
The Different Temozolomide Effects on Tumorigenesis Mechanisms of Pediatric Glioblastoma PBT24 and SF8628 Cell Tumor in CAM Model and on Cells In Vitro

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. The Biomicroscopy of PBT24 and SF8628 Xenograft on CAM
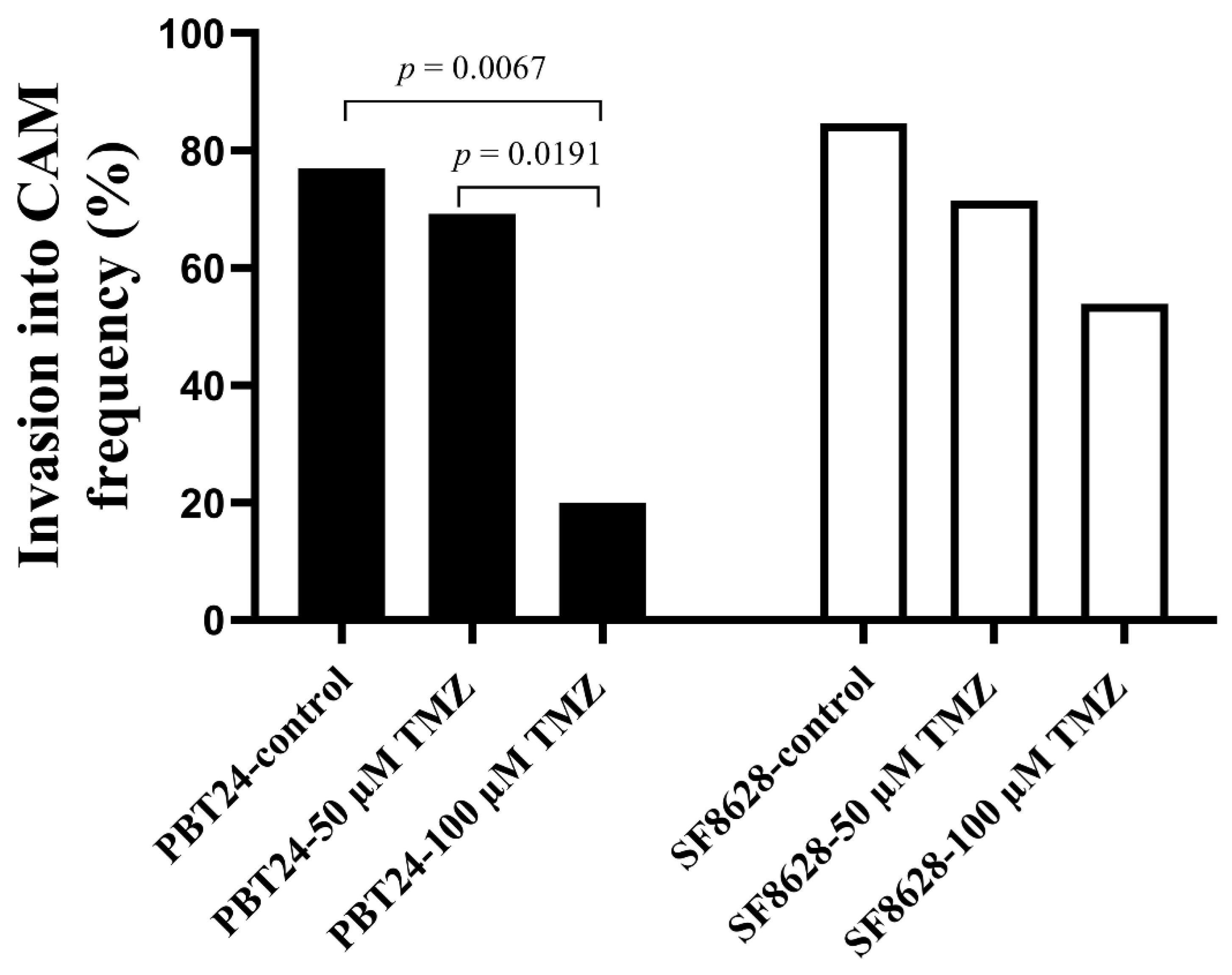
2.2. The PBT24 and SF8628 Growth, Invasion into CAM Frequency, the CAM Thickness and the Number of Blood Vessels in CAM under the Tumors of the Study Groups
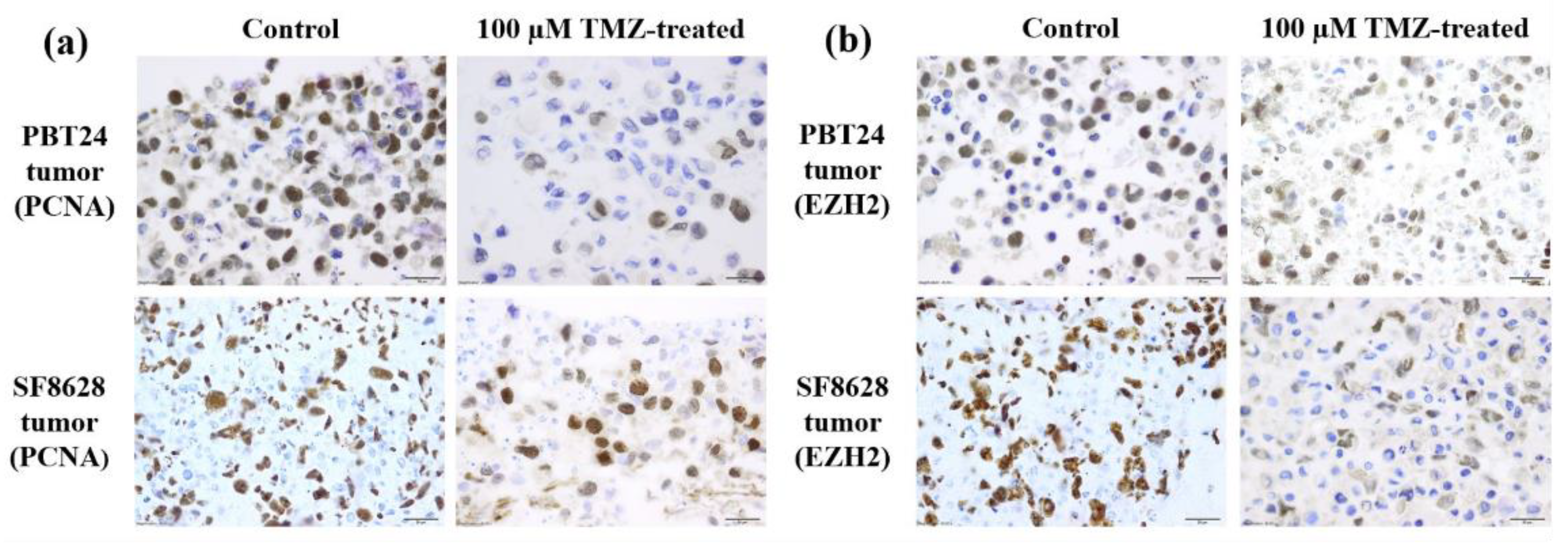
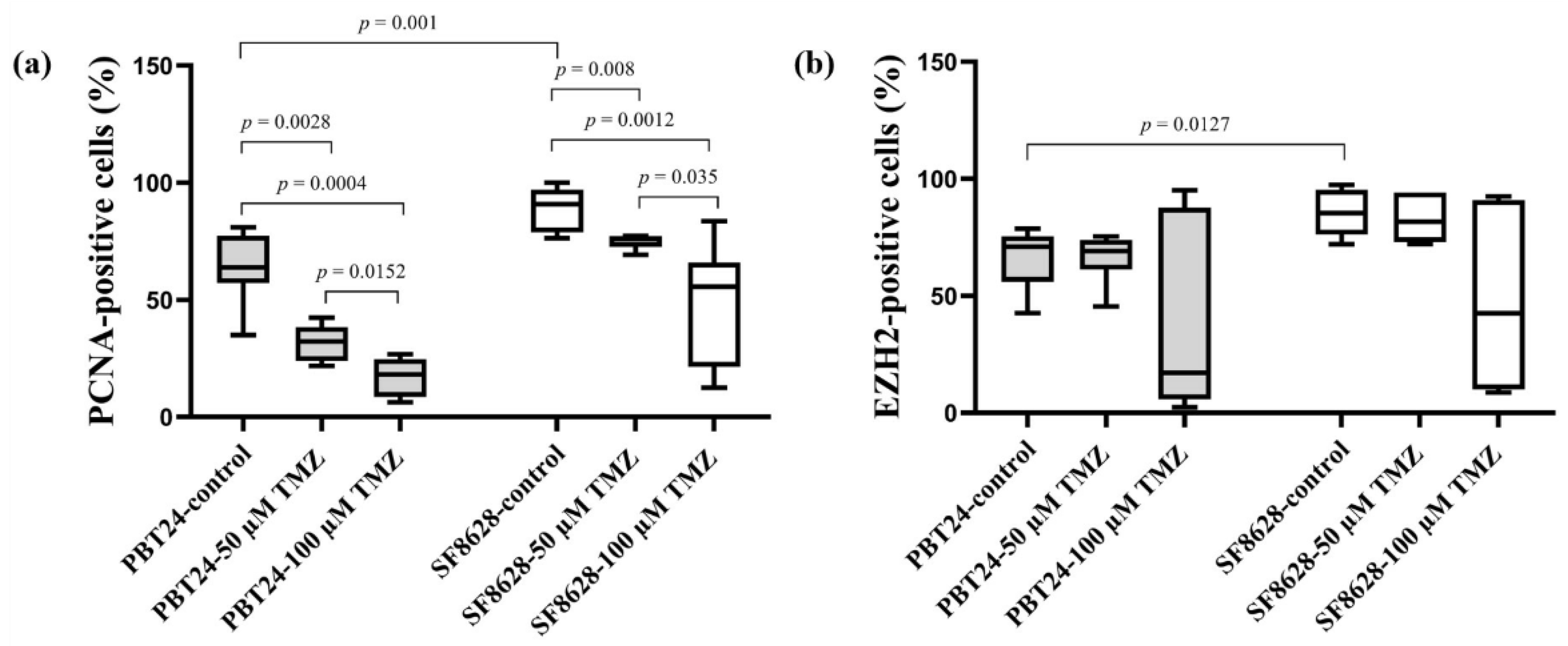
2.3. The PCNA and EZH2 Expression in PBT24 and SF8628 Tumors
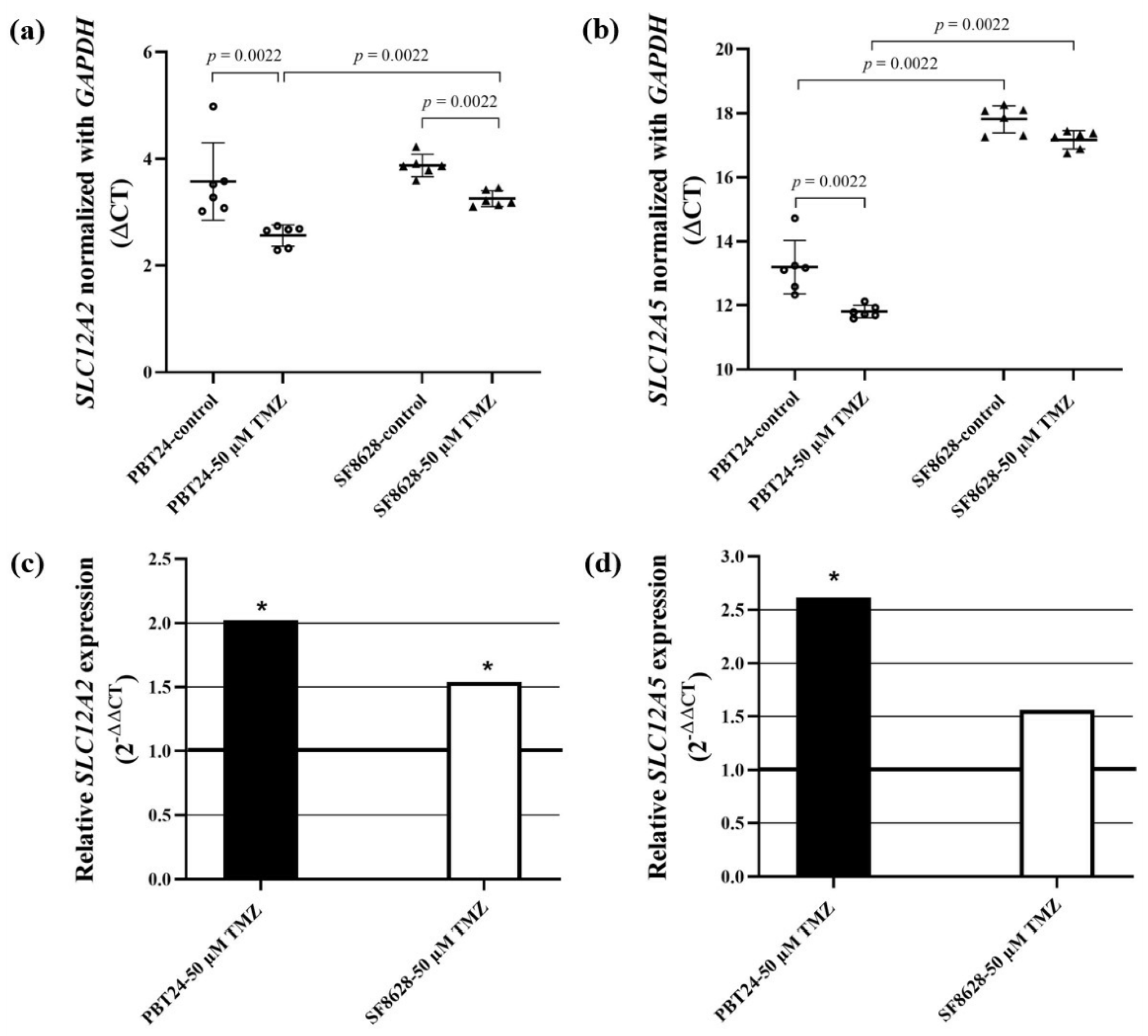
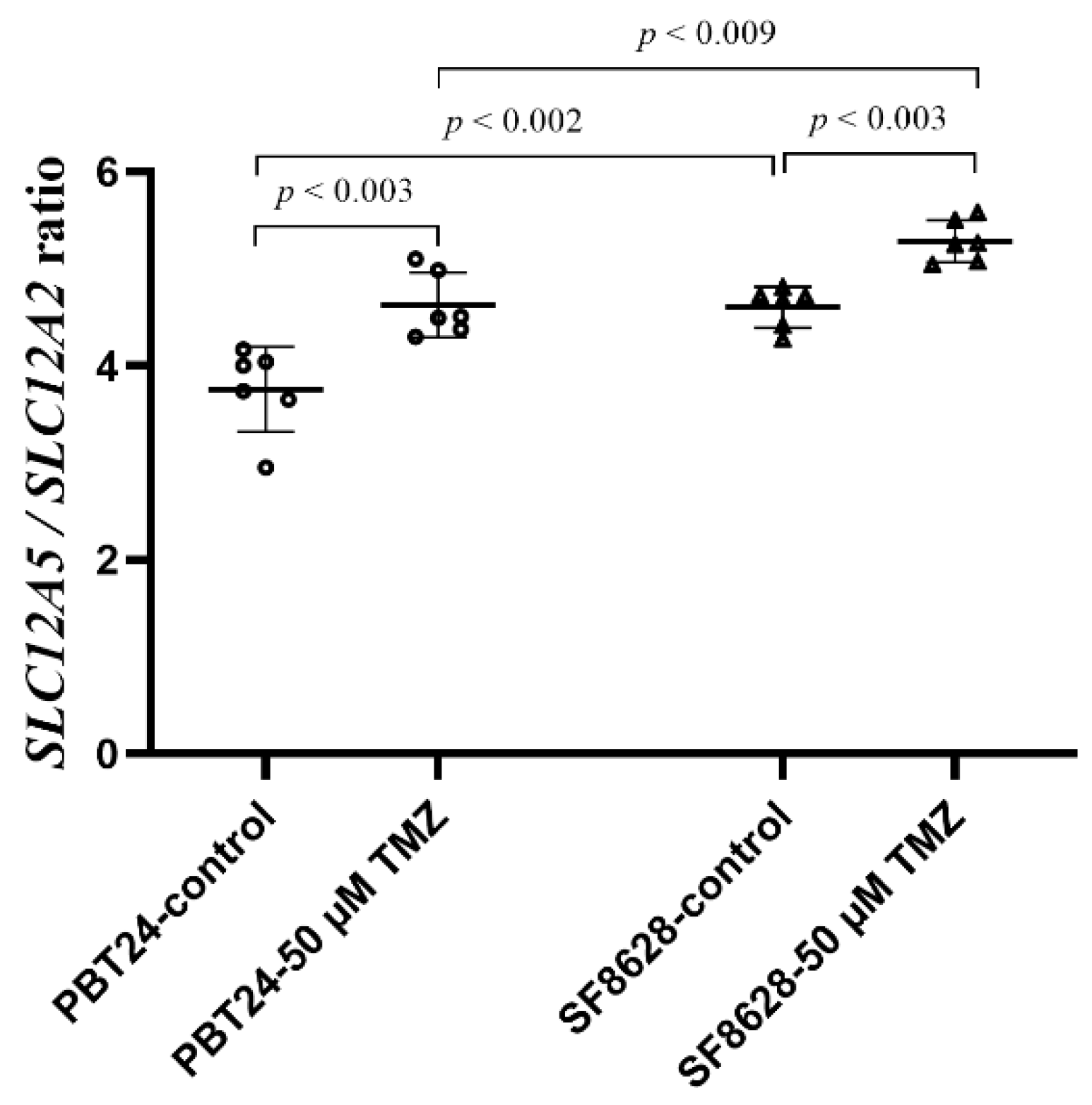
2.4. The Expression of SLC12A2 (NKCC1 Co-Transporter) and SLC12A5 (KCC2 Co-Transporter) Gene in PBT24 and SF8628 Cell Study Groups
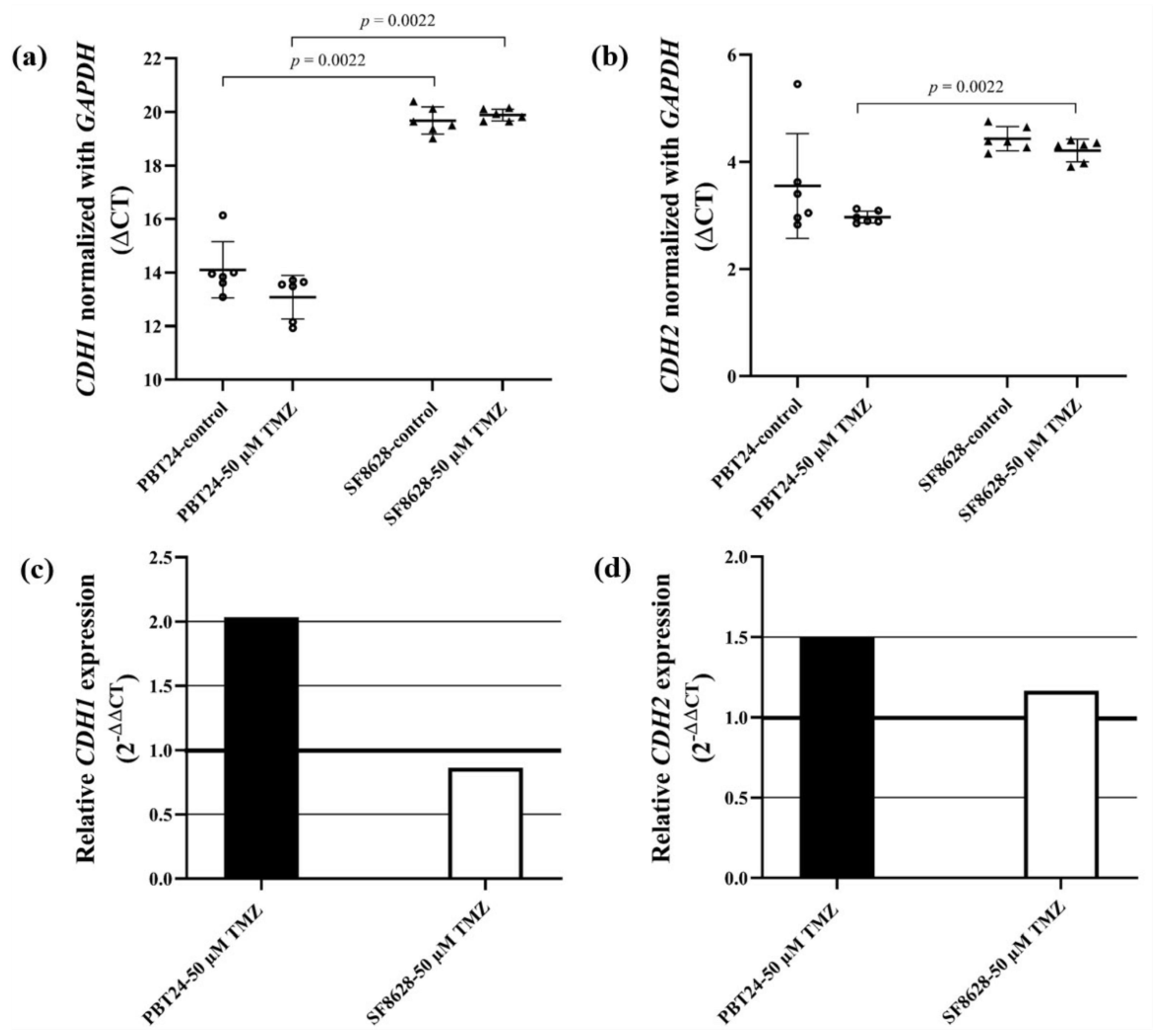
2.5. The Expression of CDH1 (E-Cadherin) and CDH2 (N-Cadherin) Gene in PBT24 and SF8628 Cell Study Groups
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. The CAM Model
4.3. The PBT24 and SF8628 Tumor Study Groups
4.4. Biomicroscopy Data to Assess Tumor Growth and Drug Efficacy
4.5. Tumor Grafting on CAM In Vivo
4.6. Histological Study of the Tumor
4.7. Assessment of the CAM Thickness and the Number of Blood Vessels in CAM
4.8. Immunohistochemical Study
4.9. Extraction of RNA from PBT24 and SF8628 Cell Line Cells
4.10. Determination of the SLC12A5, SLC12A2, CDH1 and CDH2 Gene Expression in PBT24 and SF8628 Cell Line Cells
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Durno, C.A.; Aronson, M.; Tabori, U.; Malkin, D.; Gallinger, S.; Chan, H.S.L. Oncologic surveillance for subjects with biallelic mismatch repair gene mutations: 10 year follow-up of a kindred. Pediatr. Blood Cancer 2012, 59, 652–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, C.; Stoller, S.; Grotzer, M.; Stucklin, A.G.; Nazarian, J.; Mueller, S. Pediatric hemispheric high-grade glioma: Targeting the future. Cancer Metastasis Rev. 2020, 39, 245–260. [Google Scholar] [CrossRef] [PubMed]
