, G. Endoplasmic Reticulum Stress in Doxorubicin-Induced Cardiotoxicity May Be Therapeutically Targeted by Natural and Chemical Compounds: A Review. Pharmacol. Res. 2021, 164, 105383. [Google Scholar] [CrossRef]
- Shore, G.C.; Papa, F.R.; Oakes, S.A. Signaling Cell Death from the Endoplasmic Reticulum Stress Response. Curr. Opin. Cell Biol. 2011, 23, 143–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tewey, K.M.; Rowe, T.C.; Yang, L.; Halligan, B.D.; Liu, L.F. Adriamycin-Induced DNA Damage Mediated by Mammalian DNA Topoisomerase II. Science 1984, 226, 466–468. [Google Scholar] [CrossRef] [PubMed]
- Schellenberg, M.J.; Lieberman, J.A.; Herrero-Ruiz, A.; Butler, L.R.; Williams, J.G.; Muñoz-Cabello, A.M.; Mueller, G.A.; London, R.E.; Cortés-Ledesma, F.; Williams, R.S. ZATT (ZNF451)–Mediated Resolution of Topoisomerase 2 DNA-Protein Cross-Links. Science 2017, 357, 1412–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capranico, G.; Tinelli, S.; Austin, C.A.; Fisher, M.L.; Zunino, F. Different Patterns of Gene Expression of Topoisomerase II Isoforms in Differentiated Tissues during Murine Development. Biochim. Biophys. Acta-Gene Struct. Expr. 1992, 1132, 43–48. [Google Scholar] [CrossRef]
- Murabito, A.; Hirsch, E.; Ghigo, A. Mechanisms of Anthracycline-Induced Cardiotoxicity: Is Mitochondrial Dysfunction the Answer? Front. Cardiovasc. Med. 2020, 7, 35. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T.H. Identification of the Molecular Basis of Doxorubicin-Induced Cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef]
- Classen, S.; Olland, S.; Berger, J.M. Structure of the Topoisomerase II ATPase Region and Its Mechanism of Inhibition by the Chemotherapeutic Agent ICRF-187. Proc. Natl. Acad. Sci. USA 2003, 100, 10629–10634. [Google Scholar] [CrossRef] [Green Version]
- Berthiaume, J.M.; Wallace, K.B. Persistent Alterations to the Gene Expression Profile of the Heart Subsequent to Chronic Doxorubicin Treatment. Cardiovasc. Toxicol. 2007, 7, 178–191. [Google Scholar] [CrossRef]
- Smith, R.A.J.; Hartley, R.C.; Cochemé, H.M.; Murphy, M.P. Mitochondrial Pharmacology. Trends Pharmacol. Sci. 2012, 33, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Ouyang, J.; Hu, N.; Li, G.; Wang, H. Protective Effect of Two Alkaloids from Hippophae Rhamnoides Linn. against Doxorubicin-Induced Toxicity in H9c2 Cardiomyoblasts. Molecules 2021, 26, 1946. [Google Scholar] [CrossRef]
- Adachi, K.; Fujiura, Y.; Mayumi, F.; Nozuhara, A.; Sugiu, Y.; Sakanashi, T.; Hidaka, T.; Toshima, H. A Deletion of Mitochondrial DNA in Murine Doxorubicin-Induced Cardiotoxicity. Biochem. Biophys. Res. Commun. 1993, 195, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Lebrecht, D.; Kokkori, A.; Ketelsen, U.P.; Setzer, B.; Walker, U.A. Tissue-Specific MtDNA Lesions and Radical-Associated Mitochondrial Dysfunction in Human Hearts Exposed to Doxorubicin. J. Pathol. 2005, 207, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Fan, W.; Procaccio, V. Mitochondrial Energetics and Therapeutics. Annu. Rev. Pathol.-Mech Dis. 2010, 5, 297–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golforoush, P.A.; Narasimhan, P.; Chaves-Guerrero, P.P.; Lawrence, E.; Newton, G.; Yan, R.; Harding, S.E.; Perrior, T.; Chapman, K.L.; Schneider, M.D. Selective Protection of Human Cardiomyocytes from Anthracycline Cardiotoxicity by Small Molecule Inhibitors of MAP4K4. Sci. Rep. 2020, 10, 12060. [Google Scholar] [CrossRef]
- Liang, X.; Wang, S.; Wang, L.; Ceylan, A.F.; Ren, J.; Zhang, Y. Mitophagy Inhibitor Liensinine Suppresses Doxorubicin-Induced Cardiotoxicity through Inhibition of Drp1-Mediated Maladaptive Mitochondrial Fission. Pharmacol. Res. 2020, 157, 104846. [Google Scholar] [CrossRef]
- Arinno, A.; Maneechote, C.; Khuanjing, T.; Ongnok, B.; Prathumsap, N.; Chunchai, T.; Arunsak, B.; Kerdphoo, S.; Shinlapawittayatorn, K.; Chattipakorn, S.C.; et al. Cardioprotective Effects of Melatonin and Metformin against Doxorubicin-Induced Cardiotoxicity in Rats Are through Preserving Mitochondrial Function and Dynamics. Biochem. Pharmacol. 2021, 192, 114743. [Google Scholar] [CrossRef]
- Yu, J.L.; Jin, Y.; Cao, X.Y.; Gu, H.H. Dexmedetomidine Alleviates Doxorubicin Cardiotoxicity by Inhibiting Mitochondrial Reactive Oxygen Species Generation. Hum. Cell 2020, 33, 47–56. [Google Scholar] [CrossRef]
