1. Introduction
Sarcoid is one of the most common skin tumor types in equids. It does not belong to the metastasizing tumors but is considered to be locally invasive [
1,
2,
3]. Moreover, the high severity rates that have been found to result from sarcoid-dependent oncogenic transformation of epidermal and dermal tissues seem to be low. However, sarcoids contribute to lowering the value of the animal and the overall deterioration of the animal’s welfare by occurring mainly in places exposed to movement [
2]. This location exposes the possibility of mechanical damage, which can lead to transformations of minor forms into severe forms characterized by ulceration [
2].
So far, it has been possible to link the presence of sarcoids with the infection of bovine papillomavirus types 1 and 2 (BPV-1, BPV-2) and, less frequently, type 13 (BPV-13), which has been confirmed at the DNA, miRNA, and protein levels [
3,
4,
5]. These viruses belong to a species-specific family of
Papillomaviridae, which means they can only infect specific species of animals. BPV is so far the only documented case of a natural species barrier breach [
6]. In cattle, it typically attacks the differentiated cells stemming from epithelial and connective tissues such as epidermal keratinocytes and dermal fibroblasts [
6,
7], causing mainly skin lesions in the form of papillomas, warts, and various neoplasms [
8]. In the family
Equidae, the infection does not produce new virus particles [
9], but leads to changes in the level of gene expression, leading to neoplastic changes.
The genome of
Papillomaviridae is highly conserved. It consists of seven early genes (
E1–E7), two late genes (
L1 and
L2), and a noncoding region (NCR, also known as the upstream regulatory region or URR) containing regions that control viral replication. The early genes are responsible for the replication activities of the virus (
E1), the regulation of transcription (
E2), and the coding of individual viral proteins (cytoplasmic—
E3, and transforming—
E4–
E7). Late genes encode viral capsid proteins [
10,
11]. Among the transforming proteins, the
E5–E7 proteins are found in the genomes of all known carcinogenic viruses [
12].
There are many known treatment methods for dealing with sarcoids. Unfortunately, some of them are only effective for a specific type of sarcoid and only for a specific tumor site (such as the BCG vaccine) [
13]. Surgical methods have a high probability (up to 30%) of the disease’s recurrence in a more aggressive form [
14]. There are also methods with good prognosis, but due to the need to apply them directly to the skin lesion, they can be very painful; also some sites, like ears, are more sensitive, which requires general anesthesia in certain cases [
15]. For this reason, further efforts are needed to develop new treatments for this condition, which could be amended by the development of new models that can study that neoplasm at the molecular level.
The molecular mechanisms underlying sarcoid-dependent neoplastic transformation are not yet fully understood. Previous studies have focused on the analysis of differences in gene expression between sarcoid and normal skin tissues, comparing the transcriptional activities of genes in the cell lines established from these tissues [
16,
17]. Some studies have aimed to devise in vitro models of murine fibroblast-derived cancerous cell lines generated by transfection with
BPV transgenes or to create mouse models of dermal sarcoid-related neoplasia [
18]. However, so far there have been no studies that target the development of an ex vivo model of sarcoid-dependent tumorigenesis in equine adult cutaneous fibroblasts cell (ACFC) lines, whose oncogenic transformation has been accomplished by transfection with
BPV fusion genes. Moreover, there has been a lack of data confirming which virus genes are responsible for the neoplastic transformation of ACFCs into dermal sarcoid-like cells. That model could contribute to a more comprehensive understanding of the molecular changes in equine ACFCs undergoing sarcoid-related cancerogenesis due to viral infection.
Multifaceted transcriptomic characterization of mitotically stable cancerous cell lines stemming from
BPV-E4 and
BPV-E4^E1 transgenic equine ACFCs that have undergone nucleofection-mediated neoplastic transformation into nonmalignant sarcoid-like tumor cells is a sine qua non for accomplishing somatic cell cloning. The use of ACFC-derived sarcoid-like cells as a completely new source of nuclear donor cells (NDCs) to create equine cloned embryos and progeny by somatic cell nuclear transfer (SCNT) has not yet been realized. On the other hand, efforts by Li et al. [
19] and Shao et al. [
20] have confirmed that successful transcriptional reprogramming and molecular dedifferentiation of genomes inherited from NDCs that originated, respectively, from such highly metastatic neoplasms as cerebellum-specific medulloblastoma and breast cancer have sustainably contributed to promoting the epigenetically controlled remission of their typically cancerous markers and malignancy-related attributes in cloned mouse embryos.
To the best of our knowledge, transgenization and simultaneous co-transfection of the ex vivo expanded equine ACFCs that have been created by nucleofection according to the approaches formerly devised and adapted by Skrzyszowska et al. [
21] and Samiec et al. [
22] to generate genetically modified cloned pig embryos have not yet been reported. Additionally, the aforementioned strategies have been applied, for the first time, to research targeted at not only cell culture engineering but also experimental and preclinical attempts, with the use of in vitro transgenic models designed to examine the molecular nature of sarcoid-dependent oncogenic transformation (carcinogenesis) of equine ACFCs. These extracorporeal models have also been developed to explore the genetic and epigenetic determinants of procancerous tumorigenesis of epidermal and dermal provenance in horses and phylogenetically consanguineous taxa (i.e., other equids).
Furthermore, it is also noteworthy that, thus far, approaches focused on utilizing BPV-E4 and BPV-E4^E1 transgenic ACFC derivatives, which have undergone oncogenic transformation into sarcoid-like cells as a result of nucleofection, have been conceptualized for the needs of SCNT-based cloning in horses and a variety of members of Equidae family for the first time. For all these reasons, elaborating the abovementioned approaches seems to be strongly justified by the scientific thesis assuming profound amelioration of epigenomic plasticity in the ex vivo immortalized nonmalignant cancerous derivatives of ACFCs, which are characterized by an unlimited lifespan. This, in turn, might result in the enhanced susceptibility of genetically modulated ACFC-derived sarcoid-like cell nuclei to being epigenomically dedifferentiated and transcriptionally reprogrammed in equine cloned embryos generated by SCNT-mediated assisted reproductive technologies (ARTs).
