
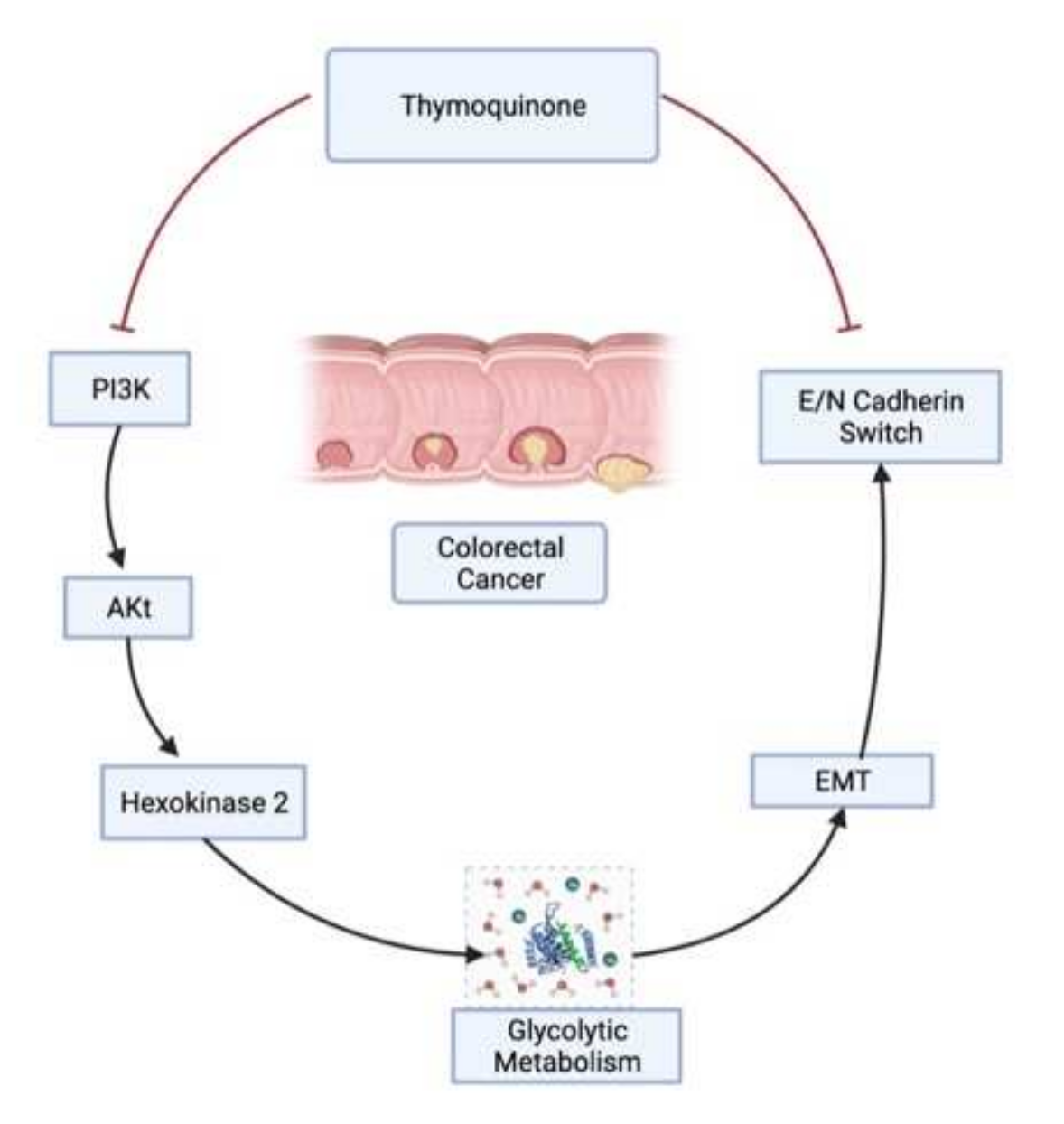
PI3K-AKT Pathway Modulation by Thymoquinone Limits Tumor