
Genome-Wide Association Analysis and Genomic Prediction of Thyroglobulin Plasma Levels

 ,
,  , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Results
2.1. Genome-Wide Association Analyses
2.2. SNP Heritability Estimation
2.3. Genetic Prediction of Thyroglobulin Levels (Polygenic Score PGS Analysis)
2.4. Meta-Analysis
2.5. Colocalization Analysis
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Genotyping and Imputation
4.3. Biochemical Measurements
4.4. Genome-Wide Association Analyses
4.4.1. Linear Mixed Model (LMM)
4.4.2. Bayesian Framework
4.4.3. Bayesian Sparse Linear Mixed Model (BSLMM)
4.5. SNP Heritability Estimation
4.6. Genetic Prediction of Thyroglobulin Levels (Polygenic Score PGS Analysis)
4.7. Meta-Analysis
4.8. GTEx Project
4.9. Colocalization Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Citterio, C.E.; Targovnik, H.M.; Arvan, P. The role of thyroglobulin in thyroid hormonogenesis. Nat. Rev. Endocrinol. 2019, 15, 323–338. [Google Scholar] [CrossRef]
- Dunn, J.T.; Dunn, A.D. The importance of thyroglobulin structure for thyroid hormone biosynthesis. Biochimie 1999, 81, 505–509. [Google Scholar] [CrossRef]
- Rousset, B.; Dupuy, C.; Miot, F.; Dumont, J. Chapter 2 Thyroid Hormone Synthesis and Secretion. In Endotext [Internet]; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Marino, M.; Chiovato, L.; Mitsiades, N.; Latrofa, F.; Andrews, D.; Tseleni-Balafouta, S.; Collins, A.B.; Pinchera, A.; McCluskey, R.T. Circulating thyroglobulin transcytosed by thyroid cells is complexed with secretory components of its endocytic receptor megalin. J. Clin. Endocrinol. Metab. 2000, 85, 3458–3467. [Google Scholar] [CrossRef]
- Indrasena, B.S. Use of thyroglobulin as a tumour marker. World J. Biol. Chem. 2017, 8, 81–85. [Google Scholar] [CrossRef]
- Premawardhana, L.D.; Lo, S.S.; Phillips, D.I.; Prentice, L.M.; Rees Smith, B. Variability of serum thyroglobulin levels is determined by a major gene. Clin. Endocrinol. 1994, 41, 725–729. [Google Scholar] [CrossRef]
- Vali, M.; Rose, N.R.; Caturegli, P. Thyroglobulin as autoantigen: Structure-function relationships. Rev. Endocr. Metab. Disord. 2000, 1, 69–77. [Google Scholar] [CrossRef]
- Mallet, B.; Lejeune, P.J.; Baudry, N.; Niccoli, P.; Carayon, P.; Franc, J.L. N-Glycans Modulate in-Vivo and in-Vitro Thyroid-Hormone Synthesis - Study at the N-Terminal Domain of Thyroglobulin. J. Biol. Chem. 1995, 270, 29881–29888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verge, C.; Bouchatal, A.; Chirat, F.; Guerardel, Y.; Maftah, A.; Petit, J.M. Involvement of ST6Gal I-mediated alpha2,6 sialylation in myoblast proliferation and differentiation. FEBS Open Bio 2020, 10, 56–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grollman, E.F.; Doi, S.Q.; Weiss, P.; Ashwell, G.; Wajchenberg, B.L.; Medeiros-Neto, G. Hyposialylated thyroglobulin in a patient with congenital goiter and hypothyroidism. J. Clin. Endocrinol. Metab. 1992, 74, 43–48. [Google Scholar]