- Zhao, X.X.; Cho, H.; Lee, S.; Woo, J.S.; Song, M.Y.; Cheng, X.W.; Lee, K.H.; Kim, W. BAY60-2770 Attenuates Doxorubicin-Induced Cardiotoxicity by Decreased Oxidative Stress and Enhanced Autophagy. Chem.-Biol. Interact. 2020, 328, 109190. [Google Scholar] [CrossRef]
- Russo, M.; Guida, F.; Paparo, L.; Trinchese, G.; Aitoro, R.; Avagliano, C.; Fiordelisi, A.; Napolitano, F.; Mercurio, V.; Sala, V.; et al. The Novel Butyrate Derivative Phenylalanine-Butyramide Protects from Doxorubicin-Induced Cardiotoxicity. Eur. J. Heart Fail. 2019, 21, 519–528. [Google Scholar] [CrossRef]
- Zheng, D.; Zhang, Y.; Zheng, M.; Cao, T.; Wang, G.; Zhang, L.; Ni, R.; Brockman, J.; Zhong, H.; Fan, G.C.; et al. Nicotinamide Riboside Promotes Autolysosome Clearance in Preventing Doxorubicin-Induced Cardiotoxicity. Clin. Sci. 2019, 133, 1505–1521. [Google Scholar] [CrossRef]
- Pan, J.A.; Tang, Y.; Yu, J.Y.; Zhang, H.; Zhang, J.F.; Wang, C.Q.; Gu, J. miR-146a Attenuates Apoptosis and Modulates Autophagy by Targeting TAF9b/P53 Pathway in Doxorubicin-Induced Cardiotoxicity. Cell Death Dis. 2019, 10, 668. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Yang, J.; Jiang, L.; Chen, J.; Wang, H. microRNA-29b Regulates the Mitochondria-Dependent Apoptotic Pathway by Targeting Bax in Doxorubicin Cardiotoxicity. Cell. Physiol. Biochem. 2018, 48, 692–704. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Q.; Wei, C.; Zhao, L.; Guo, X.; Cui, X.; Shao, L.; Long, J.; Gu, J.; Zhao, M. miR-378 Modulates Energy Imbalance and Apoptosis of Mitochondria Induced by Doxorubicin. Am. J. Transl. Res. 2018, 10, 3600–3609. [Google Scholar] [PubMed]
- Zhao, L.; Qi, Y.; Xu, L.; Tao, X.; Han, X.; Yin, L.; Peng, J. microRNA-140-5p Aggravates Doxorubicin-Induced Cardiotoxicity by Promoting Myocardial Oxidative Stress via Targeting Nrf2 and Sirt2. Redox Biol. 2018, 15, 284–296. [Google Scholar] [CrossRef]
- Shi, W.; Deng, H.; Zhang, J.; Zhang, Y.; Zhang, X.; Cui, G. Mitochondria-Targeting Small Molecules Effectively Prevent Cardiotoxicity Induced by Doxorubicin. Molecules 2018, 23, 1486. [Google Scholar] [CrossRef] [Green Version]
- Fiedler, L.R.; Chapman, K.; Xie, M.; Maifoshie, E.; Jenkins, M.; Golforoush, P.A.; Bellahcene, M.; Noseda, M.; Faust, D.; Jarvis, A.; et al. MAP4K4 Inhibition Promotes Survival of Human Stem Cell-Derived Cardiomyocytes and Reduces Infarct Size In Vivo. Cell Stem Cell 2019, 24, 579–591. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Ma, Z.; Di, S.; Yang, Y.; Yang, J.; Xu, L.; Reiter, R.J.; Qiao, S.; Yuan, J. AMPK/PGC1α Activation by Melatonin Attenuates Acute Doxorubicin Cardiotoxicity via Alleviating Mitochondrial Oxidative Damage and Apoptosis. Free Radic. Biol. Med. 2018, 129, 59–72. [Google Scholar] [CrossRef]
- Tesoriere, L.; Ciaccio, M.; Valenza, M.; Bongiorno, A.; Maresi, E.; Albiero, R.; Livrea, M.A. Effect of Vitamin A Administration on Resistance of Rat Heart against Doxorubicin-Induced Cardiotoxicity and Lethality. J. Pharmacol. Exp. Ther. 1994, 269, 430. [Google Scholar]
- Doroshow, J.H.; Locker, G.Y.; Ifrim, I.; Myers, C.E. Prevention of Doxorubicin Cardiac Toxicity in the Mouse by N-Acetylcysteine. J. Clin. Investig. 1981, 68, 1053–1064. [Google Scholar] [CrossRef]
- van Dalen, E.C.; Caron, H.N.; Dickinson, H.O.; Kremer, L.C.M. Cardioprotective Interventions for Cancer Patients Receiving Anthracyclines. Cochrane Database Syst. Rev. 2011, 2011, CD003917. [Google Scholar] [CrossRef]
- Myers, C.; Bonow, R.; Palmeri, S.; Jenkins, J.; Corden, B.; Locker, G.; Doroshow, J.; Epstein, S. A Randomized Controlled Trial Assessing the Prevention of Doxorubicin Cardiomyopathy by N-Acetylcysteine. Semin. Oncol. 1983, 10, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.H.; Kim, L.S.; Kim, S.A.; Kim, H.S.; Han, S.J.; Park, W.J.; Choi, Y.J. Evaluation of Short-Term Use of N-Acetylcysteine as a Strategy for Prevention of Anthracycline-Induced Cardiomyopathy: EPOCH Trial—A Prospective Randomized Study. Korean Circ. J. 2013, 43, 174–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dresdale, A.R.; Barr, L.H.; Myers, C.E. Prospective Randomized Study of the Role of N-Acetylcysteine in Reversing Doxorubicin-Induced Cardiomyopathy. Am. J. Clin. Oncol.-Cancer Clin. Trials 1982, 5, 657–663. [Google Scholar] [CrossRef]
