
TAMpepK Suppresses Metastasis through the Elimination of M2-Like Tumor-Associated Macrophages in Triple-Negative Breast Cancer

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
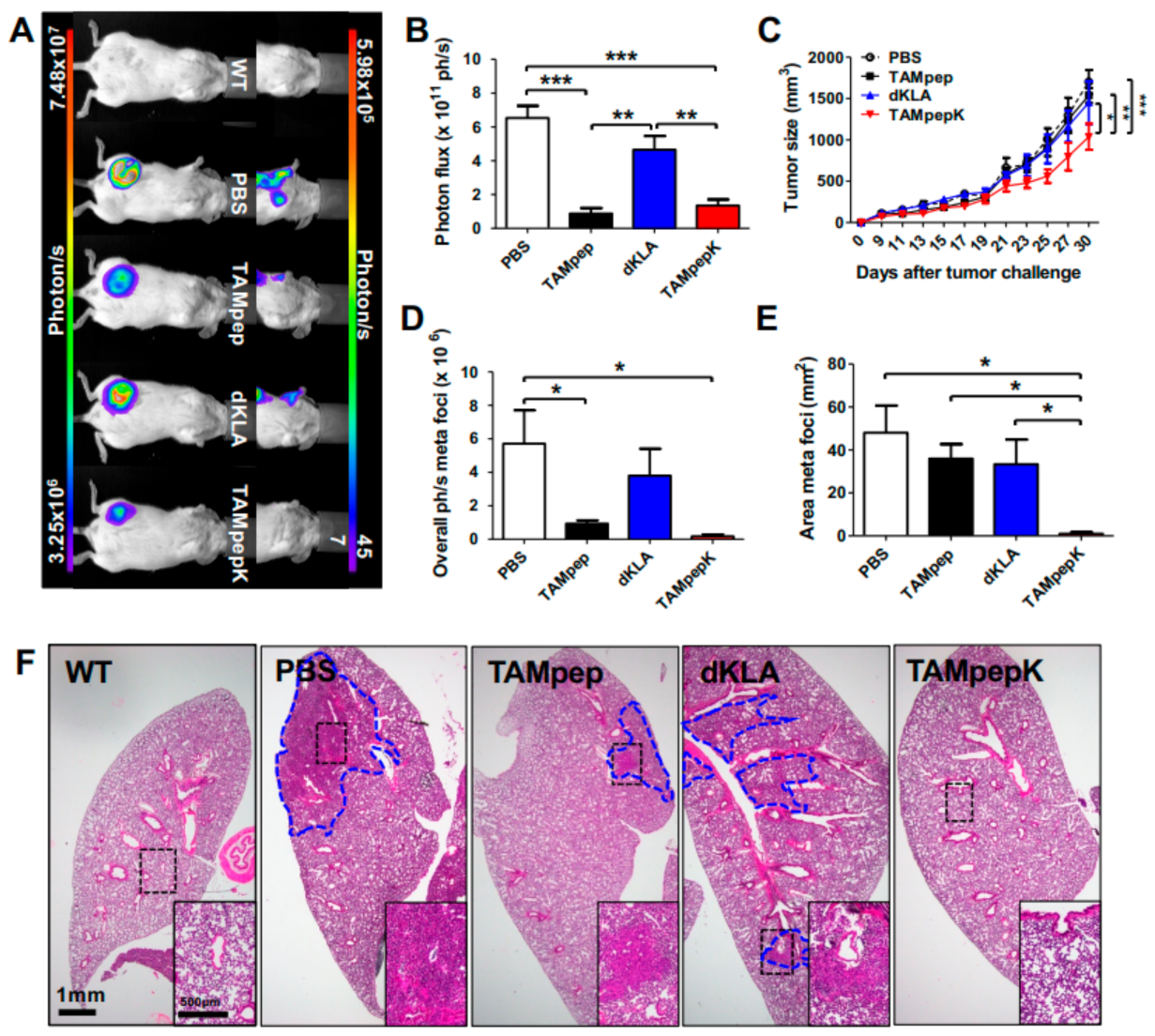
2.1. Inhibition of Breast Cancer Metastasis by TAMpepK in the Orthotopic Model
2.2. Elimination of M2-Like TAMs by TAMpepK in the Primary Tumor and Lymph Node of the Orthotopic Model
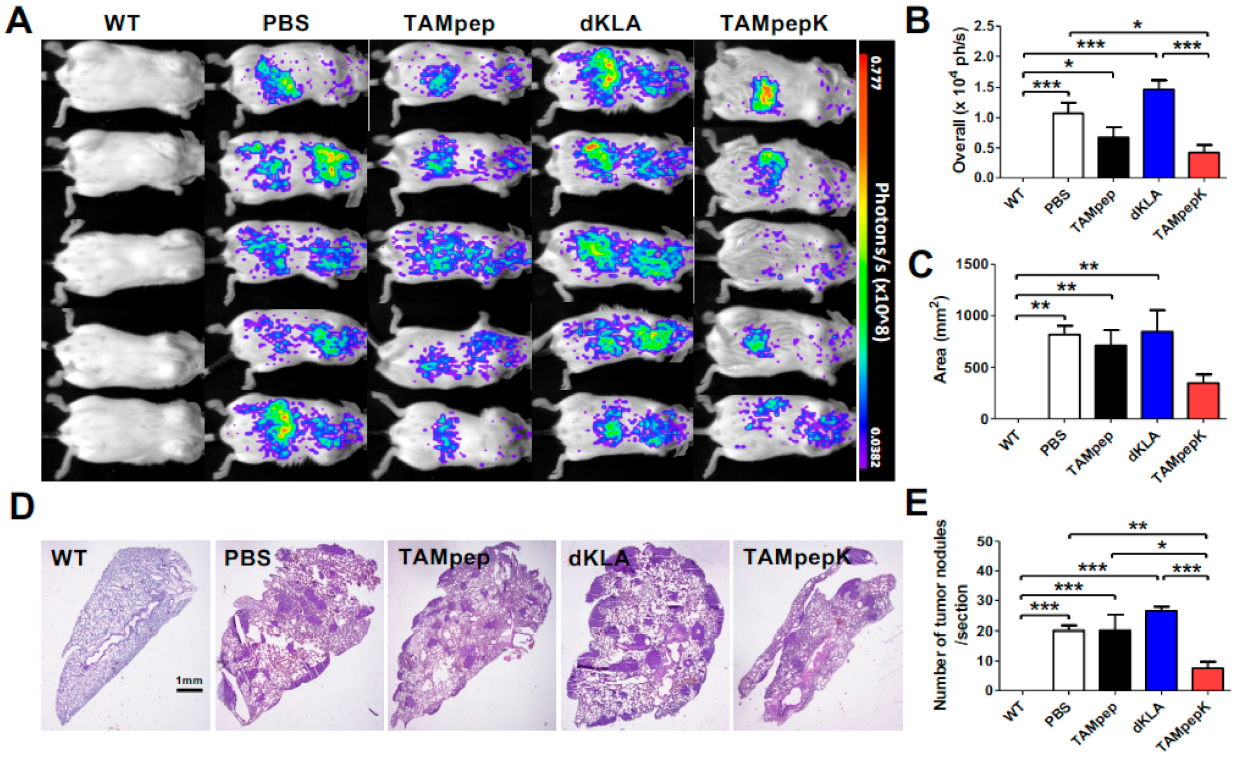
2.3. Inhibition of Metastatic Pulmonary Colonization by TAMpepK in the 4T1-Luc2 Breast Cancer Tail Vein Injection Model
2.4. Depletion of Metastatic M2 TAMs and Related Genes by TAMpepK in the Tail Vein Injection Model
2.5. Depletion of CD8+ T Cells Related to TAMs by TAMpepK in the Tumor Stroma
3. Discussion
4. Materials and Methods
4.1. Peptides
4.2. Cell Cultures
4.3. Experimental Animals
4.4. Measurement of Tumor Growth
4.5. Quantitative Real-Time PCR
4.6. Hematoxylin and Eosin (H&E) Staining
4.7. Immunohistochemistry
4.8. Flow Cytometry
4.9. Immunofluorescence
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Fang, C.; Xu, X.; Li, A.; Cai, Q.; Long, X. Androgen receptor, EGFR, and BRCA1 as biomarkers in triple-negative breast cancer: A meta-analysis. BioMed Res. Int. 2015, 2015, 357485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gluz, O.; Liedtke, C.; Gottschalk, N.; Pusztai, L.; Nitz, U.; Harbeck, N. Triple-negative breast cancer—Current status and future directions. Ann. Oncol. 2009, 20, 1913–1927. [Google Scholar] [CrossRef]
- Chaudhary, L.N.; Wilkinson, K.H.; Kong, A. Triple-Negative Breast Cancer: Who Should Receive Neoadjuvant Chemotherapy? Surg. Oncol. Clin. N. Am. 2018, 27, 141–153. [Google Scholar] [CrossRef]
- Keren, L.; Bosse, M.; Marquez, D.; Angoshtari, R.; Jain, S.; Varma, S.; Yang, S.R.; Kurian, A.; Van Valen, D.; West, R.; et al. A Structured Tumor-Immune Microenvironment in Triple Negative Breast Cancer Revealed by Multiplexed Ion Beam Imaging. Cell 2018, 174, 1373–1387.e19. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, T.; Qian, B.Z.; Soong, D.; Cassetta, L.; Noy, R.; Sugano, G.; Kato, Y.; Li, J.; Pollard, J.W. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 2015, 212, 1043–1059. [Google Scholar] [CrossRef]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.; Deng, Y.; Im, J.H.; Muschel, R.J.; Zou, Y.; Li, J.; Lang, R.A.; Pollard, J.W. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS ONE 2009, 4, e6562. [Google Scholar] [CrossRef] [Green Version]