- Grollman, E.F.; Saji, M.; Shimura, Y.; Lau, J.T.; Ashwell, G. Thyrotropin regulation of sialic acid expression in rat thyroid cells. J. Biol. Chem. 1993, 268, 3604–3609. [Google Scholar] [CrossRef]
- Kiljanski, J.; Ambroziak, M.; Pachucki, J.; Jazdzewski, K.; Wiechno, W.; Stachlewska, E.; Gornicka, B.; Bogdanska, M.; Nauman, J.; Bartoszewicz, Z. Thyroid sialyltransferase mRNA level and activity are increased in Graves’ disease. Thyroid 2005, 15, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Thul, P.J.; Akesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Bjork, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, M.; Ng, E.; Varki, A.; Varki, N.M. Alpha 2-6-linked sialic acids on N-glycans modulate carcinoma differentiation in vivo. Cancer Res. 2008, 68, 388–394. [Google Scholar] [CrossRef] [Green Version]
- Fenouillet, E.; Fayet, G.; Hovsepian, S.; Bahraoui, E.M.; Ronin, C. Immunochemical Evidence for a Role of Complex Carbohydrate Chains in Thyroglobulin Antigenicity. J. Biol. Chem. 1986, 261, 15153–15158. [Google Scholar] [CrossRef]
- Salabe, H.; Dominici, R.; Salabe, G.B. Immunological Properties of Tg Carbohydrates—Enhancement of Tg Immunoreaction by Removal of Sialic-Acid. Clin. Exp. Immunol. 1976, 25, 234–243. [Google Scholar]
- Sue, M.; Hayashi, M.; Kawashima, A.; Akama, T.; Tanigawa, K.; Yoshihara, A.; Hara, T.; Ishido, Y.; Ito, T.; Takahashi, S.; et al. Thyroglobulin (Tg) activates MAPK pathway to induce thyroid cell growth in the absence of TSH, insulin and serum. Biochem. Biophys. Res. Commun. 2012, 420, 611–615. [Google Scholar] [CrossRef]
- Matana, A.; Popović, M.; Boutin, T.; Torlak, V.; Brdar, D.; Gunjača, I.; Kolčić, I.; Boraska Perica, V.; Punda, A.; Rudan, I.; et al. Genetic Variants in the ST6GAL1 Gene Are Associated with Thyroglobulin Plasma Level in Healthy Individuals. Thyroid 2019, 29, 886–893. [Google Scholar] [CrossRef]
- Pruim, R.J.; Welch, R.P.; Sanna, S.; Teslovich, T.M.; Chines, P.S.; Gliedt, T.P.; Boehnke, M.; Abecasis, G.R.; Willer, C.J. LocusZoom: Regional visualization of genome-wide association scan results. Bioinformatics 2010, 26, 2336–2337. [Google Scholar] [CrossRef] [Green Version]
- Altshuler, D.M.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; Flicek, P.; Gabriel, S.B.; et al. An integrated map of genetic variation from 1092 human genomes. Nature 2012, 491, 56–65. [Google Scholar]