- Weitzman, S.A.; Lorell, B.; Carey, R.W. Prospective Study of Tocopherol Prophylaxis for Anthracycline Cardiac Toxicity. Curr. Ther. Res.-Clin. Exp. 1980, 28, 682–686. [Google Scholar]
- Legha, S.S.; Wang, Y.-M.; Mackay, B.; Ewer, M.; Hortobagyi, G.N.; Benjamin, R.S.; Ali, M.K. Clinical and Pharmacologic Investigation of the Effects of Alpha-Tocopherol on Adriamycin Cardiotoxicity. Ann. N. Y. Acad. Sci. 1982, 393, 411–418. [Google Scholar] [CrossRef]
- Elitok, A.; Oz, F.; Ahmet, Y.; Kilic, L.; Ciftci, R.; Sen, F.; Bugra, Z.; Mercanoglu, F.; Oncul, A.; Oflaz, H. Effect of Carvedilol on Silent Anthracycline-Induced Cardiotoxicity Assessed by Strain Imaging: A Prospective Randomized Controlled Study with Six-Month Follow-Up. Cardiol. J. 2014, 21, 509–515. [Google Scholar] [CrossRef] [Green Version]
- Avila, M.S.; Ayub-Ferreira, S.M.; de Barros Wanderley, M.R.; das Dores Cruz, F.; Gonçalves Brandão, S.M.; Rigaud, V.O.C.; Higuchi-dos-Santos, M.H.; Hajjar, L.A.; Kalil Filho, R.; Hoff, P.M.; et al. Carvedilol for Prevention of Chemotherapy-Related Cardiotoxicity: The CECCY Trial. J. Am. Coll. Cardiol. 2018, 71, 2281–2290. [Google Scholar] [CrossRef]
- Huang, S.; Zhao, Q.; Yang, Z.-G.; Diao, K.-Y.; He, Y.; Shi, K.; Shen, M.-T.; Fu, H.; Guo, Y.-K. Protective Role of Beta-Blockers in Chemotherapy-Induced Cardiotoxicity—A Systematic Review and Meta-Analysis of Carvedilol. Heart Fail. Rev. 2019, 24, 325–333. [Google Scholar] [CrossRef] [Green Version]
- Demetrius, L. Aging in Mouse and Human Systems: A Comparative Study. Ann. N. Y. Acad. Sci. 2006, 1067, 66–82. [Google Scholar] [CrossRef]
- Trnka, J.; Blaikie, F.H.; Smith, R.A.J.; Murphy, M.P. A Mitochondria-Targeted Nitroxide Is Reduced to Its Hydroxylamine by Ubiquinol in Mitochondria. Free Radic. Biol. Med. 2008, 44, 1406–1419. [Google Scholar] [CrossRef]
- Skulachev, V.P. Cationic Antioxidants as a Powerful Tool against Mitochondrial Oxidative Stress. Biochem. Biophys. Res. Commun. 2013, 441, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Smith, R.A.J. Targeting Antioxidants to Mitochondria by Conjugation to Lipophilic Cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [Google Scholar] [CrossRef] [PubMed]
- Sacks, B.; Onal, H.; Martorana, R.; Sehgal, A.; Harvey, A.; Wastella, C.; Ahmad, H.; Ross, E.; Pjetergjoka, A.; Prasad, S.; et al. Mitochondrial Targeted Antioxidants, Mitoquinone and SKQ1, Not Vitamin C, Mitigate Doxorubicin-Induced Damage in H9c2 Myoblast: Pretreatment vs. Co-Treatment. BMC Pharmacol. Toxicol. 2021, 22, 49. [Google Scholar] [CrossRef] [PubMed]
- Chandran, K.; Aggarwal, D.; Migrino, R.Q.; Joseph, J.; McAllister, D.; Konorev, E.A.; Antholine, W.E.; Zielonka, J.; Srinivasan, S.; Avadhani, N.G.; et al. Doxorubicin Inactivates Myocardial Cytochrome c Oxidase in Rats: Cardioprotection by Mito-Q. Biophys. J. 2009, 96, 1388–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, V.C.J.; França, L.S.D.A.; de Araújo, C.F.; Ng, A.M.; de Andrade, C.M.; Andrade, A.C.; Santos, E.D.S.; Borges-Silva, M.D.C.; Macambira, S.G.; Noronha-Dutra, A.A.; et al. Protective Effects of Mito-TEMPO against Doxorubicin Cardiotoxicity in Mice. Cancer Chemother. Pharmacol. 2016, 77, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Poon, E.N.Y.; Hao, B.; Guan, D.; Li, J.M.; Lu, J.; Yang, Y.; Wu, B.; Wu, S.C.M.; Webb, S.E.; Liang, Y.; et al. Integrated Transcriptomic and Regulatory Network Analyses Identify microRNA-200c as a Novel Repressor of Human Pluripotent Stem Cell-Derived Cardiomyocyte Differentiation and Maturation. Cardiovasc. Res. 2018, 114, 894–906. [Google Scholar] [CrossRef]
- Poon, E.; Lieu, D.K.; Li, R.A. microRNA and Pluripotent Stem Cell-Based Heart Therapies: The Electrophysiological Perspective. In Heart Rate and Rhythm; Tripathi, O., Ravens, U., Sanguinetti, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 365–383. [Google Scholar]
- Daneri-Becerra, C.; Valeiras, B.; Gallo, L.I.; Lagadari, M.; Galigniana, M.D. Cyclophilin A Is a Mitochondrial Factor That Forms Complexes with P23—Correlative Evidence for an Anti-Apoptotic Action. J. Cell Sci. 2021, 134, jcs253401. [Google Scholar] [CrossRef]