Therefore, in our current investigation, efforts were undertaken to generate equine ACFC lines that had been genetically transformed into sarcoid-like cells as a result of their nucleofection with BPV transgenes encoding recombinant representatives of the transforming protein family, designated as either BPV-E4 or BPV-E1^E4. Our study also sought to thoroughly unravel the modifications arising in genomic signatures that have incurred sarcoid-dependent alterations in transcriptomic profiles of horse ACFC-derived neoplastic cells.
3. Discussion
To date, there is still limited information about equine sarcoid genetics as well as about the etiology of sarcoids’ occurrence at the molecular level. The identification of such mechanisms related with neoplasia formation seems to be critical for prophylaxis or future treatment. Little research has been done comparing the sarcoid cell transcript with that of healthy horse skin cells. These studies were mainly performed on microarrays, so they were limited by the selected panel of genes [
16]. Transfected horse skin cells are proposed as a new model to conduct research on the effect of individual viral genes on changes inside the cell. In this study, we compared the overall transcriptome using the RNA-seq technique, which enabled the detection of DEGs on a much larger scale. We tried to answer the question of which viral genes affect the cell transcriptome, directing changes toward neoplastic transformations, and how. So far, a model of transfected skin cells has been developed in sarcoid research, but it included transfected mouse skin cells [
18]. Moreover, this model was not conducted fully in vitro due to the introduction of altered cells into living organisms. An additional advantage of the present research was using two variants of the studied transcript. Such an approach made it possible to approximate the functions performed by the added fragment of the E1 protein. So far, it has been argued that the effect of the papillomavirus E4 protein is mainly related to viral replication by controlling cellular processes towards the return of differentiated cells to the cell cycle [
25,
26,
27]. Our research has demonstrated that the presence of the BPV-E1^E4 protein also influences changes in the expression of other host cell genes and may play a role in carcinogenesis.
Following
BPV-E4 transgene-mediated nucleofection of equine ACFCs, a total of 1640 DEGs were identified, out of which 62% were found to be downregulated and 38% upregulated. In contrast, after