- Panjwani, N.; Wang, F.; Mastromatteo, S.; Bao, A.; Wang, C.; He, G.; Gong, J.; Rommens, J.M.; Sun, L.; Strug, L.J. LocusFocus: Web-based colocalization for the annotation and functional follow-up of GWAS. PLoS Comput. Biol. 2020, 16, e1008336. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.; Fan, B.; Zhao, Y.; Zhang, H.; Wang, L.; Yu, X.; Yuan, Q.; Yang, D.; Wang, S. ST6Gal-I overexpression facilitates prostate cancer progression via the PI3K/Akt/GSK-3beta/beta-catenin signaling pathway. Oncotarget 2016, 7, 65374–65388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert-Sirieix, M.; Makoukji, J.; Kimura, S.; Talbot, M.; Caillou, B.; Massaad, C.; Massaad-Massade, L. Wnt/beta-catenin signaling pathway is a direct enhancer of thyroid transcription factor-1 in human papillary thyroid carcinoma cells. PLoS ONE 2011, 6, e22280. [Google Scholar] [CrossRef] [Green Version]
- Civitareale, D.; Lonigro, R.; Sinclair, A.J.; Di Lauro, R. A thyroid-specific nuclear protein essential for tissue-specific expression of the thyroglobulin promoter. EMBO J. 1989, 8, 2537–2542. [Google Scholar] [CrossRef] [PubMed]
- Frenzel, R.; Krohn, K.; Eszlinger, M.; Tonjes, A.; Paschke, R. Sialylation of human thyrotropin receptor improves and prolongs its cell-surface expression. Mol. Pharmacol. 2005, 68, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Korta, P.; Pochec, E. Glycosylation of thyroid-stimulating hormone receptor. Endokrynol. Pol. 2019, 70, 86–100. [Google Scholar] [CrossRef]
- Huang, H.; Shi, Y.; Liang, B.; Cai, H.; Cai, Q. Iodinated TG in Thyroid Follicular Lumen Regulates TTF-1 and PAX8 Expression via TSH/TSHR Signaling Pathway. J. Cell Biochem. 2017, 118, 3444–3451. [Google Scholar] [CrossRef]
- Suzuki, K.; Lavaroni, S.; Mori, A.; Ohta, M.; Saito, J.; Pietrarelli, M.; Singer, D.S.; Kimura, S.; Katoh, R.; Kawaoi, A.; et al. Autoregulation of thyroid-specific gene transcription by thyroglobulin. Proc. Natl. Acad. Sci. USA 1998, 95, 8251–8256. [Google Scholar] [CrossRef] [Green Version]
- Sellitti, D.F.; Suzuki, K. Intrinsic Regulation of Thyroid Function by Thyroglobulin. Thyroid 2014, 24, 625–638. [Google Scholar] [CrossRef] [Green Version]
- Ulianich, L.; Suzuki, K.; Mori, A.; Nakazato, M.; Pietrarelli, M.; Goldsmith, P.; Pacifico, F.; Consiglio, E.; Formisano, S.; Kohn, L.D. Follicular thyroglobulin (TG) suppression of thyroid-restricted genes involves the apical membrane asialoglycoprotein receptor and TG phosphorylation. J. Biol. Chem. 1999, 274, 25099–25107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Ishido, Y.; Hiroi, N.; Ishii, N.; Suzuki, K. The Emerging Roles of Thyroglobulin. Adv. Endocrinol. 2014, 2014, 1–7. [Google Scholar] [CrossRef]
- Marino, M.; McCluskey, R.T. Role of thyroglobulin endocytic pathways in the control of thyroid hormone release. Am. J. Physiol. Cell Physiol. 2000, 279, C1295–C1306. [Google Scholar] [CrossRef] [PubMed]
- Onagoruwa, O.T.; Pal, G.; Ochu, C.; Ogunwobi, O.O. Oncogenic Role of PVT1 and Therapeutic Implications. Front. Oncol. 2020, 10, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Chen, J.; Feng, J.; Wang, J. Long noncoding RNA PVT1 modulates thyroid cancer cell proliferation by recruiting EZH2 and regulating thyroid-stimulating hormone receptor (TSHR). Tumor Biol. 2016, 37, 3105–3113. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudzińska, M.; Gaweł, D.; Sikorska, J.; Karpińska, K.M.; Kiedrowski, M.; Stępień, T.; Marchlewska, M.; Czarnocka, B. The Role of Podoplanin in the Biology of Differentiated Thyroid Cancers. PLoS ONE 2014, 9, e96541. [Google Scholar] [CrossRef] [Green Version]