- Xu, C.; Liu, C.H.; Zhang, D.L. microRNA-22 Inhibition Prevents Doxorubicin-Induced Cardiotoxicity via Upregulating SIRT1. Biochem. Biophys. Res. Commun. 2020, 521, 485–491. [Google Scholar] [CrossRef]
- Wang, R.; Xu, Y.; Niu, X.; Fang, Y.; Guo, D.; Chen, J.; Zhu, H.; Dong, J.; Zhao, R.; Wang, Y.; et al. miR-22 Inhibition Alleviates Cardiac Dysfunction in Doxorubicin-Induced Cardiomyopathy by Targeting the Sirt1/PGC-1α Pathway. Front. Physiol. 2021, 12, 646903. [Google Scholar] [CrossRef]
- Chen, J.; Zhong, J.; Wang, L.; Chen, Y. Mitochondrial Transfer in Cardiovascular Disease: From Mechanisms to Therapeutic Implications. Front. Cardiovasc. Med. 2021, 8, 771298. [Google Scholar] [CrossRef]
- Robicsek, O.; Ene, H.M.; Karry, R.; Ytzhaki, O.; Asor, E.; McPhie, D.; Cohen, B.M.; Ben-Yehuda, R.; Weiner, I.; Ben-Shachar, D. Isolated Mitochondria Transfer Improves Neuronal Differentiation of Schizophrenia-Derived Induced Pluripotent Stem Cells and Rescues Deficits in a Rat Model of the Disorder. Schizophr. Bull. 2018, 44, 432–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanmughapriya, S.; Langford, D.; Natarajaseenivasan, K. Inter and Intracellular Mitochondrial Trafficking in Health and Disease. Ageing Res. Rev. 2020, 62, 101128. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of Mitochondria from Astrocytes to Neurons after Stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, C.G.; Ozen, M.O.; Ikeda, G.; Vaskova, E.; Jung, J.H.; Bayardo, N.; Santoso, M.R.; Shi, L.; Wahlquist, C.; Jiang, Z.; et al. Mitochondria-Rich Extracellular Vesicles Rescue Patient-Specific Cardiomyocytes From Doxorubicin Injury. JACC CardioOncol. 2021, 3, 428–440. [Google Scholar] [CrossRef]
- Cowan, D.B.; Yao, R.; Thedsanamoorthy, J.K.; Zurakowski, D.; del Nido, P.J.; McCully, J.D. Transit and Integration of Extracellular Mitochondria in Human Heart Cells. Sci. Rep. 2017, 7, 17450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Li, H.; Yao, Y.; Zhao, T.; Chen, Y.Y.; Shen, Y.L.; Wang, L.L.; Zhu, Y. Stem Cell-Derived Mitochondria Transplantation: A Novel Strategy and the Challenges for the Treatment of Tissue Injury. Stem Cell Res. Ther. 2018, 9, 106. [Google Scholar] [CrossRef] [Green Version]
- Luz-Crawford, P.; Hernandez, J.; Djouad, F.; Luque-Campos, N.; Caicedo, A.; Carrère-Kremer, S.; Brondello, J.M.; Vignais, M.L.; Pène, J.; Jorgensen, C. Mesenchymal Stem Cell Repression of Th17 Cells Is Triggered by Mitochondrial Transfer. Stem Cell Res. Ther. 2019, 10, 232. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, Y.; Yeung, S.C.; Liang, Y.; Liang, X.; Ding, Y.; Ip, M.S.M.; Tse, H.F.; Mak, J.C.W.; Lian, Q. Mitochondrial Transfer of Induced Pluripotent Stem Cell-Derived Mesenchymal Stem Cells to Airway Epithelial Cells Attenuates Cigarette Smoke-Induced Damage. Am. J. Respir. Cell Mol. Biol. 2014, 51, 455–465. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, Z.; Jiang, D.; Liang, X.; Liao, S.; Zhang, Z.; Yue, W.; Li, X.; Chiu, S.M.; Chai, Y.H.; et al. IPSC-MSCs with High Intrinsic MIRO1 and Sensitivity to TNF-α Yield Efficacious Mitochondrial Transfer to Rescue Anthracycline-Induced Cardiomyopathy. Stem Cell Rep. 2016, 7, 749–763. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.Y.; Biswas, S.; Li, J.; Mao, C.J.; Chechneva, O.; Chen, J.; Li, K.; Li, J.; Zhang, J.R.; Liu, C.F.; et al. Human IPSCs Derived Astrocytes Rescue Rotenone-Induced Mitochondrial Dysfunction and Dopaminergic Neurodegeneration in Vitro by Donating Functional Mitochondria. Transl. Neurodegener. 2020, 9, 13. [Google Scholar] [CrossRef]
- Ikeda, G.; Santoso, M.R.; Tada, Y.; Li, A.M.; Vaskova, E.; Jung, J.H.; O’Brien, C.; Egan, E.; Ye, J.; Yang, P.C. Mitochondria-Rich Extracellular Vesicles From Autologous Stem Cell–Derived Cardiomyocytes Restore Energetics of Ischemic Myocardium. J. Am. Coll. Cardiol. 2021, 77, 1073–1088. [Google Scholar] [CrossRef] [PubMed]
