
Dysregulation of S-adenosylmethionine Metabolism in Nonalcoholic Steatohepatitis Leads to Polyamine Flux and Oxidative Stress

Abstract
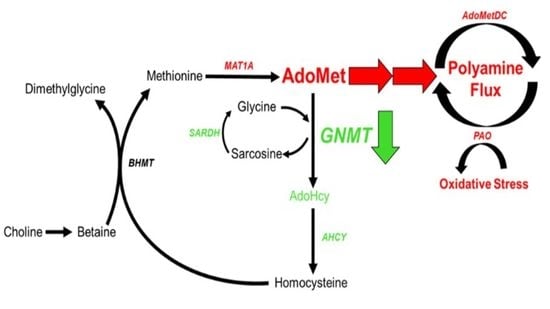
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. GNMT Is Downregulated in Human NASH
2.2. Modified Amylin Diet Induces NASH in Mice
2.3. Proteomic Characterization of NASH Animal Model Resembles Human Pathophysiology
2.4. Dysregulated AdoMet Metabolism in NASH
2.5. Polyamine Metabolism Is Activated in NASH, Causing Flux and Oxidative Stress
3. Discussion
4. Materials and Methods
4.1. Human Liver Samples
4.2. Animal Studies
4.3. Histology and NAS Scoring
4.4. Global Mass Spectrometry Analysis
4.5. Bioinformatic Analysis
4.6. Polyamine Metabolomic Analysis
4.7. Targeted Mass Spectrometry Analysis
4.8. HNE Immunofluorescence
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Marchesini, G.; Pinto-Cortez, H.; Petta, S. Epidemiology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis: Implications for Liver Transplantation. Transplantation 2019, 103, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Henry, L. The Impact of Obesity and Type 2 Diabetes on Chronic Liver Disease. Am. J. Gastroenterol. 2019, 114, 1714–1715. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Mudd, S.H.; Brosnan, J.T.; Brosnan, M.E.; Jacobs, R.L.; Stabler, S.P.; Allen, R.H.; Vance, D.E.; Wagner, C. Methyl balance and transmethylation fluxes in humans. Am. J. Clin. Nutr. 2007, 85, 19–25. [Google Scholar] [CrossRef]
- Mato, J.M.; Martínez-Chantar, M.L.; Lu, S.C. S-adenosylmethionine metabolism and liver disease. Ann. Hepatol. 2013, 12, 183–189. [Google Scholar] [CrossRef]
- Martínez-Chantar, M.L.; Vázquez-Chantada, M.; Ariz, U.; Martínez, N.; Varela, M.; Luka, Z.; Capdevila, A.; Rodríguez, J.; Aransay, A.M.; Matthiesen, R.; et al. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology 2008, 47, 1191–1199. [Google Scholar] [CrossRef]
- Hughey, C.C.; Trefts, E.; Bracy, D.P.; James, F.D.; Donahue, E.P.; Wasserman, D.H. Glycine N-methyltransferase deletion in mice diverts carbon flux from gluconeogenesis to pathways that utilize excess methionine cycle intermediates. J. Biol. Chem. 2018, 293, 11944–11954. [Google Scholar] [CrossRef]
- Teufel, A.; Itzel, T.; Erhart, W.; Brosch, M.; Wang, X.Y.; Kim, Y.O.; von Schönfels, W.; Herrmann, A.; Brückner, S.; Stickel, F.; et al. Comparison of Gene Expression Patterns Between Mouse Models of Nonalcoholic Fatty Liver Disease and Liver Tissues from Patients. Gastroenterology 2016, 151, 513–525.e0. [Google Scholar] [CrossRef]
- Ryaboshapkina, M.; Hammar, M. Human hepatic gene expression signature of non-alcoholic fatty liver disease progression, a meta-analysis. Sci. Rep. 2017, 7, 12361. [Google Scholar] [CrossRef]
- Moylan, C.A.; Pang, H.; Dellinger, A.; Suzuki, A.; Garrett, M.E.; Guy, C.D.; Murphy, S.K.; Ashley-Koch, A.E.; Choi, S.S.; Michelotti, G.A.; et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 2014, 59, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Álvarez, S.; Gutiérrez-de Juan, V.; Zubiete-Franco, I.; Barbier-Torres, L.; Lahoz, A.; Parés, A.; Luka, Z.; Wagner, C.; Lu, S.C.; Mato, J.M.; et al. TRAIL-producing NK cells contribute to liver injury and related fibrogenesis in the context of GNMT deficiency. Lab. Investig. 2015, 95, 223–236. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Simile, M.M.; Cigliano, A.; Paliogiannis, P.; Daino, L.; Manetti, R.; Feo, C.F.; Calvisi, D.F.; Feo, F.; Pascale, R.M. Nuclear localization dictates hepatocarcinogenesis suppression by glycine N-methyltransferase. Transl. Oncol. 2022, 15, 101239. [Google Scholar] [CrossRef] [PubMed]