BPV-E1^E4 transgene-dependent neoplastic transformation of ACFCs into sarcoid-like cells, 3328 genes were detected, out of which 51% were shown to be downregulated and 49% upregulated. This confirms the ratio of downregulated genes to upregulated genes obtained in the microarray studies performed by Semik et al. [
16] and in other cancers such as pancreatic cancer, cervical cancer, and renal cancer [
28,
29,
30]. Attention should be paid to the differences in deregulated genes depending on the type of insert introduced. In the case of the fragment encoding the BPV-E4 protein alone, genes deregulated also by the splicing protein BPV-E1^E4 accounted for 55%, while genes common to both inserts accounted for 27% of all deregulated genes. Therefore, the conclusion is drawn that the presence of the BPV-E1 protein fragment strongly influences the change of the protein function in the process of neoplasia formation, not only by enabling modification of the expression level of new genes but also by inhibiting the deregulation of gene expression occurring in the case of the direct product of the
BPV-E4 gene.
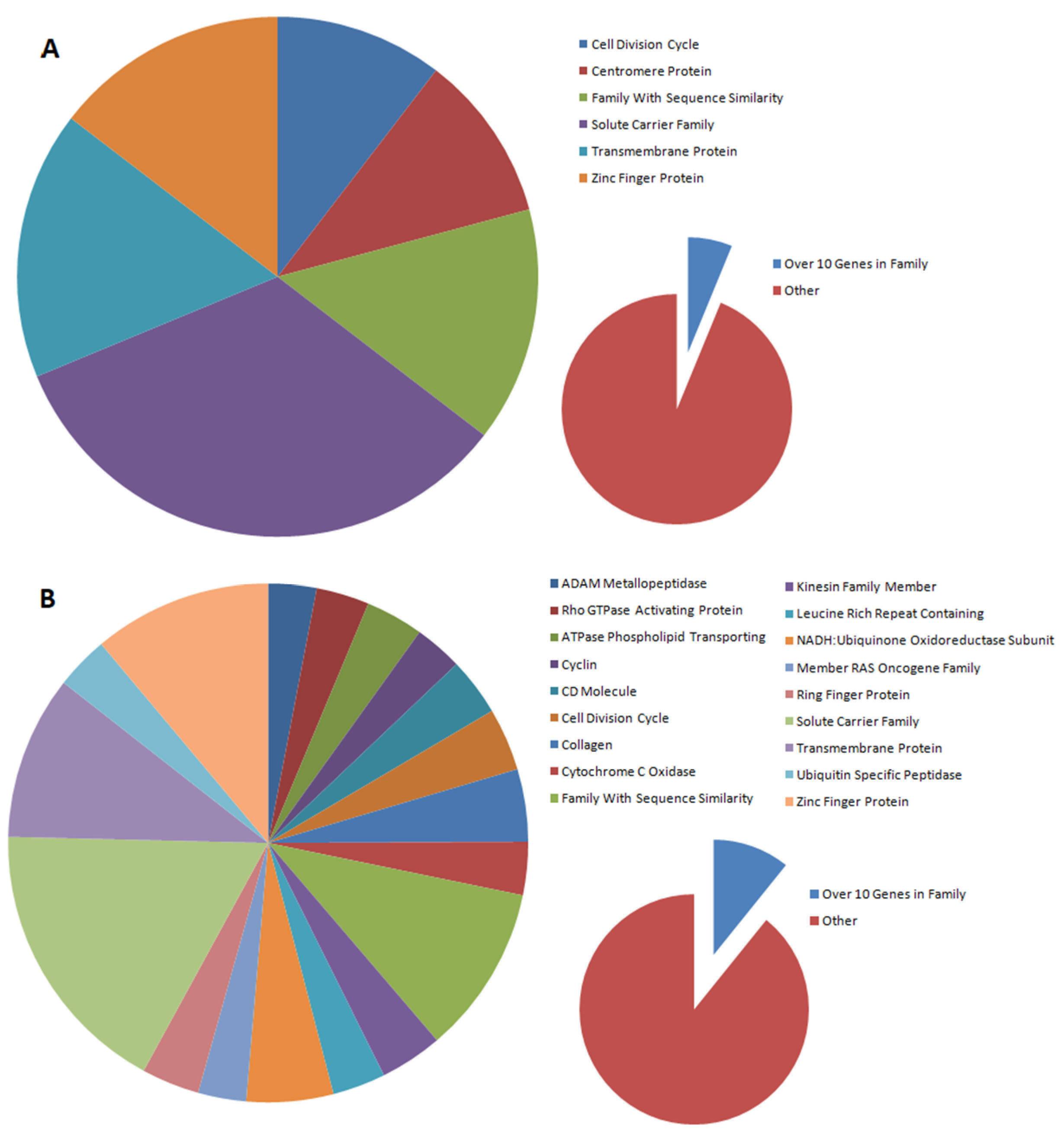
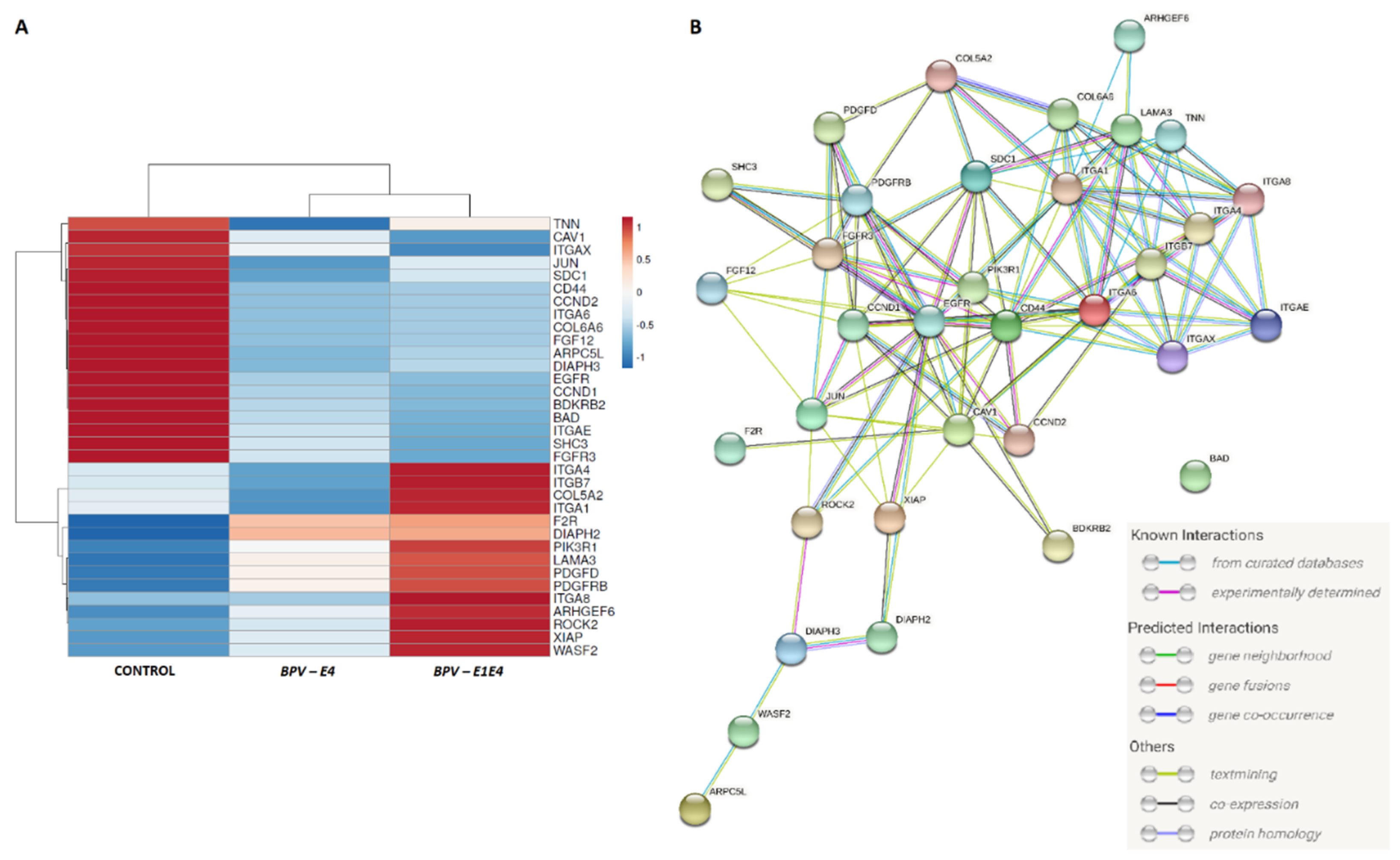
The distribution of gene families that are differentially expressed between nontransfected and transfected cells depends on the type of gene introduced. In our studies, gene families were selected in which at least 10 genes were subject to altered expression. In the case of the introduction of the BPV-E4 gene alone, six gene families were observed to be differentially expressed (cell division cycle, centromere protein, family with sequence similarity, solute carrier family, transmembrane protein, and zinc finger protein), while for the spliced insert, three times as many families were detected (ADAM metallopeptidase, Rho GTPase-activating protein, phospholipid-transporting ATPase, cyclin, CD molecule, cell division cycle, collagen, cytochrome c oxidase, family with sequence similarity, kinesin family member, leucine-rich repeat-containing protein, NADH:ubiquinone oxidoreductase subunit, member of RAS oncogene family, ring finger protein, solute carrier family, transmembrane protein, ubiquitin-specific peptidase, and zinc finger protein). Moreover, all families (with the exception of centromere proteins) designated for the BPV-E4 insert were also among the families designated for the BPV-E1^E4 insert. This may indicate that, despite the differences in DEGs, the major pathways regulated by this protein are not altered by the introduction of the BPV-E1 protein fragment to cells, but the number of such families is increasing.