- Masuzawa, A.; Black, K.M.; Pacak, C.A.; Ericsson, M.; Barnett, R.J.; Drumm, C.; Seth, P.; Bloch, D.B.; Levitsky, S.; Cowan, D.B.; et al. Transplantation of Autologously Derived Mitochondria Protects the Heart from Ischemia-Reperfusion Injury. Am. J. Physiol.-Heart Circul. Physiol. 2013, 304, H966–H982. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.; Saeed, M.Y.; Esch, J.J.; Guariento, A.; Blitzer, D.; Moskowitzova, K.; Ramirez-Barbieri, G.; Orfany, A.; Thedsanamoorthy, J.K.; Cowan, D.B.; et al. A Novel Biological Strategy for Myocardial Protection by Intracoronary Delivery of Mitochondria: Safety and Efficacy. JACC-Basic Transl. Sci. 2019, 4, 871–888. [Google Scholar] [CrossRef] [PubMed]
- Emani, S.M.; Piekarski, B.L.; Harrild, D.; del Nido, P.J.; McCully, J.D. Autologous Mitochondrial Transplantation for Dysfunction after Ischemia-Reperfusion Injury. J. Thorac. Cardiovasc. Surg. 2017, 154, 286–289. [Google Scholar] [CrossRef] [Green Version]
- Yip, H.K.; Shao, P.L.; Wallace, C.G.; Sheu, J.J.; Sung, P.H.; Lee, M.S. Early Intramyocardial Implantation of Exogenous Mitochondria Effectively Preserved Left Ventricular Function in Doxorubicin-Induced Dilated Cardiomyopathy Rat. Am. J. Transl. Res. 2020, 12, 4612–4627. [Google Scholar]
- Tokudome, T.; Mizushige, K.; Noma, T.; Manabe, K.; Murakami, K.; Tsuji, T.; Nozaki, S.; Tomohiro, A.; Matsuo, H. Prevention of Doxorubicin (Adriamycin)-Induced Cardiomyopathy by Simultaneous Administration of Angiotensin-Converting Enzyme Inhibitor Assessed by Acoustic Densitometry. J. Cardiovasc. Pharmacol. 2000, 36, 361–368. [Google Scholar] [CrossRef]
- Sacco, G.; Bigioni, M.; Evangelista, S.; Goso, C.; Manzini, S.; Maggi, C.A. Cardioprotective Effects of Zofenopril, a New Angiotensin-Converting Enzyme Inhibitor, on Doxorubicin-Induced Cardiotoxicity in the Rat. Eur. J. Pharmacol. 2001, 414, 71–78. [Google Scholar] [CrossRef]
- Hiona, A.; Lee, A.S.; Nagendran, J.; Xie, X.; Connolly, A.J.; Robbins, R.C.; Wu, J.C. Pretreatment with Angiotensin-Converting Enzyme Inhibitor Improves Doxorubicin-Induced Cardiomyopathy via Preservation of Mitochondrial Function. J. Thorac. Cardiovasc. Surg. 2011, 142, 396–403.e3. [Google Scholar] [CrossRef] [Green Version]
- Cardinale, D.; Colombo, A.; Sandri, M.T.; Lamantia, G.; Colombo, N.; Civelli, M.; Martinelli, G.; Veglia, F.; Fiorentini, C.; Cipolla, C.M. Prevention of High-Dose Chemotherapy-Induced Cardiotoxicity in High-Risk Patients by Angiotensin-Converting Enzyme Inhibition. Circulation 2006, 114, 2474–2481. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Chen, Z.; Chen, A.; Fu, M.; Dong, Z.; Hu, K.; Yang, X.; Zou, Y.; Sun, A.; Qian, J.; et al. LCZ696 Improves Cardiac Function via Alleviating Drp1-Mediated Mitochondrial Dysfunction in Mice with Doxorubicin-Induced Dilated Cardiomyopathy. J. Mol. Cell. Cardiol. 2017, 108, 138–148. [Google Scholar] [CrossRef]
- Metra, M.; Nodari, S.; Bordonali, T.; Milani, P.; Fracassi, F.; Dei Cas, L. β-Blocker Therapy of Heart Failure: An Update. Expert Opin. Pharmacother. 2007, 8, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, H.; Ryomoto, T.; Ishikawa, K. Effect of Beta-Blocker on Metabolism and Contraction of Doxorubicin-Induced Cardiotoxicity in the Isolated Perfused Rabbit Heart. Angiology 2000, 51, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Kalay, N.; Basar, E.; Ozdogru, I.; Er, O.; Cetinkaya, Y.; Dogan, A.; Inanc, T.; Oguzhan, A.; Eryol, N.K.; Topsakal, R.; et al. Protective Effects of Carvedilol Against Anthracycline-Induced Cardiomyopathy. J. Am. Coll. Cardiol. 2006, 48, 2258–2262. [Google Scholar] [CrossRef] [Green Version]
- Bosch, X.; Rovira, M.; Sitges, M.; Domènech, A.; Ortiz-Pérez, J.T.; de Caralt, T.M.; Morales-Ruiz, M.; Perea, R.J.; Monzó, M.; Esteve, J. Enalapril and Carvedilol for Preventing Chemotherapy-Induced Left Ventricular Systolic Dysfunction in Patients with Malignant Hemopathies: The OVERCOME Trial (Prevention of Left Ventricular Dysfunction with Enalapril and CaRvedilol in Patients Submitted to Intensive ChemOtherapy for the Treatment of Malignant HEmopathies). J. Am. Coll. Cardiol. 2013, 61, 2355–2362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhorawat, R.; Kumari, S.; Varma, S.C.; Rohit, M.K.; Narula, N.; Suri, V.; Malhotra, P.; Jain, S. Preventive Role of Carvedilol in Adriamycin-Induced Cardiomyopathy. Indian J. Med. Res. 2016, 144, 725–729. [Google Scholar] [CrossRef] [Green Version]
- Kheiri, B.; Abdalla, A.; Osman, M.; Haykal, T.; Chahine, A.; Ahmed, S.; Osman, K.; Hassan, M.; Bachuwa, G.; Bhatt, D.L. Meta-Analysis of Carvedilol for the Prevention of Anthracycline-Induced Cardiotoxicity. Am. J. Cardiol. 2018, 122, 1959–1964. [Google Scholar] [CrossRef]