- Jell, J.; Merali, S.; Hensen, M.L.; Mazurchuk, R.; Spernyak, J.A.; Diegelman, P.; Kisiel, N.D.; Barrero, C.; Deeb, K.K.; Alhonen, L.; et al. Genetically altered expression of spermidine/spermine N1-acetyltransferase affects fat metabolism in mice via acetyl-CoA. J. Biol. Chem. 2007, 282, 8404–8413. [Google Scholar] [CrossRef] [PubMed]
- Casero, R.A.; Murray Stewart, T.; Pegg, A.E. Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat. Rev. Cancer 2018, 18, 681–695. [Google Scholar] [CrossRef]
- Liu, C.; Perez-Leal, O.; Barrero, C.; Zahedi, K.; Soleimani, M.; Porter, C.; Merali, S. Modulation of polyamine metabolic flux in adipose tissue alters the accumulation of body fat by affecting glucose homeostasis. Amino Acids 2014, 46, 701–715. [Google Scholar] [CrossRef]
- Kramer, D.L.; Diegelman, P.; Jell, J.; Vujcic, S.; Merali, S.; Porter, C.W. Polyamine acetylation modulates polyamine metabolic flux, a prelude to broader metabolic consequences. J. Biol. Chem. 2008, 283, 4241–4251. [Google Scholar] [CrossRef]
- Charlton, M.; Krishnan, A.; Viker, K.; Sanderson, S.; Cazanave, S.; McConico, A.; Masuoko, H.; Gores, G. Fast food diet mouse: Novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G825–G834. [Google Scholar] [CrossRef]
- Kohli, R.; Kirby, M.; Xanthakos, S.A.; Softic, S.; Feldstein, A.E.; Saxena, V.; Tang, P.H.; Miles, L.; Miles, M.V.; Balistreri, W.F.; et al. High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 2010, 52, 934–944. [Google Scholar] [CrossRef]
- Machado, M.V.; Michelotti, G.A.; Xie, G.; Almeida Pereira, T.; de Almeida, T.P.; Boursier, J.; Bohnic, B.; Guy, C.D.; Diehl, A.M. Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS ONE 2015, 10, e0127991. [Google Scholar] [CrossRef]
- Liang, W.; Menke, A.L.; Driessen, A.; Koek, G.H.; Lindeman, J.H.; Stoop, R.; Havekes, L.M.; Kleemann, R.; van den Hoek, A.M. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS ONE 2014, 9, e115922. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.J.; Bao, L.; Keefer, K.; Shanmughapriya, S.; Chen, L.; Lee, J.; Wang, J.; Zhang, X.Q.; Hirschler-Laszkiewicz, I.; Merali, S.; et al. Transient receptor potential ion channel TRPM2 promotes AML proliferation and survival through modulation of mitochondrial function, ROS, and autophagy. Cell Death Dis. 2020, 11, 247. [Google Scholar] [CrossRef]
- McBrearty, N.; Arzumanyan, A.; Bichenkov, E.; Merali, S.; Merali, C.; Feitelson, M. Short chain fatty acids delay the development of hepatocellular carcinoma in HBx transgenic mice. Neoplasia 2021, 23, 529–538. [Google Scholar] [CrossRef]
- Molina-Franky, J.; Plaza, D.F.; Merali, C.; Merali, S.; Barrero, C.; Arevalo-Pinzon, G.; Patarroyo, M.E.; Patarroyo, M.A. A novel platform for peptide-mediated affinity capture and LC-MS/MS identification of host receptors involved in Plasmodium invasion. J. Proteom. 2021, 231, 104002. [Google Scholar] [CrossRef]
- Zhang, H.; Pandey, S.; Travers, M.; Sun, H.; Morton, G.; Madzo, J.; Chung, W.; Khowsathit, J.; Perez-Leal, O.; Barrero, C.A.; et al. Targeting CDK9 Reactivates Epigenetically Silenced Genes in Cancer. Cell 2018, 175, 1244–1258.e26. [Google Scholar] [CrossRef]