- Tseng, C.P.; Leong, K.K.; Liou, M.J.; Hsu, H.L.; Lin, H.C.; Chen, Y.A.; Lin, J.D. Circulating epithelial cell counts for monitoring the therapeutic outcome of patients with papillary thyroid carcinoma. Oncotarget 2017, 8, 77453–77464. [Google Scholar] [CrossRef] [Green Version]
- Sikorska, J.; Gaweł, D.; Domek, H.; Rudzińska, M.; Czarnocka, B. Podoplanin (PDPN) affects the invasiveness of thyroid carcinoma cells by inducing ezrin, radixin and moesin (E/R/M) phosphorylation in association with matrix metalloproteinases. BMC Cancer 2019, 19, 85. [Google Scholar] [CrossRef]
- Zhu, X.; Morales, F.C.; Agarwal, N.K.; Dogruluk, T.; Gagea, M.; Georgescu, M.M. Moesin Is a Glioma Progression Marker That Induces Proliferation and Wnt/β-Catenin Pathway Activation via Interaction with CD44. Cancer Res. 2013, 73, 1142–1155. [Google Scholar] [CrossRef] [Green Version]
- Müller, J.; Ritt, D.A.; Copeland, T.D.; Morrison, D.K. Functional analysis of C-TAK1 substrate binding and identification of PKP2 as a new C-TAK1 substrate. EMBO J. 2003, 22, 4431–4442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatzfeld, M.; Wolf, A.; Keil, R. Plakophilins in Desmosomal Adhesion and Signaling. Cell Commun. Adhes. 2014, 21, 25–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niell, N.; Larriba, M.J.; Ferrer-Mayorga, G.; Sánchez-Pérez, I.; Cantero, R.; Real, F.X.; Del Peso, L.; Muñoz, A.; González-Sancho, J.M. The human PKP2/plakophilin-2 gene is induced by Wnt/β-catenin in normal and colon cancer-associated fibroblasts. Int. J. Cancer 2018, 142, 792–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoftijzer, H.C.; Liu, Y.Y.; Morreau, H.; van Wezel, T.; Pereira, A.M.; Corssmit, E.P.M.; Romijn, J.A.; Smit, J.W.A. Retinoic acid receptor and retinoid X receptor subtype expression for the differential diagnosis of thyroid neoplasms. Eur. J. Endocrinol. 2009, 160, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Czajka, A.A.; Wójcicka, A.; Kubiak, A.; Kotlarek, M.; Bakuła-Zalewska, E.; Koperski, L.; Wiechno, W.; Jażdżewski, K. Family of microRNA-146 Regulates RARβ in Papillary Thyroid Carcinoma. PLoS ONE 2016, 11, e0151968. [Google Scholar] [CrossRef] [PubMed]
- Namba, H.; Yamashita, S.; Morita, S.; Villadolid, M.C.; Kimura, H.; Yokoyama, N.; Izumi, M.; Ishikawa, N.; Ito, K.; Nagataki, S. Retinole acid inhibits human thyroid peroxidase and thyroglobulin gene expression in cultured human thyrocytes. J. Endocrinol. Investig. 1993, 16, 87–93. [Google Scholar] [CrossRef]