- Rivera, F.B.; Magalong, J.V.; Sachiko, E.; Chiu, H.H.; Magno, J.D.; John, A. Efficacy of Carvedilol in Preventing Anthracycline-Induced Cardiotoxicity: A Meta-Analysis of Randomized Controlled Trials. J. Card. Fail. 2020, 26, S80. [Google Scholar] [CrossRef]
- Wang, C.Y.; Chen, C.C.; Lin, M.H.; Su, H.T.; Ho, M.Y.; Yeh, J.K.; Tsai, M.L.; Hsieh, I.C.; Wen, M.S. TLR9 Binding to Beclin 1 and Mitochondrial SIRT3 by a Sodium-glucose Co-transporter 2 Inhibitor Protects the Heart from Doxorubicin Toxicity. Biology 2020, 9, 369. [Google Scholar] [CrossRef]
- Liu, G.; Liu, Y.; Wang, R.; Hou, T.; Chen, C.; Zheng, S.; Dong, Z. Spironolactone Attenuates Doxorubicin-Induced Cardiotoxicity in Rats. Cardiovasc. Ther. 2016, 34, 216–224. [Google Scholar] [CrossRef]
- Lother, A.; Bergemann, S.; Kowalski, J.; Huck, M.; Gilsbach, R.; Bode, C.; Hein, L. Inhibition of the Cardiac Myocyte Mineralocorticoid Receptor Ameliorates Doxorubicin-Induced Cardiotoxicity. Cardiovasc. Res. 2018, 114, 282–290. [Google Scholar] [CrossRef]
- Musunuru, K.; Sheikh, F.; Gupta, R.M.; Houser, S.R.; Maher, K.O.; Milan, D.J.; Terzic, A.; Wu, J.C. Induced Pluripotent Stem Cells for Cardiovascular Disease Modeling and Precision Medicine: A Scientific Statement From the American Heart Association. Circ.-Genom. Precis. Med. 2018, 11, e000043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paik, D.T.; Chandy, M.; Wu, J.C. Patient and Disease–Specific Induced Pluripotent Stem Cells for Discovery of Personalized Cardiovascular Drugs and Therapeutics. Pharmacol. Rev. 2020, 72, 320–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gintant, G.; Burridge, P.; Gepstein, L.; Harding, S.; Herron, T.; Hong, C.; Jalife, J.; Wu, J.C. Use of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes in Preclinical Cancer Drug Cardiotoxicity Testing: A Scientific Statement From the American Heart Association. Circ. Res. 2019, 125, e75–e92. [Google Scholar] [CrossRef]
- Kwok, M.; Lee, C.; Li, H.S.; Deng, R.; Tsoi, C.; Ding, Q.; Tsang, S.Y.; Leung, K.T.; Yan, B.P.; Poon, E.N. Remdesivir Induces Persistent Mitochondrial and Structural Damage in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Circ. Res. 2021, cvab311. [Google Scholar] [CrossRef] [PubMed]
- Maillet, A.; Tan, K.; Chai, X.; Sadananda, S.N.; Mehta, A.; Ooi, J.; Hayden, M.R.; Pouladi, M.A.; Ghosh, S.; Shim, W.; et al. Modeling Doxorubicin-Induced Cardiotoxicity in Human Pluripotent Stem Cell Derived-Cardiomyocytes. Sci. Rep. 2016, 6, 25333. [Google Scholar] [CrossRef] [Green Version]
- Louisse, J.; Wüst, R.C.I.; Pistollato, F.; Palosaari, T.; Barilari, M.; Macko, P.; Bremer, S.; Prieto, P. Assessment of Acute and Chronic Toxicity of Doxorubicin in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Toxicol. Vitro 2017, 42, 182–190. [Google Scholar] [CrossRef]
- Poon, E.N.Y.; Luo, X.-L.; Webb, S.E.; Yan, B.; Zhao, R.; Wu, S.C.M.; Yang, Y.; Zhang, P.; Bai, H.; Shao, J.; et al. The Cell Surface Marker CD36 Selectively Identifies Matured, Mitochondria-Rich HPSC-Cardiomyocytes. Cell Res. 2020, 30, 626–629. [Google Scholar] [CrossRef] [Green Version]
- McSweeney, K.M.; Bozza, W.P.; Alterovitz, W.L.; Zhang, B. Transcriptomic Profiling Reveals P53 as a Key Regulator of Doxorubicin-Induced Cardiotoxicity. Cell Death Discov. 2019, 5, 102. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, P.Y.; Long, N.A.; Zhuang, J.; Springer, D.A.; Zou, J.; Lin, Y.; Bleck, C.K.E.; Park, J.H.; Kang, J.G.; et al. P53 Prevents Doxorubicin Cardiotoxicity Independently of Its Prototypical Tumor Suppressor Activities. Proc. Natl. Acad. Sci. USA 2019, 116, 19626–19634. [Google Scholar] [CrossRef] [Green Version]
- Holmgren, G.; Sartipy, P.; Andersson, C.X.; Lindahl, A.; Synnergren, J. Expression Profiling of Human Pluripotent Stem Cell-Derived Cardiomyocytes Exposed to Doxorubicin-Integration and Visualization of Multi-Omics Data. Toxicol. Sci. 2018, 163, 182–195. [Google Scholar] [CrossRef]
- Gupta, S.K.; Garg, A.; Bär, C.; Chatterjee, S.; Foinquinos, A.; Milting, H.; Streckfus-Bomeke, K.; Fiedler, J.; Thum, T. Quaking Inhibits Doxorubicin-Mediated Cardiotoxicity through Regulation of Cardiac Circular RNA Expression Short Communication. Circ.Res. 2018, 122, 246–254. [Google Scholar] [CrossRef]