Growth and Glycolytic Metabolism in Colorectal Cancer

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
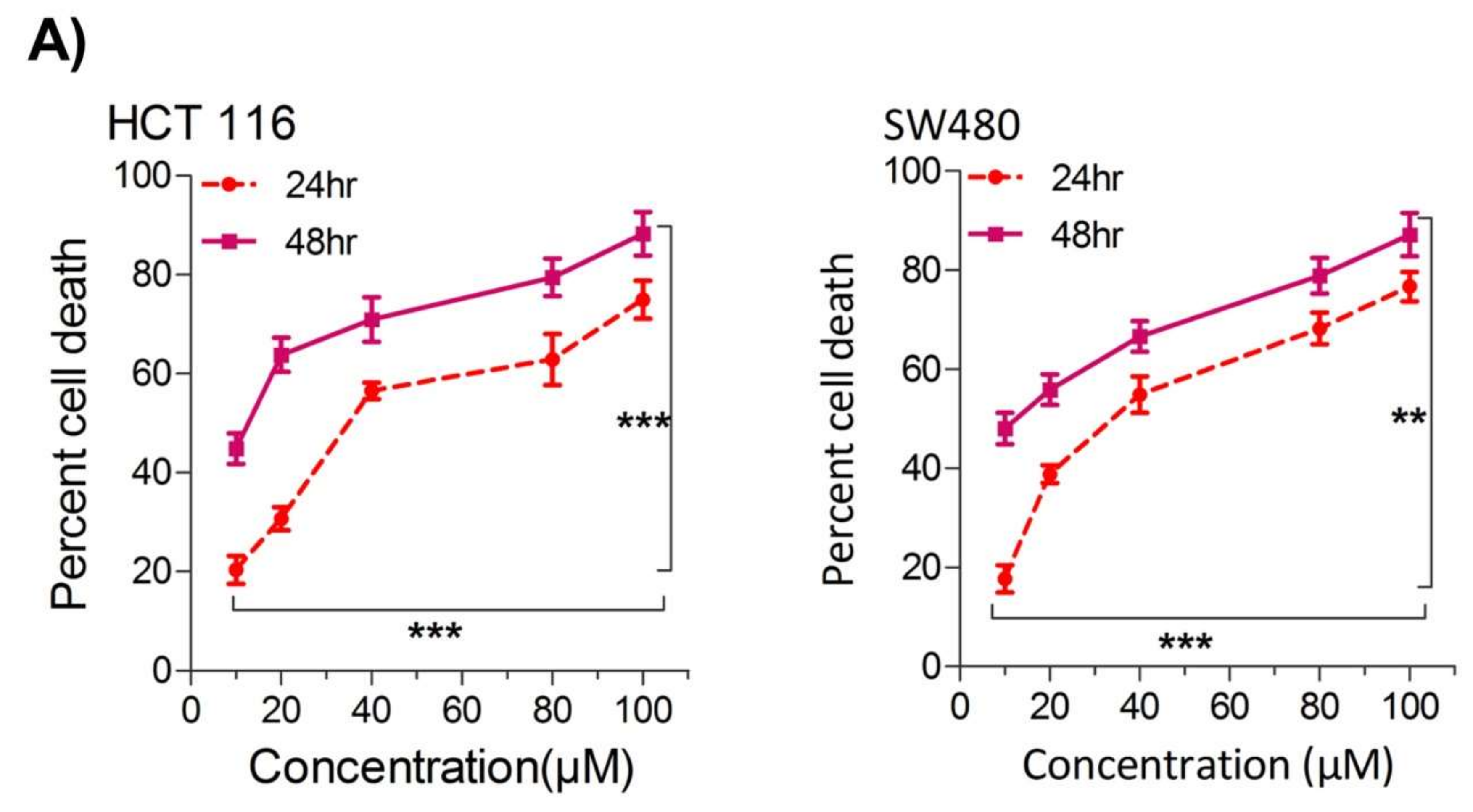
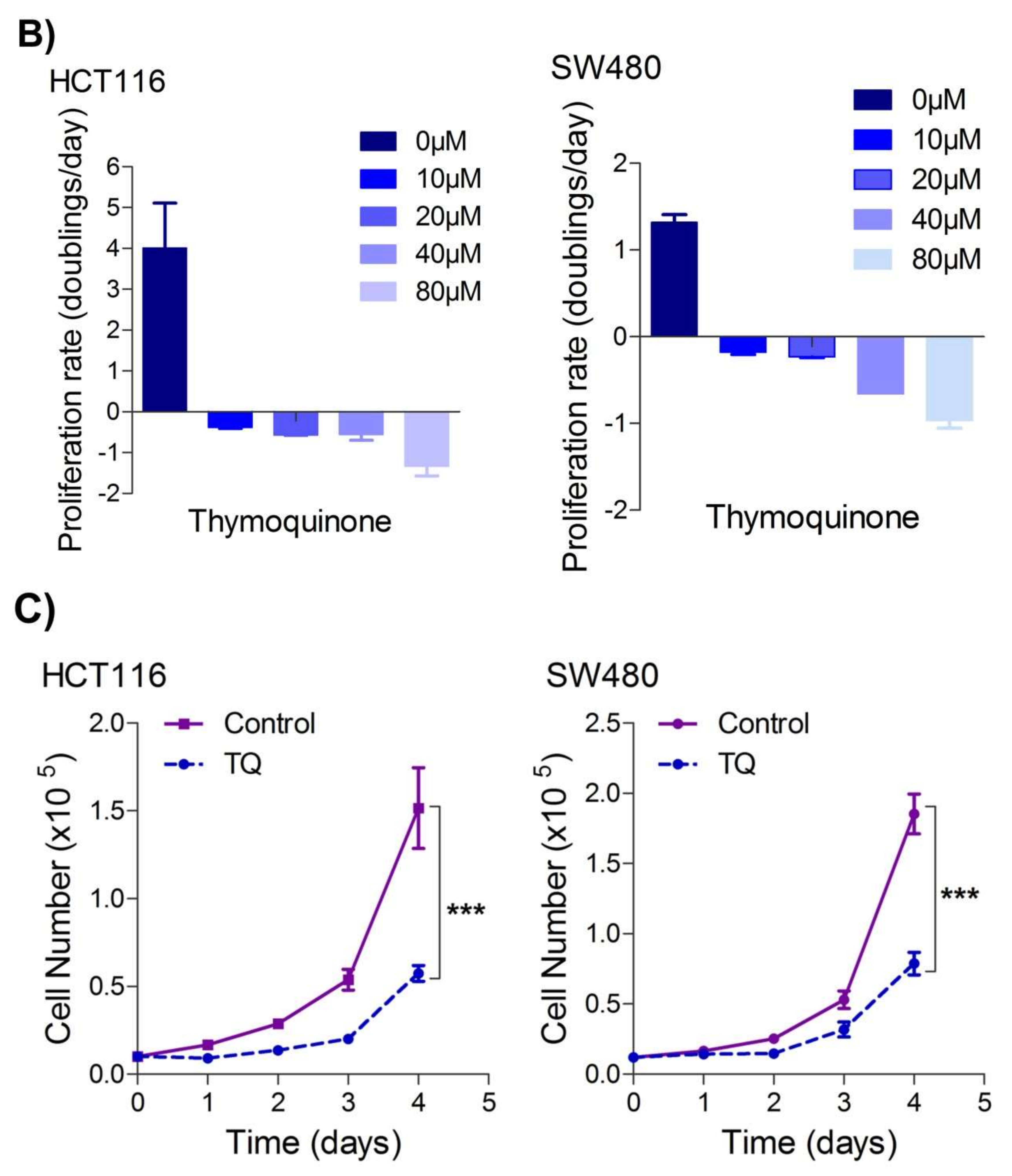