- Jihong, Z.; Malcolm, F.G.S.; Tracey, D.B. Temozolomide: Mechanisms of action, repair and resistance. Curr. Mol. Pharmacol. 2011, 5, 102–114. [Google Scholar]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Karachi, A.; Dastmalchi, F.; Mitchell, D.A.; Rahman, M. Temozolomide for immunomodulation in the treatment of glioblastoma. Neuro-Oncol. 2018, 20, 1566–1572. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.; Karajannis, M.A.; Jones, D.T.W.; Kieran, M.W.; Monje, M.; Baker, S.J.; Becher, O.J.; Cho, Y.-J.; Gupta, N.; Hawkins, C.; et al. Pediatric high-grade glioma: Biologically and clinically in need of new thinking. Neuro-Oncol. 2017, 19, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Margueron, R.; Reinberg, D. The polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Maeno, E.; Ishizaki, Y.; Kanaseki, T.; Hazama, A.; Okada, Y. Normotonic cell shrinkage because of disordered volume regulation is an early prerequisite to apoptosis. Proc. Natl. Acad. Sci. USA 2000, 97, 9487–9492. [Google Scholar] [CrossRef] [Green Version]
- Cong, D.; Zhu, W.; Kuo, J.S.; Hu, S.; Sun, D. Ion transporters in brain tumors. Curr. Med. Chem. 2015, 22, 1171–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzińska-Ustymowicz, K.; Pryczynicz, A.; Kemona, A.; Czyzewska, J. Correlation between proliferation markers: PCNA, Ki-67, MCM-2 and antiapoptotic protein Bcl-2 in colorectal cancer. Anticancer Res. 2009, 29, 3049–3052. [Google Scholar] [PubMed]
- González-Magaña, A.; Blanco, F.J. Human PCNA structure, function, and interactions. Biomolecules 2020, 10, 570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, J.A.; Lange, C.A. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 2008, 647, 21–29. [Google Scholar] [CrossRef]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [Green Version]
- Bortner, C.D.; Cidlowski, J.A. Cell shrinkage and monovalent cation fluxes: Role in apoptosis. Arch. Biochem. Biophys. 2007, 462, 176–188. [Google Scholar] [CrossRef] [Green Version]
- Pallud, J.; Le Van Quyen, M.; Bielle, F.; Pellegrino, C.; Varlet, P.; Cresto, N.; Baulac, M.; Duyckaerts, C.; Kourdougli, N.; Chazal, G.; et al. Cortical GABAergic excitation contributes to epileptic activities around human glioma. Sci. Transl. Med. 2014, 6, 244ra89. [Google Scholar] [CrossRef] [Green Version]