- Han, D.; Wang, Y.; Wang, Y.; Dai, X.; Zhou, T.; Chen, J.; Tao, B.; Zhang, J.; Cao, F. The Tumor-Suppressive Human Circular RNA CircITCH Sponges miR-330-5p to Ameliorate Doxorubicin-Induced Cardiotoxicity through Upregulating SIRT6, Survivin, and SERCA2a. Circ. Res. 2020, 127, e108–e125. [Google Scholar] [CrossRef]
- Holmgren, G.; Synnergren, J.; Andersson, C.X.; Lindahl, A.; Sartipy, P. microRNAs as Potential Biomarkers for Doxorubicin-Induced Cardiotoxicity. Toxicol. Vitro 2016, 34, 26–34. [Google Scholar] [CrossRef]
- Chaudhari, U.; Ellis, J.K.; Wagh, V.; Nemade, H.; Hescheler, J.; Keun, H.C.; Sachinidis, A. Metabolite Signatures of Doxorubicin Induced Toxicity in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Amino Acids 2017, 49, 1955–1963. [Google Scholar] [CrossRef]
- Burridge, P.W.; Li, Y.F.; Matsa, E.; Wu, H.; Ong, S.G.; Sharma, A.; Holmström, A.; Chang, A.C.; Coronado, M.J.; Ebert, A.D.; et al. Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes Recapitulate the Predilection of Breast Cancer Patients to Doxorubicin-Induced Cardiotoxicity. Nat. Med. 2016, 22, 547–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magdy, T.; Jiang, Z.; Jouni, M.; Fonoudi, H.; Lyra-Leite, D.; Jung, G.; Romero-Tejeda, M.; Kuo, H.H.; Fetterman, K.A.; Gharib, M.; et al. RARG Variant Predictive of Doxorubicin-Induced Cardiotoxicity Identifies a Cardioprotective Therapy. Cell Stem Cell 2021, 28, 2076–2089.e7. [Google Scholar] [CrossRef] [PubMed]
- Poon, E.; Keung, W.; Liang, Y.; Ramalingam, R.; Yan, B.; Zhang, S.; Chopra, A.; Moore, J.; Herren, A.; Lieu, D.K.; et al. Proteomic Analysis of Human Pluripotent Stem Cell-Derived, Fetal, and Adult Ventricular Cardiomyocytes Reveals Pathways Crucial for Cardiac Metabolism and Maturation. Circ.-Cardiovasc. Genet. 2015, 8, 427–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, E.; Yan, B.; Zhang, S.; Rushing, S.; Keung, W.; Ren, L.; Lieu, D.K.; Geng, L.; Kong, C.W.; Wang, J.; et al. Transcriptome-Guided Functional Analyses Reveal Novel Biological Properties and Regulatory Hierarchy of Human Embryonic Stem Cell-Derived Ventricular Cardiomyocytes Crucial for Maturation. PLoS ONE 2013, 8, e77784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, R.E.; Anzai, T.; Chanthra, N.; Uosaki, H. A Brief Review of Current Maturation Methods for Human Induced Pluripotent Stem Cells-Derived Cardiomyocytes. Front. Cell. Dev. Biol. 2020, 8, 178. [Google Scholar] [CrossRef] [Green Version]
- Karbassi, E.; Fenix, A.; Marchiano, S.; Muraoka, N.; Nakamura, K.; Yang, X.; Murry, C.E. Cardiomyocyte Maturation: Advances in Knowledge and Implications for Regenerative Medicine. Nat. Rev. Cardiol. 2020, 17, 341–359. [Google Scholar] [CrossRef]
- Denning, C.; Borgdorff, V.; Crutchley, J.; Firth, K.S.A.; George, V.; Kalra, S.; Kondrashov, A.; Hoang, M.D.; Mosqueira, D.; Patel, A.; et al. Cardiomyocytes from Human Pluripotent Stem Cells: From Laboratory Curiosity to Industrial Biomedical Platform. Biochim. Biophys. Acta-Mol. Cell Res. 2016, 1863, 1728–1748. [Google Scholar] [CrossRef]
- Yang, X.; Rodriguez, M.L.; Leonard, A.; Sun, L.; Fischer, K.A.; Wang, Y.; Ritterhoff, J.; Zhao, L.; Kolwicz, S.C.; Pabon, L.; et al. Fatty Acids Enhance the Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells. Stem Cell Rep. 2019, 13, 657–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Rodriguez, M.; Pabon, L.; Fischer, K.A.; Reinecke, H.; Regnier, M.; Sniadecki, N.J.; Ruohola-Baker, H.; Murry, C.E. Tri-Iodo-l-Thyronine Promotes the Maturation of Human Cardiomyocytes-Derived from Induced Pluripotent Stem Cells. J. Mol. Cell. Cardiol. 2014, 72, 296–304. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Cui, C.; Nan, H.; Yu, Y.; Xiao, Y.; Poon, E.; Yang, G.; Wang, X.; Wang, C.; Li, L.; et al. Graphene Sheet-Induced Global Maturation of Cardiomyocytes Derived from Human Induced Pluripotent Stem Cells. ACS Appl. Mater. Interfaces 2017, 9, 25929–25940. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson-Bouchard, K.; Ma, S.P.; Yeager, K.; Chen, T.; Song, L.J.; Sirabella, D.; Morikawa, K.; Teles, D.; Yazawa, M.; Vunjak-Novakovic, G. Advanced Maturation of Human Cardiac Tissue Grown from Pluripotent Stem Cells. Nature 2018, 556, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, L.; Dai, X.; Tian, Q.; Yu, M.; Agheb, M.; Chan, H.N.; Poon, E.; Guo, Z.; Boheler, K.R.; et al. Facile Formation of a Microporous Chitosan Hydrogel Based on Self-Crosslinking. J. Mat. Chem. B 2017, 5, 9291–9299. [Google Scholar] [CrossRef]