- García-Monzón, C.; Lo Iacono, O.; Crespo, J.; Romero-Gómez, M.; García-Samaniego, J.; Fernández-Bermejo, M.; Domínguez-Díez, A.; Rodríguez de Cía, J.; Sáez, A.; Porrero, J.L.; et al. Increased soluble CD36 is linked to advanced steatosis in nonalcoholic fatty liver disease. Eur. J. Clin. Investig. 2014, 44, 65–73. [Google Scholar] [CrossRef]
- Wilson, C.G.; Tran, J.L.; Erion, D.M.; Vera, N.B.; Febbraio, M.; Weiss, E.J. Hepatocyte-Specific Disruption of CD36 Attenuates Fatty Liver and Improves Insulin Sensitivity in HFD-Fed Mice. Endocrinology 2016, 157, 570–585. [Google Scholar] [CrossRef]
- Steensels, S.; Qiao, J.; Zhang, Y.; Maner-Smith, K.M.; Kika, N.; Holman, C.D.; Corey, K.E.; Bracken, W.C.; Ortlund, E.A.; Ersoy, B.A. Acyl-Coenzyme A Thioesterase 9 Traffics Mitochondrial Short-Chain Fatty Acids Toward De Novo Lipogenesis and Glucose Production in the Liver. Hepatology 2020, 72, 857–872. [Google Scholar] [CrossRef]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef]
- Iacobini, C.; Menini, S.; Ricci, C.; Blasetti Fantauzzi, C.; Scipioni, A.; Salvi, L.; Cordone, S.; Delucchi, F.; Serino, M.; Federici, M.; et al. Galectin-3 ablation protects mice from diet-induced NASH: A major scavenging role for galectin-3 in liver. J. Hepatol. 2011, 54, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Abdelmalek, M.F.; Garcia-Tsao, G.; Vuppalanchi, R.; Alkhouri, N.; Rinella, M.; Noureddin, M.; Pyko, M.; Shiffman, M.; Sanyal, A.; et al. Effects of Belapectin, an Inhibitor of Galectin-3, in Patients with Nonalcoholic Steatohepatitis with Cirrhosis and Portal Hypertension. Gastroenterology 2020, 158, 1334–1345.e5. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Wang, Q.; Wang, S.; Zhang, J.; Liu, T.; Guo, L.; Yu, Y.; Lin, J.D. Mapping the molecular signatures of diet-induced NASH and its regulation by the hepatokine Tsukushi. Mol. Metab. 2019, 20, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Zhang, B.; Zhao, J.; Feng, Q.; Peng, J.; Hu, Y.; Zhao, Y. Biological mechanisms and related natural modulators of liver X receptor in nonalcoholic fatty liver disease. Biomed. Pharmacother. 2019, 113, 108778. [Google Scholar] [CrossRef]
- Noureddin, M.; Sanyal, A.J. Pathogenesis of NASH: The Impact of Multiple Pathways. Curr. Hepatol. Rep. 2018, 17, 350–360. [Google Scholar] [CrossRef]
- Cariello, M.; Piccinin, E.; Moschetta, A. Transcriptional Regulation of Metabolic Pathways via Lipid-Sensing Nuclear Receptors PPARs, FXR, and LXR in NASH. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 1519–1539. [Google Scholar] [CrossRef]
- Day, K.; Seale, L.A.; Graham, R.M.; Cardoso, B.R. Selenotranscriptome Network in Non-Alcoholic Fatty Liver Disease. Front. Nutr. 2021, 8, 744825. [Google Scholar] [CrossRef]
- Gornicka, A.; Morris-Stiff, G.; Thapaliya, S.; Papouchado, B.G.; Berk, M.; Feldstein, A.E. Transcriptional profile of genes involved in oxidative stress and antioxidant defense in a dietary murine model of steatohepatitis. Antioxid. Redox Signal. 2011, 15, 437–445. [Google Scholar] [CrossRef]
- Hernandez, E.D.; Zheng, L.; Kim, Y.; Fang, B.; Liu, B.; Valdez, R.A.; Dietrich, W.F.; Rucker, P.V.; Chianelli, D.; Schmeits, J.; et al. Tropifexor-Mediated Abrogation of Steatohepatitis and Fibrosis Is Associated with the Antioxidative Gene Expression Profile in Rodents. Hepatol. Commun. 2019, 3, 1085–1097. [Google Scholar] [CrossRef]