- Aronica, E.; Boer, K.; Redeker, S.; Spliet, W.G.M.; van Rijen, P.C.; Troost, D.; Gorter, J.A. Differential expression patterns of chloride transporters, Na+-K+-2Cl−-cotransporter and K+-Cl−-cotransporter, in epilepsy-associated malformations of cortical development. Neuroscience 2007, 145, 185–196. [Google Scholar] [CrossRef]
- Garzon-Muvdi, T.; Schiapparelli, P.; ap Rhys, C.; Guerrero-Cazares, H.; Smith, C.; Kim, D.-H.; Kone, L.; Farber, H.; Lee, D.Y.; An, S.S.; et al. Regulation of brain tumor dispersal by NKCC1 through a novel role in focal adhesion regulation. PLoS Biol. 2012, 10, e1001320. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.R.; Cuddapah, V.A.; Watkins, S.; Rohn, K.J.; Dy, T.E.; Sontheimer, H. With-No-Lysine Kinase 3 (WNK3) stimulates glioma invasion by regulating cell volume. Am. J. Physiol. Cell Physiol. 2011, 301, C1150–C1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.R.; Sontheimer, H. Inhibition of the sodium-potassium-chloride cotransporter isoform-1 reduces glioma invasion. Cancer Res. 2010, 70, 5597–5606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L.; Guan, X.; Begum, G.; Ding, D.; Gayden, J.; Hasan, M.N.; Fiesler, V.M.; Dodelson, J.; Kohanbash, G.; Hu, B.; et al. Blockade of cell volume regulatory protein NKCC1 increases TMZ-induced glioma apoptosis and reduces astrogliosis. Mol. Cancer Ther. 2020, 19, 1550–1561. [Google Scholar] [CrossRef] [PubMed]
- Algharabil, J.; Kintner, D.B.; Wang, Q.; Begum, G.; Clark, P.A.; Yang, S.-S.; Lin, S.-H.; Kahle, K.T.; Kuo, J.S.; Sun, D. Inhibition of Na+-K+-2Cl− cotransporter isoform 1 accelerates temozolomide-mediated apoptosis in glioblastoma cancer cells. Cell. Physiol. Biochem. 2012, 30, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Bortner, C.D.; Sifre, M.I.; Cidlowski, J.A. Cationic gradient reversal and cytoskeleton-independent volume regulatory pathways define an early stage of apoptosis. J. Biol. Chem. 2008, 283, 7219–7229. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Maeno, E. Apoptosis, cell volume regulation and volume-regulatory chloride channels. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2001, 130, 377–383. [Google Scholar] [CrossRef]