- Genard, G.; Lucas, S.; Michiels, C. Reprogramming of Tumor-Associated Macrophages with Anticancer Therapies: Radiotherapy versus Chemo- and Immunotherapies. Front. Immunol. 2017, 8, 828. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zhang, S.; Wang, Q.; Zhang, X. Tumor-recruited M2 macrophages promote gastric and breast cancer metastasis via M2 macrophage-secreted CHI3L1 protein. J. Hematol. Oncol. 2017, 10, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, C.E.; Pollard, J.W. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef] [Green Version]
- Hao, N.B.; Lu, M.H.; Fan, Y.H.; Cao, Y.L.; Zhang, Z.R.; Yang, S.M. Macrophages in tumor microenvironments and the progression of tumors. Clin. Dev. Immunol. 2012, 2012, 948098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, C.; Han, J.; Bai, J.; Xia, Y.; Wang, S. Trojan-Like Peptide Drug Conjugate Design and Construction for Application in Treatment of Triple-Negative Breast Cancer. J. Biomed. Nanotechnol. 2021, 17, 1554–1563. [Google Scholar] [CrossRef]
- Demeule, M.; Charfi, C.; Currie, J.C.; Larocque, A.; Zgheib, A.; Kozelko, S.; Béliveau, R.; Marsolais, C.; Annabi, B. TH1902, a new docetaxel-peptide conjugate for the treatment of sortilin-positive triple-negative breast cancer. Cancer Sci. 2021, 112, 4317–4334. [Google Scholar] [CrossRef]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The future of peptide-based drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef]
- Cooper, B.M.; Iegre, J.; Donovan, D.H.O. Olwegard Halvarsson M, Spring DR, Peptides as a platform for targeted therapeutics for cancer: Peptide-drug conjugates (PDCs). Chem. Soc. Rev. 2021, 50, 1480–1494. [Google Scholar] [CrossRef]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Bae, S.-J.S.; Joo, H.; Bae, H. Melittin suppresses tumor progression by regulating tumor-associated macrophages in a Lewis lung carcinoma mouse model. Oncotarget 2017, 8, 54951–54965. [Google Scholar] [CrossRef] [Green Version]