Interesting results were obtained based on the Gene Ontology analysis. The function of the BP virus E4 protein is related to the reintroduction of the host cells into the cell cycle [
27]. For the
BPV-E4 insert, 34 DEGs were identified for GO regulation of cell proliferation, 11 DEGs for chromosome segregation, nine DEGs for the mitotic spindle assembly, and eight DEGs for the mitotic cytokinesis that can be associated with this function. However, we cannot unequivocally determine whether the differences noticed in the transcriptomic profiles exert a negative or positive effect on the host cell cycle. Our research also showed that a similar number of statistically significant changes in GO were seen for the DEGs associated with cell migration processes such as positive regulation of cell migration (24 DEGs), cell adhesion and cell migration (21 DEGs respectively), and cell–matrix adhesion (15 DEGs). This may indicate that the reasons for the nonmetastatic nature of the sarcoid [
2] can be found in the analyzed gene. Among the DEGs belonging to the changed GOs, the protein families of integrins and kinesin superfamily proteins had the largest share. The high proportion of integrins may indicate the neoplasmic nature of the BPV-E4 protein. Changes in the expression level of integrins have been associated with various cancers. They act as a factor controlling the migration capacity of altered cells via extracellular matrix (ECM) remodeling and modification of cell–ECM interaction [
31,
32,
33]. It has been established that integrin also plays a key role in the regulation of cancer progression through involvement in the regulation of cancer stem cells, metastasis, tumor angiogenesis, and metabolism [
33]. In turn, kinesin superfamily proteins are involved in transporting many intracellular components. Additionally, they are involved in cell division and are responsible, among other things, for the assembly of microtubule spindles and the separation of chromosomes. Their expression is tightly regulated and its disturbance can lead to increased (in the case of upregulation) or decreased (downregulation) cell proliferation [
34].
In the case of the
BPV-E1^E4 insert, the largest number of DEGs were involved in focal adhesion, with 99 genes in total, and nearly twice as many genes were upregulated. This may indicate a high involvement of the BPV-E1^E4 fusion protein in the processes related to cells migration. Another significant GO was the negative regulation of the canonical Wnt signaling pathway. Deregulation of this pathway is associated with the formation and metastasis of numerous cancers, such as colorectal cancer, breast cancer, and ovarian cancer [
35]. An example that can be drawn from our present study is the overexpression of the
SOX9 gene, which is considered a tumor progression factor [
36]. The results of the research by Aldaz et al. [
36] proved that an increased level of
SOX9 can promote tumor cell proliferation in both in vitro and in vivo models throughout
BMI1 activation and
p21 inhibition. The study by Xue et al. [
37] pinpointed the strong influence of the overexpression of
SOX9 on breast cancer stem cells, while in the report by Ma et al. [
38],
SOX9 was designated as a “master regulator” of the processes encompassing the survival and metastasis of breast cancer cells [
38].
Additionally, GOs, whose deregulation is associated with carcinogenic processes, such as apoptosis, and which are classified as related to hallmarks of cancer [
39], and the
TGF-β (transforming growth factor-β) receptor (
TGFBR) signaling pathway, have been shown to be significant. The
TGF-β gene is considered to be one of the most potent regulators of cell proliferation (usually negative), and it can also function as a promoter of the metastasis of TGF-β-resistant tumor cells [
40]. Several previous reports indicated that upregulation of the
TGFβ1 gene is characteristic during tumorigenesis and can promote cell motility and migration [
41,
42]. Moreover, in vitro studies by Zhou et al. [
43] have confirmed that the transfection of neoplastic (adenocarcinoma) cells derived from colonic and rectal epithelial cells (enterocytes) with the use of a
pCMV5-TGFBR1*6A-HA gene construct brings about TGFBR1*6A (type 1 transforming growth factor β receptor)-induced activation of the p38 MAPK pathway, followed by expedited and highly malignant oncogenic modulation of these colorectal tumor cells. This, in turn, gives rise to an enhancement of the ex vivo capabilities of colorectal cancer cells to grow unchecked, invade less invasive or noninvasive subpopulations of intestinal adenocarcinoma cells, and metastasize from primary malignant lesions (i.e., primary tumor sites) to other locations (the so-called metastatic foci) of the transgenic cell culture engineering model [
43]. The upregulation of the
TGFBR gene, which was observed in our study due to the
BPV-E1^E4 transgene-mediated oncogenic transformation of equine ACFCs into sarcoid-like cells, may also indicate a pivotal role of TGF-β receptors in the onset and progression of the processes responsible for the migration and metastasis of neoplastic cells. The other upregulated gene, which represents the GOs related to
TGF-β receptors, is the
c-Fos proto-oncogene, widely recognized as one of the most important predictors determining carcinoma’s progression [
44]. The exact role of the
FOS gene in tumorigenesis and metastasis is still unclear, but the overexpression of this gene has been hypothesized to trigger tumorigenesis and, thereby, has been potentially found to be a poor prognostic factor for oncology patients. The increased expression of the
FOS gene can trigger the
VEGF (vascular endothelial growth factor) and enhance
NANOG and
c-myc genes in head and neck squamous cell carcinoma [
45]. On the other hand, downregulation of the
Fos proto-oncogene can be related to tumor suppression [
46].