- Kurebayashi, J.; Tanaka, K.; Otsuki, T.; Moriya, T.; Kunisue, H.; Uno, M.; Sonoo, H. All-Trans-Retinoic Acid Modulates Expression Levels of Thyroglobulin and Cytokines in a New Human Poorly Differentiated Papillary Thyroid Carcinoma Cell Line, KTC-11. J. Clin. Endocrinol. Metab. 2000, 85, 2889–2896. [Google Scholar]
- Wu, Y.; Liu, H.; Gong, Y.; Zhang, B.; Chen, W. ANKRD22 enhances breast cancer cell malignancy by activating the Wnt/β-catenin pathway via modulating NuSAP1 expression. Bosn. J. Basic Med. Sci. 2021, 21, 294–304. [Google Scholar] [CrossRef]
- Pointer, T.C.; Gorelick, F.S.; Desir, G.V. Renalase: A Multi-Functional Signaling Molecule with Roles in Gastrointestinal Disease. Cells 2021, 10, 2006. [Google Scholar] [CrossRef]
- Thompson, M.; Nejak-Bowen, K.; Monga, S.P.S. Crosstalk of the Wnt Signaling Pathway. In Targeting the Wnt Pathway in Cancer; Goss, K.H., Kahn, M., Eds.; Springer: New York, NY, USA, 2011; pp. 51–80. [Google Scholar]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [Green Version]
- Rudan, I.; Marusic, A.; Jankovic, S.; Rotim, K.; Boban, M.; Lauc, G.; Grkovic, I.; Dogas, Z.; Zemunik, T.; Vatavuk, Z.; et al. “10001 Dalmatians:” Croatia launches its national biobank. Croat. Med. J. 2009, 50, 4–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durbin, R. Efficient haplotype matching and storage using the positional Burrows-Wheeler transform (PBWT). Bioinformatics 2014, 30, 1266–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, S.; Das, S.; Kretzschmar, W.; Delaneau, O.; Wood, A.R.; Teumer, A.; Kang, H.M.; Fuchsberger, C.; Danecek, P.; Sharp, K.; et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 2016, 48, 1279–1283. [Google Scholar] [PubMed] [Green Version]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing.; R Foundation for Statistical Computing: Vienna, Austria, 2016. [Google Scholar]
- Zhou, X.; Carbonetto, P.; Stephens, M. Polygenic modeling with bayesian sparse linear mixed models. PLoS Genet. 2013, 9, e1003264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel-accelerated tool for Genome-Wide Association Study. Genomics Proteom. Bioinform. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhou, X. Genetic prediction of complex traits with polygenic scores: A statistical review. Trends Genet. 2021, 37, 995–1011. [Google Scholar] [CrossRef]
- Turner, S.D. qqman: An R package for visualizing GWAS results using Q-Q and manhattan plots. J. Open Source Softw. 2018, 3, 731. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.F.; Wang, F.; Xiao, B.W.; Panjwani, N.; Lin, F.; Keenan, K.; Avolio, J.; Esmaeili, M.; Zhang, L.; He, G.M.; et al. Genetic association and transcriptome integration identify contributing genes and tissues at cystic fibrosis modifier loci. PLoS Genet. 2019, 15, e1008007. [Google Scholar] [CrossRef]
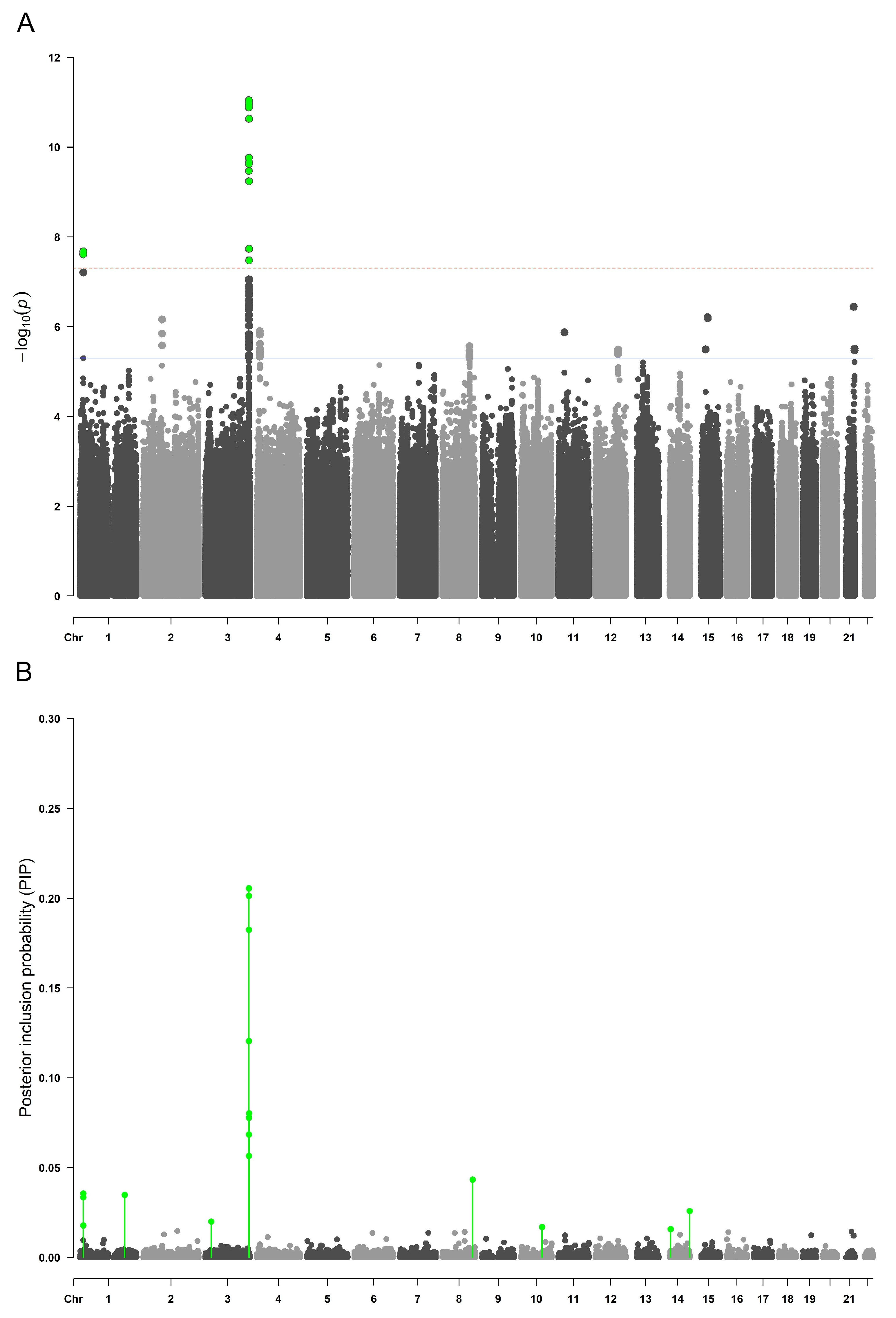