- Mai, J.C.; Mi, Z.; Kim, S.H.; Ng, B.; Robbins, P.D. A proapoptotic peptide for the treatment of solid tumors. Cancer Res. 2001, 61, 7709–7712. [Google Scholar] [PubMed]
- Lee, C.; Jeong, H.; Bae, Y.; Shin, K.; Kang, S.; Kim, H.; Oh, J.; Bae, H. Targeting of M2-like tumor-associated macrophages with a melittin-based pro-apoptotic peptide. J. Immunother. Cancer 2019, 7, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brutkiewicz, S.; Mendonca, M.; Stantz, K.; Comerford, K.; Bigsby, R.; Hutchins, G.; Goebl, M.; Harrington, M. The expression level of luciferase within tumour cells can alter tumour growth upon in vivo bioluminescence imaging. Lumin. J. Biol. Chem. Lumin. 2007, 22, 221–228. [Google Scholar] [CrossRef]
- Baklaushev, V.P.; Kilpelainen, A.; Petkov, S.; Abakumov, M.A.; Grinenko, N.F.; Yusubalieva, G.M.; Latanova, A.A.; Gubskiy, I.L.; Zabozlaev, F.G.; Starodubova, E.S.; et al. Luciferase Expression Allows Bioluminescence Imaging But Imposes Limitations on the Orthotopic Mouse (4T1) Model of Breast Cancer. Sci. Rep. 2017, 7, 7715. [Google Scholar] [CrossRef]
- Leung, C.S.; Li, J.; Xu, F.; Wong, A.S.L.; Lui, K.O. Ectopic expression of recipient CD47 inhibits mouse macrophage-mediated immune rejection against human stem cell transplants. FASEB J. 2019, 33, 484–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azad, A.K.; Rajaram, M.V.; Metz, W.L.; Cope, F.O.; Blue, M.S.; Vera, D.R.; Schlesinger, L.S. gamma-Tilmanocept, a New Radiopharmaceutical Tracer for Cancer Sentinel Lymph Nodes, Binds to the Mannose Receptor (CD206). J. Immunol. 2015, 195, 2019–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gobert, M.; Treilleux, I.; Bendriss-Vermare, N.; Bachelot, T.; Goddard-Leon, S.; Arfi, V.; Biota, C.; Doffin, A.C.; Durand, I.; Olive, D.; et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res. 2009, 69, 2000–2009. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Goodison, S.; Urquidi, V.; Tarin, D. CD44 cell adhesion molecules. Mol. Pathol. 1999, 52, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.Y.; Jones, J.G.; Li, P.; Zhu, L.; Whitney, K.D.; Muller, W.J.; Pollard, J.W. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am. J. Pathol. 2003, 163, 2113–2126. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.Y.; Li, J.F.; Gnatovskiy, L.; Deng, Y.; Zhu, L.; Grzesik, D.A.; Qian, H.; Xue, X.-N.; Pollard, J.W. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006, 66, 11238–11246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.W.; Min, H.S.; Lee, K.H.; Kim, Y.J.; Oh, D.Y.; Jeon, Y.K.; Lee, S.-H.; Im, S.-A.; Chung, D.H.; Kim, T.-Y.; et al. High tumour islet macrophage infiltration correlates with improved patient survival but not with EGFR mutations, gene copy number or protein expression in resected non-small cell lung cancer. Br. J. Cancer 2008, 98, 1118–1124. [Google Scholar] [CrossRef]
- van der Bij, G.J.; Bogels, M.; Oosterling, S.J.; Kroon, J.; Schuckmann, D.T.; de Vries, H.E.; Meijer, S.; Beelen, R.H.; van Egmond, M. Tumor infiltrating macrophages reduce development of peritoneal colorectal carcinoma metastases. Cancer Lett. 2008, 262, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Leek, R.D.; Lewis, C.E.; Whitehouse, R.; Greenall, M.; Clarke, J.; Harris, A.L. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 1996, 56, 4625–4629. [Google Scholar] [PubMed]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. J. Pathol. Soc. Great Br. Irel. 2002, 196, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Leek, R.D.; Hunt, N.C.; Landers, R.J.; Lewis, C.E.; Royds, J.A.; Harris, A.L. Macrophage infiltration is associated with VEGF and EGFR expression in breast cancer. J. Pathol. J. Pathol. Soc. Great Br. Irel. 2000, 190, 430–436. [Google Scholar] [CrossRef]
- Deng, J.; Liu, Y.; Lee, H.; Herrmann, A.; Zhang, W.; Zhang, C.; Shen, S.; Priceman, S.J.; Kujawski, M.; Pal, S.K.; et al. S1PR1-STAT3 signaling is crucial for myeloid cell colonization at future metastatic sites. Cancer Cell 2012, 21, 642–654. [Google Scholar] [CrossRef] [Green Version]
- Rashid, O.M.; Nagahashi, M.; Ramachandran, S.; Dumur, C.I.; Schaum, J.C.; Yamada, A.; Aoyagi, T.; Milstien, S.; Spiegel, S.; Takabe, K. Is tail vein injection a relevant breast cancer lung metastasis model? J. Thorac. Dis. 2013, 5, 385. [Google Scholar]
- Denkert, C.; Loibl, S.; Noske, A.; Roller, M.; Muller, B.; Komor, M.; Budczies, J.; Darb-Esfahani, S.; Kronenwett, R.; Hanusch, C.; et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J. Clin. Oncol. 2010, 28, 105–113. [Google Scholar] [CrossRef]