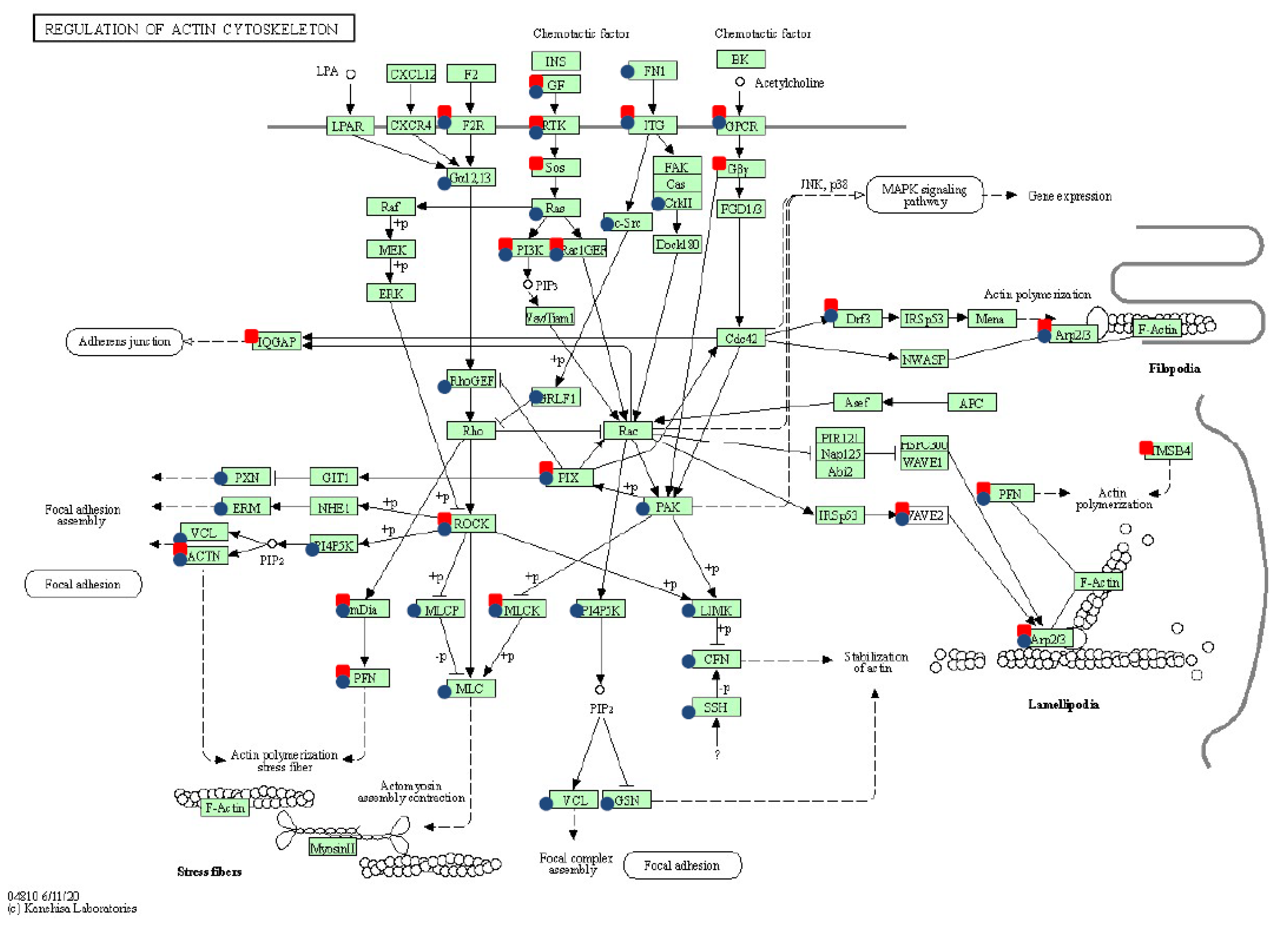
The whole transcriptome’s modification under both transfection types showed the significant overexpression of genes involved in pathways related to cytoskeleton and ECM–matrix remodeling: regulation of actin cytoskeleton, focal adhesion, and ECM–receptor interaction. These results confirmed previous findings that in cancer the ECM matrix is subject to dynamic changes that reflect progression and metastasis [
47,
48]. Together with collagens and laminins, ECM matrix modification stimulated cancer cell activity and tumor progression [
49,
50]. The present study allowed us to identify the differential expression of collagens, laminin, and integrins. The detected significantly enriched GO was due to the collagen fibril organization. The collagen family is the most exposed DEG family in this analysis, in contrast to the analysis performed for the BPV-E4 protein. The integrin and kinesin superfamily proteins had the largest share. Moreover, both
BPV-E4- and
BPV-E1^E4-mediated nucleofection of equine ACFCs brought about the upregulation of
FGFR3 and
FGF12 (fibroblast growth factor receptor 3 and fibroblast growth factor 12), which are known as factors promoting tumor growth and metastasis [
51].
Surprisingly, we have also observed differential expression of the
F2R gene (encoding coagulation factor II thrombin receptor), which, according to the literature, can stimulate the migration and invasion of cancer cells under
SOX9 influence [
52]. The other gene upregulated in nucleofected equine ACFCs was
ROCK2 (Rho associated coiled-coil containing protein kinase 2). Kaczorowski et al. [
53] indicated that both
ROCK1 and
ROCK2 genes can be critical for controlling cellular motility and cancer invasiveness, while the inhibition of
ROCK2 decreased the tumor growth based on the osteosarcoma model [
54].
The equine ACFCs nucleofected with the
BPV-E1^E4 gene construct displayed significant deregulation of s higher number of pathways than
BPV-E4 transgenic ACFCs, such as the FoxO signaling pathway, the PI3K-Akt and
TNF signaling pathways, and Proteoglycans in cancer. The study by Semik et al. [
16], which was focused on transcriptome differences between sarcoid and healthy skin tissues, showed significant over-representation of genes belonging to the PI3K-Akt signaling pathway, pathways in cancer, and cytokine–cytokine receptor interaction. Moreover, the abovementioned authors have observed differences in the expression of genes involved in actin cytoskeleton regulation, tight junction, and cell adhesion. Genes with differential expression such as
FGFR2 and
FGF10 have been identified in healthy and sarcoid-related tissues [
16] and, analogously, in both
BPV-E1^E4 transgenic ACFCs and their control, nontransgenic counterparts. Similar to in the present study, healthy skin and sarcoids were characterized by differences with regard to collagens, integrins, and tubulin genes, which can affect the cytoskeleton arrangement and cell mobility [
16].
Interestingly, in the current in vitro study, we noticed the significant downregulation of the
IGF2 (insulin-like growth factor 2) and
EGFR1 (epidermal growth factor receptor 1) genes. Such results are in contrast to the literature data, which showed overexpression of both genes in different types of cancers. The increased expression of the
IGF2 gene is strongly associated with a poor prognosis via stimulating cell proliferation [
55]. Similarly, upregulation of the
EGFR1 gene, which is closely related to the tumor stage [
56], and its overexpression means a poor prognosis of clinical outcome [
57]. The low expression of both genes is characteristic for normal cells, but not for their neoplastic counterparts. Nonetheless,
EGFR1 can be downregulated by different factors such as decorin [
58] or ubiquitin-specific peptidase 8 (
UBPY) [
59]. Such a mechanism aims to terminate cell proliferation and stop uncontrolled cell growth, which contributes to carcinogenesis. On the other hand, we observed the significant upregulation of the insulin receptor gene (
ISNR), which is responsible for stimulation of tumor cell proliferation, migration, and invasion [
60], and can be co-expressed with the
EGFR gene in tumors [
61]. Overexpression of
INSR is correlated with a poor prognosis for oncology patients [
62] and can be used as an early tumor-related marker [
63]. However, it should be highlighted that, in this study, the effect of only one gene of the
BP virus is investigated. The aforementioned differences in the achieved gene expression levels may result from the lack of interaction with other viral proteins. Thus, neoplastic changes may occur differently.