- Sun, H.; Long, S.; Wu, B.; Liu, J.; Li, G. NKCC1 Involvement in the epithelial-to-mesenchymal transition is a prognostic biomarker in gliomas. PeerJ 2020, 8, e8787. [Google Scholar] [CrossRef]
- Noh, M.-G.; Oh, S.-J.; Ahn, E.-J.; Kim, Y.-J.; Jung, T.-Y.; Jung, S.; Kim, K.-K.; Lee, J.-H.; Lee, K.-H.; Moon, K.-S. Prognostic significance of E-cadherin and N-cadherin expression in gliomas. BMC Cancer 2017, 17, 583. [Google Scholar] [CrossRef]
- Bar, J.K.; Zub, L.; Lis-Nawara, A.; Noga, L.; Jeleń, M.; Paradowski, B. Expression and interactions between cell adhesion molecules CD44v6 and E-cadherin in human gliomas. Adv. Clin. Exp. Med. 2014, 23, 827–834. [Google Scholar] [CrossRef] [Green Version]
- D’Urso, P.I.; D’Urso, O.F.; Storelli, C.; Catapano, G.; Gianfreda, C.D.; Montinaro, A.; Muscella, A.; Marsigliante, S. Retrospective protein expression and epigenetic inactivation studies of CDH1 in patients affected by low-grade glioma. J. Neurooncol. 2011, 104, 113–118. [Google Scholar] [CrossRef]
- Lewis-Tuffin, L.J.; Rodriguez, F.; Giannini, C.; Scheithauer, B.; Necela, B.M.; Sarkaria, J.N.; Anastasiadis, P.Z. Misregulated E-cadherin expression associated with an aggressive brain tumor phenotype. PLoS ONE 2010, 5, e13665. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Batista, L.F.Z.; Naumann, S.C.; Wick, W.; Weller, M.; Menck, C.F.M.; Kaina, B. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene 2007, 26, 186–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirose, Y.; Berger, M.S.; Pieper, R.O. P53 effects both the duration of G2/M arrest and the fate of temozolomide-treated human glioblastoma cells. Cancer Res. 2001, 61, 1957–1963. [Google Scholar] [PubMed]
- Beier, D.; Röhrl, S.; Pillai, D.R.; Schwarz, S.; Kunz-Schughart, L.A.; Leukel, P.; Proescholdt, M.; Brawanski, A.; Bogdahn, U.; Trampe-Kieslich, A.; et al. Temozolomide preferentially depletes cancer stem cells in glioblastoma. Cancer Res. 2008, 68, 5706–5715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribatti, D. The chick embryo chorioallantoic membrane (CAM). A multifaceted experimental model. Mech. Dev. 2016, 141, 70–77. [Google Scholar] [CrossRef]