- Lu, S.C.; Alvarez, L.; Huang, Z.Z.; Chen, L.; An, W.; Corrales, F.J.; Avila, M.A.; Kanel, G.; Mato, J.M. Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. Proc. Natl. Acad. Sci. USA 2001, 98, 5560–5565. [Google Scholar] [CrossRef]
- Cano, A.; Buqué, X.; Martínez-Uña, M.; Aurrekoetxea, I.; Menor, A.; García-Rodríguez, J.L.; Lu, S.C.; Martínez-Chantar, M.L.; Mato, J.M.; Ochoa, B.; et al. Methionine adenosyltransferase 1A gene deletion disrupts hepatic very low-density lipoprotein assembly in mice. Hepatology 2011, 54, 1975–1986. [Google Scholar] [CrossRef] [PubMed]
- Itagaki, H.; Shimizu, K.; Morikawa, S.; Ogawa, K.; Ezaki, T. Morphological and functional characterization of non-alcoholic fatty liver disease induced by a methionine-choline-deficient diet in C57BL/6 mice. Int. J. Clin. Exp. Pathol. 2013, 6, 2683–2696. [Google Scholar] [PubMed]
- Kalhan, S.C.; Edmison, J.; Marczewski, S.; Dasarathy, S.; Gruca, L.L.; Bennett, C.; Duenas, C.; Lopez, R. Methionine and protein metabolism in non-alcoholic steatohepatitis: Evidence for lower rate of transmethylation of methionine. Clin. Sci. 2011, 121, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Day, C.P. S-adenosylmethionine (SAMe) therapy in liver disease: A review of current evidence and clinical utility. J. Hepatol. 2012, 57, 1097–1109. [Google Scholar] [CrossRef] [PubMed]
- Noureddin, M.; Mato, J.M.; Lu, S.C. Nonalcoholic fatty liver disease: Update on pathogenesis, diagnosis, treatment and the role of S-adenosylmethionine. Exp. Biol. Med. 2015, 240, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R.L.; van der Veen, J.N.; Vance, D.E. Finding the balance: The role of S-adenosylmethionine and phosphatidylcholine metabolism in development of nonalcoholic fatty liver disease. Hepatology 2013, 58, 1207–1209. [Google Scholar] [CrossRef]
- Caballero, F.; Fernández, A.; Matías, N.; Martínez, L.; Fucho, R.; Elena, M.; Caballeria, J.; Morales, A.; Fernández-Checa, J.C.; García-Ruiz, C. Specific contribution of methionine and choline in nutritional nonalcoholic steatohepatitis: Impact on mitochondrial S-adenosyl-L-methionine and glutathione. J. Biol. Chem. 2010, 285, 18528–18536. [Google Scholar] [CrossRef]
- Walker, A.K.; Jacobs, R.L.; Watts, J.L.; Rottiers, V.; Jiang, K.; Finnegan, D.M.; Shioda, T.; Hansen, M.; Yang, F.; Niebergall, L.J.; et al. A conserved SREBP-1/phosphatidylcholine feedback circuit regulates lipogenesis in metazoans. Cell 2011, 147, 840–852. [Google Scholar] [CrossRef]
- Alonso, C.; Fernández-Ramos, D.; Varela-Rey, M.; Martínez-Arranz, I.; Navasa, N.; Van Liempd, S.M.; Lavín Trueba, J.L.; Mayo, R.; Ilisso, C.P.; de Juan, V.G.; et al. Metabolomic Identification of Subtypes of Nonalcoholic Steatohepatitis. Gastroenterology 2017, 152, 1449–1461.e7. [Google Scholar] [CrossRef]
- Maria Del Bas, J.; Rodríguez, B.; Puiggròs, F.; Mariné, S.; Rodríguez, M.A.; Moriña, D.; Armengol, L.; Caimari, A.; Arola, L. Hepatic accumulation of S-adenosylmethionine in hamsters with non-alcoholic fatty liver disease associated with metabolic syndrome under selenium and vitamin E deficiency. Clin. Sci. 2019, 133, 409–423. [Google Scholar] [CrossRef]