To sum up, the results of the current investigation have confirmed that BPV-E4- and BPV-E1^E4-mediated nucleofection significantly affected transcriptomic alterations, leading to sarcoid-like neoplastic transformation of equine ACFCs. Nevertheless, the changes in transcriptomic signatures arising in the cells nucleofected with BPV-E1^E4 fusion genes increasingly tended to resemble those that occurred in vivo in equine sarcoids. This finding may be justified by the onset and progression of modifications in crucial signaling pathways such as PI3K-Akt-mediated signal transduction pathway and a variety of closely related pathways. For this reason, we propose the strategy based on transgenically induced expression of BPV-E1^E4 fusion protein as a completely new ex vivo model of sarcoid-dependent oncogenic transformation in equine ACFCs. This biomedical model can be used not only to more comprehensively explore and decipher the molecular nature of dermal sarcoid-like neoplasia, but also to preclinically or clinically predict the directions and targets of anticancer therapies in specimens afflicted with epidermal and dermal sarcoids.
4. Materials and Methods
4.1. Experimental Schedule
The experimental protocol (as depicted in
Figure 7) was divided into three main steps: (1) designing and preparing the transgene sequences to be expressed in equine ACFCs; (2) nucleofection-mediated neoplastic transformation of ACFCs prompted by
BPV-E4 and
BPV-E1^E4 transgenes; and (3) analysis of transcriptome changes in
BPV-E4 and
BPV-E1^E4 transgenic cells.
In the first series of experiments, the gene sequences were designed based on information available in the biological database PaVe [
65]. The sequences were cloned by the manufacturer (GeneArt Gene Synthesis, Thermo Scientific, Waltham, MA, USA) into pMA-T plasmids, from which they were cut out with appropriately selected restriction enzymes. The excised sequences were cloned into expression plasmids (T-REx System, Invitrogen, Waltham, MA, USA, Thermo Scientific). The plasmids were propagated in competent
Escherichia coli bacteria (strain DH5α; Invitrogen).
In the second series of experiments, the sarcoid-dependent genetic transformation of ACFCs was induced by nucleofection with the use of BPV-E4 and BPV-E1^E4 transgenes. Positively transformed nucleofectants that had acquired combined immune resistance to a cocktail of select antibiotics were expanded ex vivo and subsequently assigned to a further series of experiments.
In the third series of experiments, in order to perform a transcriptome analysis, RNA samples were isolated from the control (nontransgenic) and BPV-E4 and BPV-E1^E4 transgenic cell lines, from which cDNA libraries were then derived. The assessment of transcriptomic profiles was accomplished by next-generation sequencing (NGS) on an Illumina apparatus (Illumina, San Diego, CA, USA).
4.2. Designing Gene Inserts for Further Experiments Aimed at Nucleofection of Equine ACFCs
The inserted sequence of the
BPV-E4 gene was designed on the basis of the information available in the papillomavirus database, PaVe [
65]. The sequence of the analyzed gene was designed with two variants. The first variant was based on the amino acid sequence of the
BPV1-E4 protein (gi 60965.E4) transcribed into the sequence encoding a given gene. The second variant was based on the amino acid sequence of the
BPV-E4 protein, including the amino acid sequence of the
BPV-E1 protein fragment (gi 60965), which more closely corresponds to the actual structure of the
BPV-E4 protein in vivo. In addition, the sequences were flanked with amino acid sequences characteristic of restriction enzymes (two different enzymes for each insert) enabling the creation of sticky ends. The enzymes were selected based on the MCS sequence of the plasmids of the target inserts (
pcDNA4/TO/myc-his/B; T-REx System; Invitrogen) in such a way that the sequence ends they formed were not complementary. Such selection of enzymes prevented the formation of circular structures inside the enzymatic digestion products and also ensured that the insert sequence was placed in the correct direction concerning the target plasmid sequence. Additionally, the sequences of the inserts were enriched with the consensus KOZAK sequence (gccgccaccatgg).
4.3. The Reactions of Enzymatic Restriction and Ligation
The insert sequences provided by the manufacturer were cloned into pMA-T plasmids, from which they were excised using the restriction enzymes included in the design process. The reaction mixture contained a DNA template in the form of a plasmid containing the appropriate gene and a set of specific enzymes along with a buffer matched to them (for the gene: BPV-E4-AflII, KpnI, buffer 2.1; BPV-E1^E4-SacII, AflII, Cut Smart buffer; New England BioLabs, Ipswich, MA, USA), and digestion was carried out overnight. In addition, pcDNA plasmids from the T-REx system set (pcDNA4/TO/myc-His/B; Invitrogen) were also digested by restriction enzymes corresponding to the individual sequences of the inserts. The amount of template DNA was estimated to obtain 400 ng of the actual product (cut insert sequence or linear plasmid), which corresponds to the maximum amount of DNA that could be used in one sample during the gel purification method, made in the next step.
Digestion products were separated with agarose gel electrophoresis (0.8% low melting point agarose in TBE buffer, 80 V, until DNA band separation). The DNA band containing the viral gene sequence (BPV-E4 or BPV-E1^E4) or a linear form of the digested plasmid was cut from the gel (ethidium bromide-mediated staining) and purified with a High Pure PCR Product Purification Kit (Roche, Warsaw, Poland). Purified DNA was eluted in the manufacturer’s buffer, heated to 56 °C.
The obtained fragments of the corresponding gene variants were combined with the
pcDNA 4/TO/myc-His/B plasmid in a mass ratio of 3:1. The required volumes of individual DNA were calculated using an online calculator [
66]. According to the manufacturer’s guidelines, the reaction was performed with a Rapid DNA Ligation Kit (Roche, Basel, Switzerland).