- Kavaliauskaitė, D.; Stakišaitis, D.; Martinkutė, J.; Šlekienė, L.; Kazlauskas, A.; Balnytė, I.; Lesauskaitė, V.; Valančiūtė, A. The effect of sodium valproate on the glioblastoma U87 cell line tumor development on the chicken embryo chorioallantoic membrane and on EZH2 and P53 expression. BioMed Res. Int. 2017, 2017, 6326053. [Google Scholar] [CrossRef]
- Ribatti, D. The chick embryo chorioallantoic membrane (CAM) assay. Reprod. Toxicol. 2017, 70, 97–101. [Google Scholar] [CrossRef]
- Zhao, Z.; Bauer, N.; Aleksandrowicz, E.; Yin, L.; Gladkich, J.; Gross, W.; Kaiser, J.; Hackert, T.; Strobel, O.; Herr, I. Intraductal papillary mucinous neoplasm of the pancreas rapidly xenografts in chicken eggs and predicts aggressiveness. Int. J. Cancer 2018, 142, 1440–1452. [Google Scholar] [CrossRef]
- Valiulytė, I.; Curkūnavičiūtė, R.; Ribokaitė, L.; Kazlauskas, A.; Vaitkevičiūtė, M.; Skauminas, K.; Valančiūtė, A. The anti-tumorigenic activity of Sema3C in the chick embryo chorioallantoic membrane model. Int. J. Mol. Sci. 2019, 20, 5672. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Velez, N.; Marigil, M.; García-Moure, M.; Gonzalez-Huarriz, M.; Aristu, J.J.; Ramos-García, L.-I.; Tejada, S.; Díez-Valle, R.; Patiño-García, A.; Becher, O.J.; et al. Delta-24-RGD combined with radiotherapy exerts a potent antitumor effect in diffuse intrinsic pontine glioma and pediatric high grade glioma models. Acta Neuropathol. Commun. 2019, 7, 64. [Google Scholar] [CrossRef] [Green Version]
- Efremov, L.; Abera, S.F.; Bedir, A.; Vordermark, D.; Medenwald, D. Patterns of glioblastoma treatment and survival over a 16-years period: Pooled data from the german cancer registries. J. Cancer Res. Clin. Oncol. 2021, 147, 3381–3390. [Google Scholar] [CrossRef] [PubMed]
- Ene, C.I.; Holland, E.C. Personalized medicine for gliomas. Surg. Neurol. Int. 2015, 6, S89–S95. [Google Scholar] [CrossRef] [PubMed]