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP | Chr | Position | Gene | Ref. Allele | Effect Allele | EAF | Single-SNP LMM Analysis in Cohorts Korcula2 and Korcula3 | Multi-SNP BSLMM Analysis in Cohorts Korcula2 and Korcula3 |
|---|---|---|---|---|---|---|---|---|
| (p-Value) | (PIP) | |||||||
| rs10937280 | 3 | 186738033 | ST6GAL1 | G | A | 0.35 | −0.31 () | −0.29 (0.21) |
| rs5001409 | 3 | 186735690 | ST6GAL1 | A | C | 0.35 | −0.31 () | −0.295 (0.07) |
| rs9863411 | 3 | 186737820 | ST6GAL1 | C | T | 0.35 | −0.31 () | −0.283 (0.2) |
| rs7634389 | 3 | 186738421 | ST6GAL1 | T | C | 0.35 | −0.31 () | −0.292 (0.08) |
| rs967367 | 3 | 186734466 | ST6GAL1 | G | A | 0.35 | −0.31 () | −0.29 (0.12) |
| rs3821819 | 3 | 186732725 | ST6GAL1 | G | A | 0.35 | −0.31 () | −0.292 (0.06) |
| rs4686838 | 3 | 186743053 | ST6GAL1 | A | G | 0.45 | −0.3() | −0.27 (0.08) |
| rs10212190 | 3 | 186731157 | ST6GAL1 | A | T | 0.34 | −0.29 () | −0.28 (0.003) |
| rs4012172 | 3 | 186741511 | ST6GAL1 | C | T | 0.36 | −0.29 () | −0.27 (0.0003) |
| rs3872724 | 3 | 186741221 | ST6GAL1 | C | T | 0.37 | −0.28 () | −0.27 (0.001) |
| rs3872723 | 3 | 186741131 | ST6GAL1 | C | T | 0.36 | −0.28 () | 0 (0) |
| rs28674898 | 3 | 186744563 | ST6GAL1 | G | A | 0.39 | 0.28 () | −0.28 (0.003) |
| rs4686844 | 3 | 186765135 | ST6GAL1 | G | A | 0.56 | −0.25 () | −0.15 (0.0007) |
| rs78946539 | 1 | 13921500 | PDPN | A | G | 0.04 | −0.63 () | −0.51 (0.03) |
| rs143154928 | 1 | 13921447 | PDPN | G | A | 0.04 | −0.63 () | −0.5 (0.03) |
| rs12566684 | 1 | 13922117 | PDPN | A | G | 0.04 | −0.64 () | −0.5 (0.02) |
| rs257104 | 3 | 186775807 | ST6GAL1 | G | A | 0.4 | 0.24 () | 0.17 (0.002) |
| Cohort | Split | Korcula 1 | Korcula 2 | Korcula 3 |
|---|---|---|---|---|
| n | 605 | 489 | 593 | 505 |
| Women | 321 (53%) | 297 (61%) | 328 (55.3%) | 294 (58.2%) |
| Age | 51 (39, 61) | 56 (46, 67) | 54 (40, 65) | 54 (39, 65) |
| Tg | 9.20 (4.80, 14.50) | 10.20 (6.40, 15.70) | 10.1 (5.6, 16.4) | 10.6 (7.5, 16.1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pleić, N.; Babić Leko, M.; Gunjača, I.; Boutin, T.; Torlak, V.; Matana, A.; Punda, A.; Polašek, O.; Hayward, C.; Zemunik, T. Genome-Wide Association Analysis and Genomic Prediction of Thyroglobulin Plasma Levels. Int. J. Mol. Sci. 2022, 23, 2173. https://doi.org/10.3390/ijms23042173
Pleić N, Babić Leko M, Gunjača I, Boutin T, Torlak V, Matana A, Punda A, Polašek O, Hayward C, Zemunik T. Genome-Wide Association Analysis and Genomic Prediction of Thyroglobulin Plasma Levels. International Journal of Molecular Sciences. 2022; 23(4):2173. https://doi.org/10.3390/ijms23042173
Chicago/Turabian StylePleić, Nikolina, Mirjana Babić Leko, Ivana Gunjača, Thibaud Boutin, Vesela Torlak, Antonela Matana, Ante Punda, Ozren Polašek, Caroline Hayward, and Tatijana Zemunik. 2022. "Genome-Wide Association Analysis and Genomic Prediction of Thyroglobulin Plasma Levels" International Journal of Molecular Sciences 23, no. 4: 2173. https://doi.org/10.3390/ijms23042173