4.4. Molecular Cloning of DNA Plasmid Constructs with Inserted BPV-E4 or BPV-E1^E4 Gene Sequences
Plasmids were cloned with Subcloning Efficiency DH5α Competent Cells (Invitrogen). Ten nanograms of plasmid DNA were introduced into bacteria by the heat-shock method. After the addition of DNA, the bacteria were held at 42 °C for 20 s after 30 min of incubation on ice, and then the bacteria were put on ice again for 2 min. Transformed bacteria were incubated in 1 mL low-salt Luria-Bertani Broth (Sigma-Aldrich, Merck Life Sciences, Poznań, Poland) for 1 h at 225 rpm and 37 °C. The bacteria were seeded on a low-salt LB broth with the addition of agarose (Sigma-Aldrich) and 120 µg/mL ampicillin (Gibco, Thermo Scientific), which served as a selective antibiotic, and incubated overnight at 37 °C. The genetically transformed bacterial cells that had been found to display immune resistance to ampicillin were positive for the occurrence of plasmid DNA. Obtained bacterial colonies were tested for positive recombination with Quick Screen PCR. Fragments of picked bacterial colonies were suspended in 15 µL of 0.1% Triton X-100 (Sigma-Aldrich) in TE buffer, then incubated in 100 °C for 5 min and centrifuged (13,000× g; 10 min). The supernatant was sequenced (Sanger method; Genetic Analyzer XL, Applied Biosystems, Thermo Fisher Scientific) to confirm the presence and quality of plasmids. Bacterial colonies that were positive for plasmid presence were grown for 14 h at 37 °C (250 rpm) in 100 mL of low-salt LB broth (Sigma-Aldrich) enriched with 120 µg/mL ampicillin. Suspended bacteria were centrifuged (4500× g; 20 min; 4 °C) followed by removal of supernatants. Plasmid DNA was isolated with a Qiagen Plasmid Midi Kit (Qiagen, Wroclaw, Poland), according to the manufacturer’s protocol. Plasmid DNA was eluted in 50 mL of TC-treated water.
4.5. Establishment of Primary Cultures and Mitotically Stable Lines of Equine Adult Cutaneous Fibroblast Cells (ACFCs)
Adult skin tissue-derived biopsies (n = 4) were collected postmortem from the lower eyelid regions of horses slaughtered in the local abattoir. Dermal tissue samples were deposited into tubes filled with Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco), HEPES (Gibco) and primocin (InvivoGen, Alab, Warsaw, Poland). Tubes were stored at 4 °C for no longer than 24 h after the recovery of cutaneous tissue explants.
Primary cell cultures were generated according to the modified procedures described in the study by Tomasek et al. [
67]. Briefly, dermal tissue samples were disinfected with 70% ethanol and washed thrice in a 1× solution of Dulbecco’s phosphate-buffered saline (DPBS; pH 7.2; Gibco), followed by cutting into smaller pieces (approximately 2 mm × 2 mm), which were placed into cell culture flasks containing DMEM (Gibco) enriched with 10% FBS and primocin. Cutaneous tissue fragments were incubated at 37 °C in an atmosphere of 5% CO
2 and 100% humidity for two weeks, until the cells spontaneously migrated from the tissue explants. The culture medium was changed two times per week and passages were performed immediately after the ex vivo proliferating cells had reached 90% confluence. The first passages were characterized by the presence of epidermal keratinocytes in culture. Therefore, during the passaging procedure, the cells were trypsinized until the adherent equine adult cutaneous fibroblast cells (ACFCs) were efficiently detached. Keratinocytes, as less detachable epidermal cells [
68], were still attached to the bottom of the culture dishes. For that reason, these cell subpopulations have not been replated. The homogenous ACFC lines, in the subpopulations of which the disappearance of epidermal keratinocytes was clearly confirmed, were successfully established at the third passage.
4.6. Genetic Transformation of Equine ACFCs Mediated by Nucleofection
The approaches that were applied both to prepare the equine ACFCs prior to nucleofection and to nucleofect them were accomplished according to the methods used for the transgenization of porcine dermal fibroblast cells (NDCs for SCNT), as comprehensively described in studies by Skrzyszowska et al. [
21] and Samiec et al. [
22]. Briefly, the ex vivo expanded equine ACFCs that had previously reached approximately 90% confluence were prepared for the nuclefection procedure by trypsinization and subsequent resuspension in HEPES-buffered Tissue Culture Medium 199 (TCM 199; Sigma-Aldrich) supplemented with 5% FBS (Sigma-Aldrich), followed by centrifugation at 200×
g for 10 min. Afterwards, the centrifugation pools of cells (each at a concentration ranging from 4 × 10
5 to 5 × 10
5) were subjected to co-transfection nucleofection using the Amaxa
TM Normal Human Dermal Fibroblast– Adult (NHDF-Adult) Nucleofector
TM Kit (Lonza, CELLLAB, Warsaw, Poland) and a mixture of two pcDNA plasmids included in the T-REx kit (Invitrogen). The aforementioned mixture of two plasmids (a total amount of 2.8 µg and in a mass ratio of 6:1) was comprised of pcDNA™
6/TR and pcDNA™
4/TO/myc-His/B with the appropriate transgene variant inserted (either
BPV-E4 or
BPV-E1^E4). The co-transfection of equine ACFCs was carried out within the Amaxa nucleofection cuvettes inserted into the holder of the Amaxa Nucleofector
TM II Device (Amaxa Biosystems, Lonza, Medianus, Kraków, Poland). The nucleofection process was triggered by the U-023 program intended for transgenization of human dermal fibroblasts and resulted in high transfection efficiency. The U-023 program was delivered by Amaxa Nucleofector
TM Technology (Amaxa Biosystems).
4.7. Treatment of Cell Nucleofectants Leading to Positive Antibiotic-Dependent Selection of BPV-E4 or BPV-E1^E4 Transgenic Equine ACFCs and Their Subsequent Tetracycline-Induced Neoplastic Transformation into Sarcoid-like Cells
After nucleofection, the equine ACFCs were seeded into collagen-coated culture dishes (Greiner Bio-One GmbH, BIOKOM Systems, Janki near Warsaw, Poland) and incubated for 48 h in DMEM enriched with recombinant human basic fibroblast growth factor (rh-bFGF; Sigma-Aldrich). The culture medium was subsequently changed to a medium supplemented with 200 µg/mL zeocin (Invitrogen) and 6 µg/mL blasticidin S (Thermo Scientific, Waltham, MA, USA). As a consequence of zeocin- and blasticidin S-dependent negative selection, the nontransgenic (TG–) cells that had not effectively undergone BPV-E4- or BPV-E1^E4-induced oncogenic transformation were eliminated from subpopulations encompassing cell nucleofectants due to the lack of immune resistance to selective antibiotics (i.e., combined resistance to zeocin and blasticidin S). The ACFCs that had undergone efficient transgenization were found to display antibiotic resistance. Seven days later, the selection was complete and the remaining positively selected transgenic (TG+) cells were cultured under standard conditions in the medium supplemented with 10% Tet-System Approved FBS (Gibco). Plasmid expression was induced by the addition of 1 µg/mL tetracycline (Invitrogen) to the culture medium 24 h before accomplishing further procedures.