- Murray Stewart, T.; Dunston, T.T.; Woster, P.M.; Casero, R.A. Polyamine catabolism and oxidative damage. J. Biol. Chem. 2018, 293, 18736–18745. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Sun, D.; Zhang, J.; Xue, R.; Janssen, H.L.A.; Tang, W.; Dong, L. Spermine oxidase is upregulated and promotes tumor growth in hepatocellular carcinoma. Hepatol. Res. 2018, 48, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, O.A.; Keinanen, T.A.; Ivanova, O.N.; Hyvonen, M.T.; Khomutov, A.R.; Kochetkov, S.N.; Bartosch, B.; Ivanov, A.V. Hepatitis C virus alters metabolism of biogenic polyamines by affecting expression of key enzymes of their metabolism. Biochem. Biophys. Res. Commun. 2017, 483, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Saul, A.B.; Pichavaram, P.; Xu, Z.; Rudraraju, M.; Somanath, P.R.; Smith, S.B.; Caldwell, R.B.; Narayanan, S.P. Pharmacological Inhibition of Spermine Oxidase Reduces Neurodegeneration and Improves Retinal Function in Diabetic Mice. J. Clin. Med. 2020, 9, 340. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.P.; Xu, Z.; Putluri, N.; Sreekumar, A.; Lemtalsi, T.; Caldwell, R.W.; Caldwell, R.B. Arginase 2 deficiency reduces hyperoxia-mediated retinal neurodegeneration through the regulation of polyamine metabolism. Cell Death Dis. 2014, 5, e1075. [Google Scholar] [CrossRef]
- Kee, K.; Vujcic, S.; Merali, S.; Diegelman, P.; Kisiel, N.; Powell, C.T.; Kramer, D.L.; Porter, C.W. Metabolic and antiproliferative consequences of activated polyamine catabolism in LNCaP prostate carcinoma cells. J. Biol. Chem. 2004, 279, 27050–27058. [Google Scholar] [CrossRef]
- Sumida, Y.; Niki, E.; Naito, Y.; Yoshikawa, T. Involvement of free radicals and oxidative stress in NAFLD/NASH. Free Radic. Res. 2013, 47, 869–880. [Google Scholar] [CrossRef]
- Ore, A.; Akinloye, O.A. Oxidative Stress and Antioxidant Biomarkers in Clinical and Experimental Models of Non-Alcoholic Fatty Liver Disease. Medicina 2019, 55, 26. [Google Scholar] [CrossRef]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Merali, S.; Clarkson, A.B. Polyamine analysis using N-hydroxysuccinimidyl-6-aminoquinoyl carbamate for pre-column derivatization. J. Chromatogr. B Biomed. Appl. 1996, 675, 321–326. [Google Scholar] [CrossRef]
- Gessulat, S.; Schmidt, T.; Zolg, D.P.; Samaras, P.; Schnatbaum, K.; Zerweck, J.; Knaute, T.; Rechenberger, J.; Delanghe, B.; Huhmer, A.; et al. Prosit: Proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods 2019, 16, 509–518. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quinn, C.; Rico, M.C.; Merali, C.; Merali, S. Dysregulation of S-adenosylmethionine Metabolism in Nonalcoholic Steatohepatitis Leads to Polyamine Flux and Oxidative Stress. Int. J. Mol. Sci. 2022, 23, 1986. https://doi.org/10.3390/ijms23041986
Quinn C, Rico MC, Merali C, Merali S. Dysregulation of S-adenosylmethionine Metabolism in Nonalcoholic Steatohepatitis Leads to Polyamine Flux and Oxidative Stress. International Journal of Molecular Sciences. 2022; 23(4):1986. https://doi.org/10.3390/ijms23041986
Chicago/Turabian StyleQuinn, Connor, Mario C. Rico, Carmen Merali, and Salim Merali. 2022. "Dysregulation of S-adenosylmethionine Metabolism in Nonalcoholic Steatohepatitis Leads to Polyamine Flux and Oxidative Stress" International Journal of Molecular Sciences 23, no. 4: 1986. https://doi.org/10.3390/ijms23041986
APA StyleQuinn, C., Rico, M. C., Merali, C., & Merali, S. (2022). Dysregulation of S-adenosylmethionine Metabolism in Nonalcoholic Steatohepatitis Leads to Polyamine Flux and Oxidative Stress. International Journal of Molecular Sciences, 23(4), 1986. https://doi.org/10.3390/ijms23041986