- Ali, H.R.; Provenzano, E.; Dawson, S.-J.; Blows, F.; Liu, B.; Shah, M.; Earl, H.M.; Poole, C.J.; Hiller, L.; Dunn, J.A.; et al. Association between CD8+ T-cell infiltration and breast cancer survival in 12,439 patients. Ann. Oncol. 2014, 25, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Lostao, L.; Anel, A.; Pardo, J. How do Cytotoxic Lymphocytes Kill Cancer Cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar]
- Hosoya, T.; Sato-Kaneko, F.; Ahmadi, A.; Yao, S.; Lao, F.; Kitaura, K.; Matsutani, T.; Carson, D.A.; Hayashi, T. Induction of oligoclonal CD8 T cell responses against pulmonary metastatic cancer by a phospholipid-conjugated TLR7 agonist. Proc. Natl. Acad. Sci. USA 2018, 115, E6836–E6844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasportas, L.S.; Gambhir, S.S. Imaging circulating tumor cells in freely moving awake small animals using a miniaturized intravital microscope. PLoS ONE 2014, 9, e86759. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.; Kim, S.; Jeong, C.; Cho, I.; Jo, J.; Han, I.-H.; Bae, H. TAMpepK Suppresses Metastasis through the Elimination of M2-Like Tumor-Associated Macrophages in Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2022, 23, 2157. https://doi.org/10.3390/ijms23042157
Lee C, Kim S, Jeong C, Cho I, Jo J, Han I-H, Bae H. TAMpepK Suppresses Metastasis through the Elimination of M2-Like Tumor-Associated Macrophages in Triple-Negative Breast Cancer. International Journal of Molecular Sciences. 2022; 23(4):2157. https://doi.org/10.3390/ijms23042157
Chicago/Turabian StyleLee, Chanju, Soyoung Kim, Chanmi Jeong, Inhee Cho, Juyeon Jo, Ik-Hwan Han, and Hyunsu Bae. 2022. "TAMpepK Suppresses Metastasis through the Elimination of M2-Like Tumor-Associated Macrophages in Triple-Negative Breast Cancer" International Journal of Molecular Sciences 23, no. 4: 2157. https://doi.org/10.3390/ijms23042157
APA StyleLee, C., Kim, S., Jeong, C., Cho, I., Jo, J., Han, I. -H., & Bae, H. (2022). TAMpepK Suppresses Metastasis through the Elimination of M2-Like Tumor-Associated Macrophages in Triple-Negative Breast Cancer. International Journal of Molecular Sciences, 23(4), 2157. https://doi.org/10.3390/ijms23042157