2.1. Thymoquinone Induces Cell Death in CRC Cells
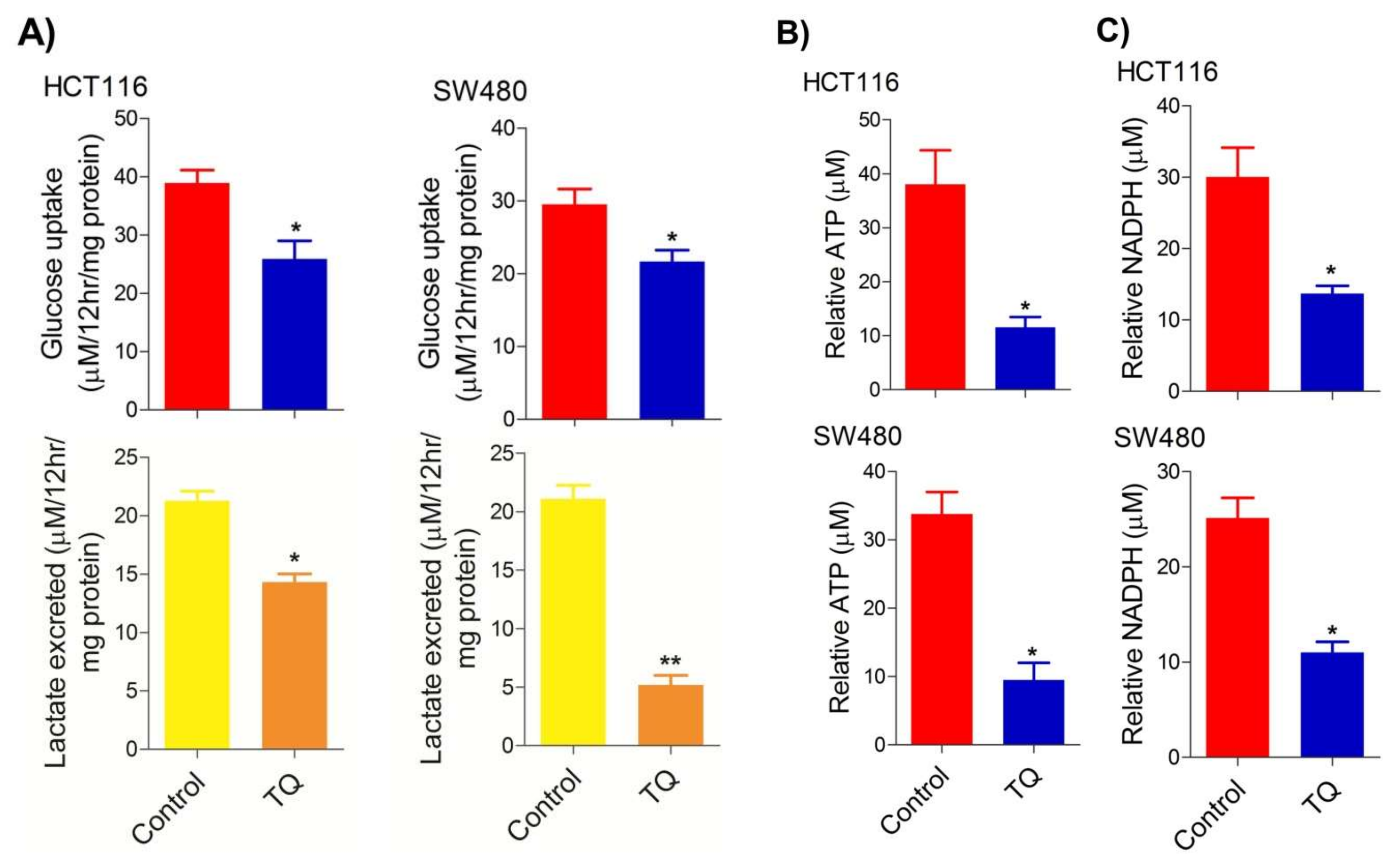
2.2. Thymoquinone