- Gu, X.; Yeung, S.Y.; Chadda, A.; Poon, E.N.Y.; Boheler, K.R.; Hsing, I.M. Organic Electrochemical Transistor Arrays for In Vitro Electrophysiology Monitoring of 2D and 3D Cardiac Tissues. Adv. Biosyst. 2019, 3, e1800248. [Google Scholar] [CrossRef]
- Kamakura, T.; Makiyama, T.; Sasaki, K.; Yoshida, Y.; Wuriyanghai, Y.; Chen, J.; Hattori, T.; Ohno, S.; Kita, T.; Horie, M.; et al. Ultrastructural Maturation of Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes in a Long-Term Culture. Circ. J. 2013, 77, 1307–1314. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Function | Model | Reference |
|---|---|---|---|
| N-acetylcysteine | ↓ Apoptosis | Neonatal rat cardiac myocytes | [80] |
| Alpha-tocopherol | ↓ Apoptosis | ||
| ↓ Oxidative stress | |||
| Ascorbic acid | ↓ Apoptosis | ||
| ↓ Oxidative stress | |||
| Dexrazoxane | ↓ DNA damage | H9c2 cells | [96] |
| ↑ Mitochondrial membrane potential | SD rat derived ventricular myocytes | [95] | |
| ↓ Oxidant production | |||
| ↓ Apoptosis | |||
| DMX-5804 | ↑ Cell viability | hiPSC-cardiomyocytes | [125] |
| ↑ Rescue spontaneous calcium cycling | |||
| Liensinine | ↑ Mitochondrial function | C57BL/6 mice, neonatal and adult mice cardiomyocytes | [126] |
| ↓ ROS level | |||
| ↓ Apoptosis | |||
| ↓ Cardiac dysfunction | |||
| ↓ Mechanics disorder | |||
| ↓ Ca2+ handling dysregulation | |||
| ↓ Mitochondrial fragmentation | |||
| Melatonin/metformin | ↓ Apoptosis | H9c2 cells, Wistar rat | [127] |
| ↓ Autophagy | |||
| ↓ ROS and inflammation | |||
| ↑ Cell viability | |||
| ↑ Mitochondrial function | |||
| ↑ Mitochondrial dynamics | |||
| ↑ Mitochondrial bioenergetics | |||
| Dexmedetomidine | ↓ Mitochondrial ROS level | C57BL/6 mice | [128] |
| ↓ Apoptosis | |||
| BAY60-2770 | ↓ Mitochondrial membrane potential loss ↓ Caspase-3 activation | H9c2 cells | [129] |
| ↓ Mitochondrial ROS | |||
| ↓ Apoptosis | |||
| Phenylala-nine-butyramide | ↑ State 3 and state 4 respiration rates | C57BL/6 mice | [130] |
| ↓ ROS level | |||
| Nicotinamide riboside | ↑ NAD+ level | C57BL/6 mice Mouse cardiomyocytes | [131] |
| ↑ Autolysosome clearance | |||
| ↓ Myocardial dysfunction | |||
| Berberine | ↑ Antioxidases activities | H9c2 cells, SD rat | [50] |
| ↓ Malondialdehyde level | |||
| ↓ Mitochondrial dysfunction | |||
| ↓ Apoptosis | |||
| miR-146a | ↑ Cell viability | AC16 cells | [132] |
| ↑ Autophagy flux | |||
| ↓ Apoptosis | |||
| miR-29b | ↑ Bcl-2 expression | Wistar rat, Rat cardiomyocytes | [133] |
| ↓ Mitochondrial membrane depolarization | |||
| ↓ Cytochrome c release | |||
| ↓ Caspase-3 activity | |||
| ↓ Bax expression | |||
| miR-378 | ↑ Mitochondrial membrane potential | SD rat, neonatal mice cardiomyocytes | [134] |
| ↓ LDH level | |||
| ↓ Apoptosis | |||
| miR-140-5p | ↑ Morphological damage | H9c2 cells, C57BL/6 J mice | [135] |
| ↑ ROS level | |||
| ↑ Creatine kinase and LDH levels | |||
| ↓ Superoxide dismutase level |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, B.B.; Leung, K.T.; Poon, E.N.-Y. Mitochondrial-Targeted Therapy for Doxorubicin-Induced Cardiotoxicity. Int. J. Mol. Sci. 2022, 23, 1912. https://doi.org/10.3390/ijms23031912
Wu BB, Leung KT, Poon EN-Y. Mitochondrial-Targeted Therapy for Doxorubicin-Induced Cardiotoxicity. International Journal of Molecular Sciences. 2022; 23(3):1912. https://doi.org/10.3390/ijms23031912
Chicago/Turabian StyleWu, Bin Bin, Kam Tong Leung, and Ellen Ngar-Yun Poon. 2022. "Mitochondrial-Targeted Therapy for Doxorubicin-Induced Cardiotoxicity" International Journal of Molecular Sciences 23, no. 3: 1912. https://doi.org/10.3390/ijms23031912
APA StyleWu, B. B., Leung, K. T., & Poon, E. N.-Y. (2022). Mitochondrial-Targeted Therapy for Doxorubicin-Induced Cardiotoxicity. International Journal of Molecular Sciences, 23(3), 1912. https://doi.org/10.3390/ijms23031912