- Li, C.M.; Haratipour, P.; Lingeman, R.G.; Perry, J.J.P.; Gu, L.; Hickey, R.J.; Malkas, L.H. Novel peptide therapeutic approaches for cancer treatment. Cells 2021, 10, 2908. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, F.; Weissmann, S.; Leblanc, B.; Pandey, D.P.; Højfeldt, J.W.; Comet, I.; Zheng, C.; Johansen, J.V.; Rapin, N.; Porse, B.T.; et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 2017, 23, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Son, M.J.; Woolard, K.; Donin, N.M.; Li, A.; Cheng, C.H.; Kotliarova, S.; Kotliarov, Y.; Walling, J.; Ahn, S.; et al. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell 2008, 13, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Kim, M.; Woo, D.-H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef] [Green Version]
- Fan, T.-Y.; Wang, H.; Xiang, P.; Liu, Y.-W.; Li, H.-Z.; Lei, B.-X.; Yu, M.; Qi, S.-T. Inhibition of EZH2 reverses chemotherapeutic drug TMZ chemosensitivity in glioblastoma. Int. J. Clin. Exp. Pathol. 2014, 7, 6662–6670. [Google Scholar]
- Amara, S.; Tiriveedhi, V. Inflammatory role of high salt level in tumor microenvironment (review). Int. J. Oncol. 2017, 50, 1477–1481. [Google Scholar] [CrossRef] [Green Version]
- Leslie, T.K.; James, A.D.; Zaccagna, F.; Grist, J.T.; Deen, S.; Kennerley, A.; Riemer, F.; Kaggie, J.D.; Gallagher, F.A.; Gilbert, F.J.; et al. Sodium homeostasis in the tumour microenvironment. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2019, 1872, 188304. [Google Scholar] [CrossRef]
- Kahle, K.T.; Rinehart, J.; Lifton, R.P. Phosphoregulation of the Na-K-2Cl and K-Cl cotransporters by the WNK kinases. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2010, 1802, 1150–1158. [Google Scholar] [CrossRef] [Green Version]
- Gagnon, K.B.E.; England, R.; Delpire, E. Characterization of SPAK and OSR1, regulatory kinases of the Na-K-2Cl cotransporter. Mol. Cell Biol. 2006, 26, 689–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowd, B.F.X.; Forbush, B. PASK (proline-alanine-rich STE20-related kinase), a regulatory kinase of the Na-K-Cl cotransporter (NKCC1). J. Biol. Chem. 2003, 278, 27347–27353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nico, B.; Mangieri, D.; Tamma, R.; Longo, V.; Annese, T.; Crivellato, E.; Pollo, B.; Maderna, E.; Ribatti, D.; Salmaggi, A. Aquaporin-4 contributes to the resolution of peritumoural brain oedema in human glioblastoma multiforme after combined chemotherapy and radiotherapy. Eur. J. Cancer 2009, 45, 3315–3325. [Google Scholar] [CrossRef] [PubMed]