The concentrations of the individual antibiotics were selected by establishing the lowest concentrations of the antibiotics that destroyed the cell culture within a week. For this purpose, media with different concentrations of individual antibiotics were introduced into the cultures, carried out in 96-well culture plates with 100% confluence. The concentrations were 2, 4, 6, 8, 9, 10, 11, 12, and 14 µg/mL for blasticidin S and 50, 100, 200, 400, 600, 800, 1000, 1100, and 1300 µg/mL for zeocin. One week later, the number of vial cells was measured using CellTiter Blue dye (Promega, Walldorf, Germany). The culture medium was removed from each well, and then 100 µL of culture medium with dye was added to it (in a 5:1 ratio). The cultures were then incubated for 5.5 h in an incubator (37 °C, 5% CO2, 100% humidity), protected from light. The measurement was performed on a PlateReader 2200 (Eppendorf, Warsaw, Poland) (excitation: 535 nm, emission: 595 nm). The lowest concentrations of antibiotics were selected as those for which the fluorescence level did not differ significantly from the fluorescence of empty wells.
4.8. Detection of BPV DNA in Equine ACFCs Subjected to Oncogenic Transformation with the Aid of Nucleofection
DNA was isolated with a NucleoMag Vet Kit (Macherey-Nagel, Bionovo, Legnica, Poland) according to the manufacturer’s protocol. DNA was dissolved in DEPC-treated water (Life Technologies, Ambion, Thermo Scientific, Waltham, MA, USA). The quality of isolated DNA was checked with NanoDrop 2000 (Life Technologies).
Polymerase chain reaction (PCR) was performed with AmpliTaq Gold 360 Master Mix polymerase (Applied Biosystems) with primers specific for the
BPV1 and
BPV2 consensus region [
69]. The temperature profile was set with respect to the polymerase supplier’s protocol and with a primer annealing temperature of 57–58 °C. PCR products were separated in agarose gel electrophoresis (3% agarose in TBE).
4.9. Isolation of RNA Samples from BPV-E4 and BPV-E1^E4 Transgenic Equine ACFC-Derived Neoplastic Cells
According to the producer’s protocol, RNA was directly isolated from adherent cultures of BPV-E4 and BPV-E1^E4 transgenic equine ACFC-derived neoplastic cell lines that were expanded ex vivo on the bottom of culture dishes. To extract RNA samples, a PureLink™ RNA mini kit (Invitrogen) was used. The procedure was maintained, with the addition of a DNase treatment step (PureLink™ DNase Set, Invitrogen). RNA was eluted with DEPC-treated water (Thermo Fisher Scientific, Waltham, MA, USA).
The quality and quantity of RNA were measured with a 2200 TapeStation Automated Electrophoresis System (Agilent Technologies, Santa Clara, CA, USA), as well as a nanodrop 2000 spectrophotometer (Thermo Scientific) and agarose gel (2% agarose in TBE buffer) electrophoresis.
4.10. NGS Sequencing among Oncogenically Transformed Equine ACFCs Expressing BPV-E4 and BPV-E4^E1 Transgenes
All samples were sequenced using the NGS approach. High-quality RNA (RIN value from 9.3 to 9.8) was used for cDNA libraries preparation with the TruSeq RNA Kit v2 kit (Illumina, San Diego, CA, USA) according to the attached protocol. The individual cDNA libraries were ligated with different indexes to be able to pool samples during the NGS sequencing procedure. The quality and quantity of obtained libraries were assessed using Qubit 2.0 (Qubit™ dsDNA BR AssayKit, Invitrogen, Waltham, MA, USA) and TapeStation 2200 (D100 ScreenTapes, Agilent Technologies, Santa Clara, CA, USA). In the next step, the cDNA libraries were sequenced on the NextSeq 500 Illumina platform (Illumina) and NextSeq 500/550 High Output KIT v 2.5 (75 cycles) according to the protocol.
The raw data were first checked for quality with FastQC v0.11.7 software, followed by the removal of adapters and low-quality reads (Phred quality of 20 and read length of 36). Then, the filtered reads were mapped to the EquCab3 genome with STAR software. Afterwards, the mapped reads were annotated and counted to specific gene thresholds with the usage of Ensembl GTF file version 100 (via htseq-count software). Differential expression analysis was performed with the use of Deseq2 software v3.14.
Gene Ontology enrichment and over-represented Pathways analyses (KEGG, GO) were performed using David software (version 6.8) [
70] based on the
Equus caballus reference. The significance was based on the False Discovery Rate (FDR), calculated as a
p-value after Benjamin multiple testing correction [
71]. For the visualization of gene interaction, String software v11.5 [
24] was applied with
Equus caballus as a reference.
4.11. qPCR-Assisted Validation Accomplished for Transcriptional Activity Levels of Genes in Neoplastically Transformed Equine ACFCs Expressing BPV-E4 and BPV-E1^E4 Transgenes
RNA-seq validation was performed using real-time PCR. The exact transcript levels were estimated for 10 DEGs for which specific primers were designed based on Ensemble reference (Primer3 Input (version 0.4.0) software; [
72]) (
Table S1). The cDNA samples were synthesized from 300 ng of total RNA using a High-Capacity RNA-to-cDNA™ Kit (Applied Biosystems). The qPCR reaction was carried out in triplicate for each sample with Sensitive RT HS-PCR EvaGreen Mix (A&A Biotechnology, Gdynia, Poland) according to the manufacturer’s protocol and using QuantStudio7Flex platform (Applied Biosystems). The expression was calculated using the delta–delta CT method, according to Pfaffl et al. [
73], and based on two endogenous controls, i.e.,
ACTB and
UBB genes that encode β-actin and ubiquitin B, respectively [
74].
The NGS data (normalized counts) and relative quantity (RQ) were compared using the Pearson correlation (SAS software, version 8.02).


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}