- Bortner, C.D.; Hughes, F.M.; Cidlowski, J.A. A primary role for K+ and Na+ efflux in the activation of apoptosis. J. Biol. Chem. 1997, 272, 32436–32442. [Google Scholar] [CrossRef] [Green Version]
- Lenart, B.; Kintner, D.B.; Shull, G.E.; Sun, D. Na-K-Cl cotransporter-mediated intracellular Na+ accumulation affects Ca2+ signaling in astrocytes in an in vitro ischemic model. J. Neurosci. 2004, 24, 9585–9597. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Sun, D. The role of Na-K-Cl co-transporter in cerebral ischemia. Neurol. Res. 2005, 27, 280–286. [Google Scholar] [CrossRef]
- Ben-Ari, Y. Excitatory actions of gaba during development: The nature of the nurture. Nat. Rev. Neurosci. 2002, 3, 728–739. [Google Scholar] [CrossRef]
- Yamada, J.; Okabe, A.; Toyoda, H.; Kilb, W.; Luhmann, H.J.; Fukuda, A. Cl− uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J. Physiol. 2004, 557, 829–841. [Google Scholar] [CrossRef]
- Owens, D.F.; Kriegstein, A.R. Is there more to GABA than synaptic inhibition? Nat. Rev. Neurosci. 2002, 3, 715–727. [Google Scholar] [CrossRef]
- Di Cristo, G.; Awad, P.N.; Hamidi, S.; Avoli, M. KCC2, epileptiform synchronization, and epileptic disorders. Prog. Neurobiol. 2018, 162, 1–16. [Google Scholar] [CrossRef]
- Yang, W.; Warrington, N.M.; Taylor, S.J.; Whitmire, P.; Carrasco, E.; Singleton, K.W.; Wu, N.; Lathia, J.D.; Berens, M.E.; Kim, A.H.; et al. Sex differences in GBM revealed by analysis of patient imaging, transcriptome, and survival data. Sci. Transl. Med. 2019, 11, eaao5253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SF8628 Human DIPG H3.3-K27M Cell Line SF8628 Pediatric Diffuse Intrinsic Pontine Glioma (DIPG) Cell Line Harbors the Histone H3.3 Lys 27-to-Methionine (K27M) Mutation and Can Support Research and Drug Development Efforts Targeting DIPG. |Sigma-Aldrich. Available online: http://www.sigmaaldrich.com/ (accessed on 8 December 2021).
- Mueller, S.; Hashizume, R.; Yang, X.; Kolkowitz, I.; Olow, A.K.; Phillips, J.; Smirnov, I.; Tom, M.W.; Prados, M.D.; James, C.D.; et al. Targeting wee1 for the treatment of pediatric high-grade gliomas. Neuro-Oncol. 2014, 16, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.M.; Gomez-Manzano, C.; Bekele, B.N.; Yung, W.K.A.; Fueyo, J. Adenovirus-based strategies overcome temozolomide resistance by silencing the O6-methylguanine-DNA methyltransferase promoter. Cancer Res. 2007, 67, 11499–11504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz, R.; Perazzoli, G.; Cabeza, L.; Jiménez-Luna, C.; Luque, R.; Prados, J.; Melguizo, C. Temozolomide: An updated overview of resistance mechanisms, nanotechnology advances and clinical applications. Curr. Neuropharmacol. 2021, 19, 513–537. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Group | n | Invasion Frequency (%) | CAM Thickness (µm) | Number of Blood Vessels |
|---|---|---|---|---|
| Median (Range) | ||||
| PBT24-control | 13 | 76.92 | 300.88 (65.23–700.87) | 15 (6–28) |
| PBT24-50 µM TMZ | 13 | 69.23 | 321.39 (67.02–516.85) | 9 (3–14) e |
| PBT24-100 µM TMZ | 10 | 20.0 a,b | 55.48 (38.4–275.2) c,d | 5.5 (3–13) f |
| SF8628-control | 13 | 84.61 | 282.5 (47.85–539.7) | 15 (5–21) |
| SF8628-50 µM TMZ | 14 | 71.43 | 419.4 (84.49–683.7) | 15 (5–29) |
| SF8628-100 µM TMZ | 13 | 53.85 | 252.1 (55.51–529.1) | 14 (7–19) |
| Study Group | PCNA-Positive Cells (%) | EZH2-Positive Cells (%) | ||
|---|---|---|---|---|
| n | Median (Range) | n | Median (Range) | |
| PBT24-control | 9 | 63.78 (34.87–80.95) | 6 | 71.00 (42.63–78.70) |
| PBT24-50 µM TMZ | 6 | 32.12 (21.78–42.42) a | 7 | 69.15 (45.38–75.37) |
| PBT24-100 µM TMZ | 6 | 18.15 (6.25–26.80) b,c | 7 | 17.11 (2.38–95.06) |
| SF8628-control | 8 | 90.81 (76.27–100) d | 8 | 85.36 (72.04–97.45) h |
| SF8628-50 µM TMZ | 6 | 76.17 (69.19–77.28) e | 6 | 81.71 (72.13–94.08) |
| SF8628-100 µM TMZ | 7 | 55.65 (12.45–83.57) f,g | 6 | 42.55 (8.68–92.45) |
| Study Group | n | CT Mean | ΔCT Mean ± SD | ΔΔCT | |
|---|---|---|---|---|---|
| SLC12A2 | GAPDH | ||||
| PBT24-control | 6 | 22.951 | 19.372 | 3.579 ± 0.73 | |
| PBT24-50 µM TMZ | 6 | 21.766 | 19.214 | 2.552 ± 0.2 a | −1.027 |
| SF8628-control | 6 | 22.894 | 19.017 | 3.876 ± 0.21 | |
| SF8628-50 µM TMZ | 6 | 22.215 | 18.966 | 3.249 ± 0.15 b,c | −0.628 |
| SLC12A5 | GAPDH | ΔCT mean ± SD | ΔΔCT | ||
| PBT24-control | 6 | 32.564 | 19.372 | 13.191 ± 0.83 | |
| PBT24-50 µM TMZ | 6 | 31.047 | 19.214 | 11.833 ± 0.19 d | −1.359 |
| SF8628-control | 6 | 36.831 | 19.017 | 17.814 ± 0.43 e | |
| SF8628-50 µM TMZ | 6 | 36.127 | 18.966 | 17.161 ± 0.29 f | −0.652 |
| Study Group | n | CT Mean | ΔCT Mean ± SD | ΔΔCT | |
|---|---|---|---|---|---|
| CDH1 | GAPDH | ||||
| PBT24-control | 6 | 33.476 | 19.372 | 14.104 ± 1.05 | |
| PBT24-50 µM TMZ | 6 | 32.294 | 19.214 | 13.079 ± 0.81 | −1.024 |
| SF8628-control | 6 | 38.689 | 19.017 | 19.672 ± 0.51 a | |
| SF8628-50 µM TMZ | 6 | 38.851 | 18.966 | 19.885 ± 0.22 b | 0.213 |
| CDH2 | GAPDH | ΔCT mean ± SD | ΔΔCT | ||
| PBT24-control | 6 | 22.924 | 19.372 | 3.552 ± 0.98 | |
| PBT24-50 µM TMZ | 6 | 22.182 | 19.214 | 2.968 ± 0.11 | −0.584 |
| SF8628-control | 6 | 23.449 | 19.017 | 4.432 ± 0.23 | |
| SF8628-50 µM TMZ | 6 | 23.177 | 18.966 | 4.211 ± 0.21 c | −0.221 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Damanskienė, E.; Balnytė, I.; Valančiūtė, A.; Alonso, M.M.; Preikšaitis, A.; Stakišaitis, D. The Different Temozolomide Effects on Tumorigenesis Mechanisms of Pediatric Glioblastoma PBT24 and SF8628 Cell Tumor in CAM Model and on Cells In Vitro. Int. J. Mol. Sci. 2022, 23, 2001. https://doi.org/10.3390/ijms23042001
Damanskienė E, Balnytė I, Valančiūtė A, Alonso MM, Preikšaitis A, Stakišaitis D. The Different Temozolomide Effects on Tumorigenesis Mechanisms of Pediatric Glioblastoma PBT24 and SF8628 Cell Tumor in CAM Model and on Cells In Vitro. International Journal of Molecular Sciences. 2022; 23(4):2001. https://doi.org/10.3390/ijms23042001
Chicago/Turabian StyleDamanskienė, Eligija, Ingrida Balnytė, Angelija Valančiūtė, Marta Maria Alonso, Aidanas Preikšaitis, and Donatas Stakišaitis. 2022. "The Different Temozolomide Effects on Tumorigenesis Mechanisms of Pediatric Glioblastoma PBT24 and SF8628 Cell Tumor in CAM Model and on Cells In Vitro" International Journal of Molecular Sciences 23, no. 4: 2001. https://doi.org/10.3390/ijms23042001