Modulates Glycolytic Metabolism in CRC Cells
2.3. Thymoquinone Inhibits Hexokinase 2 via Modulating the PI3K-AKT Pathway
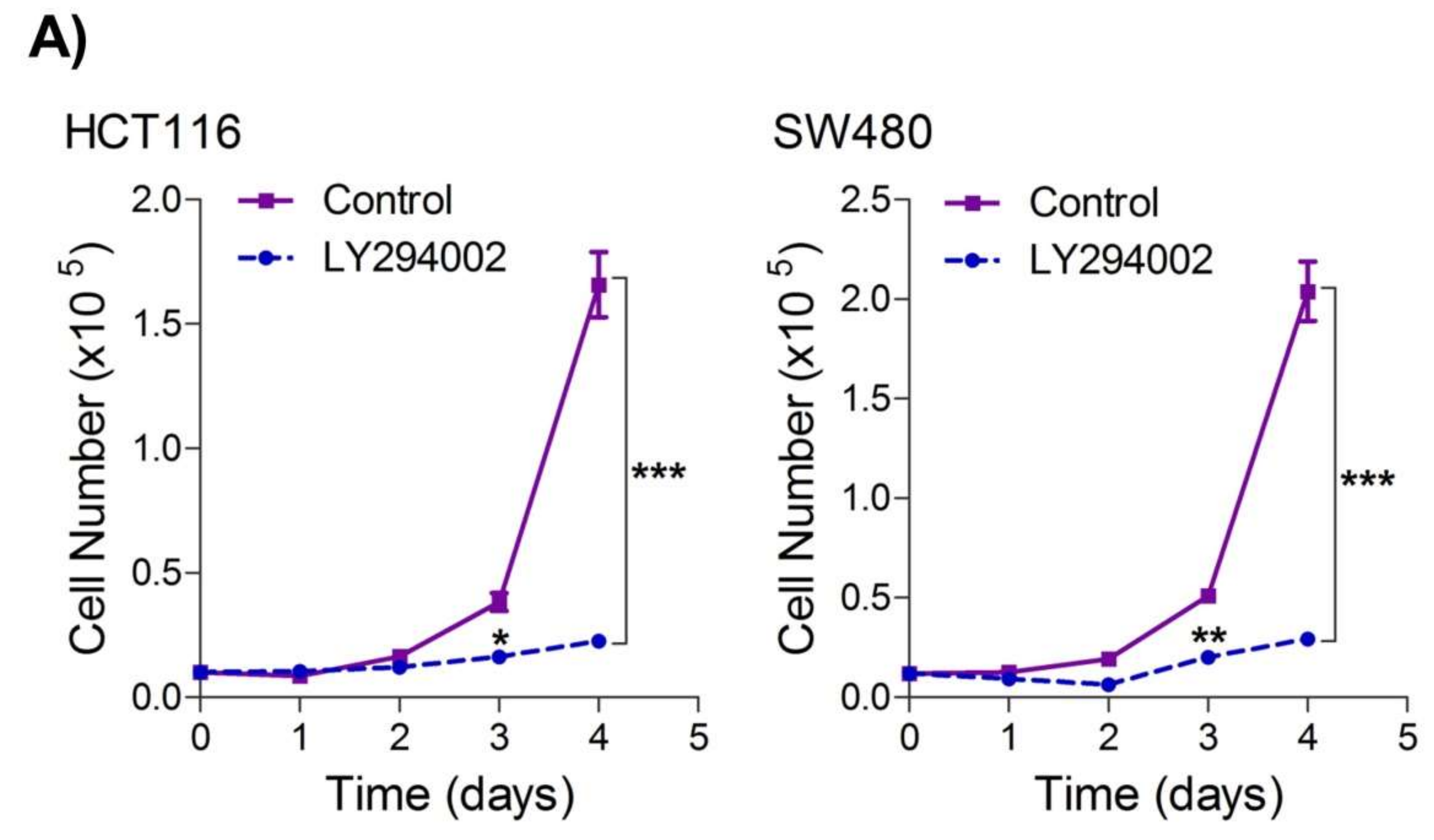
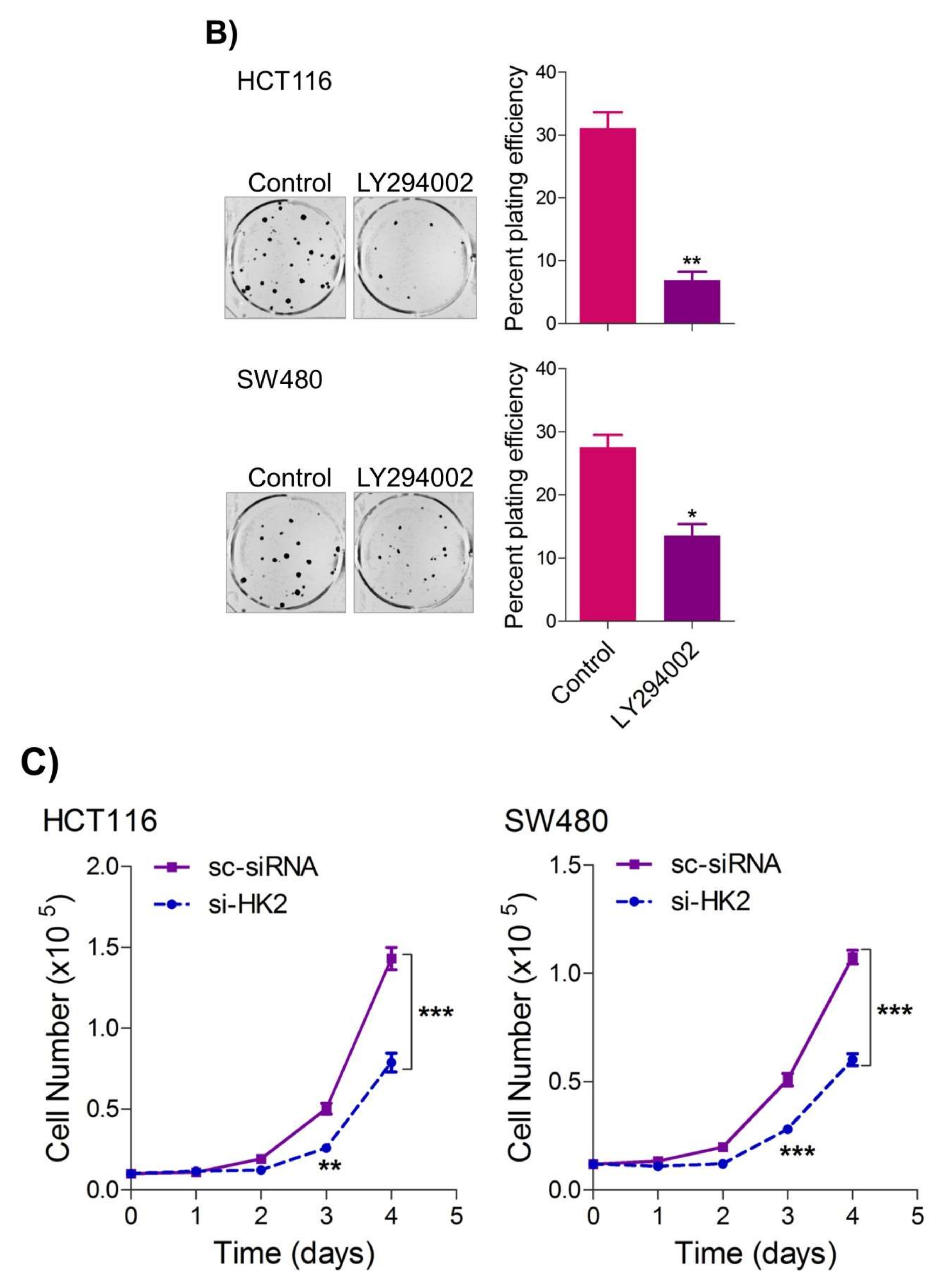
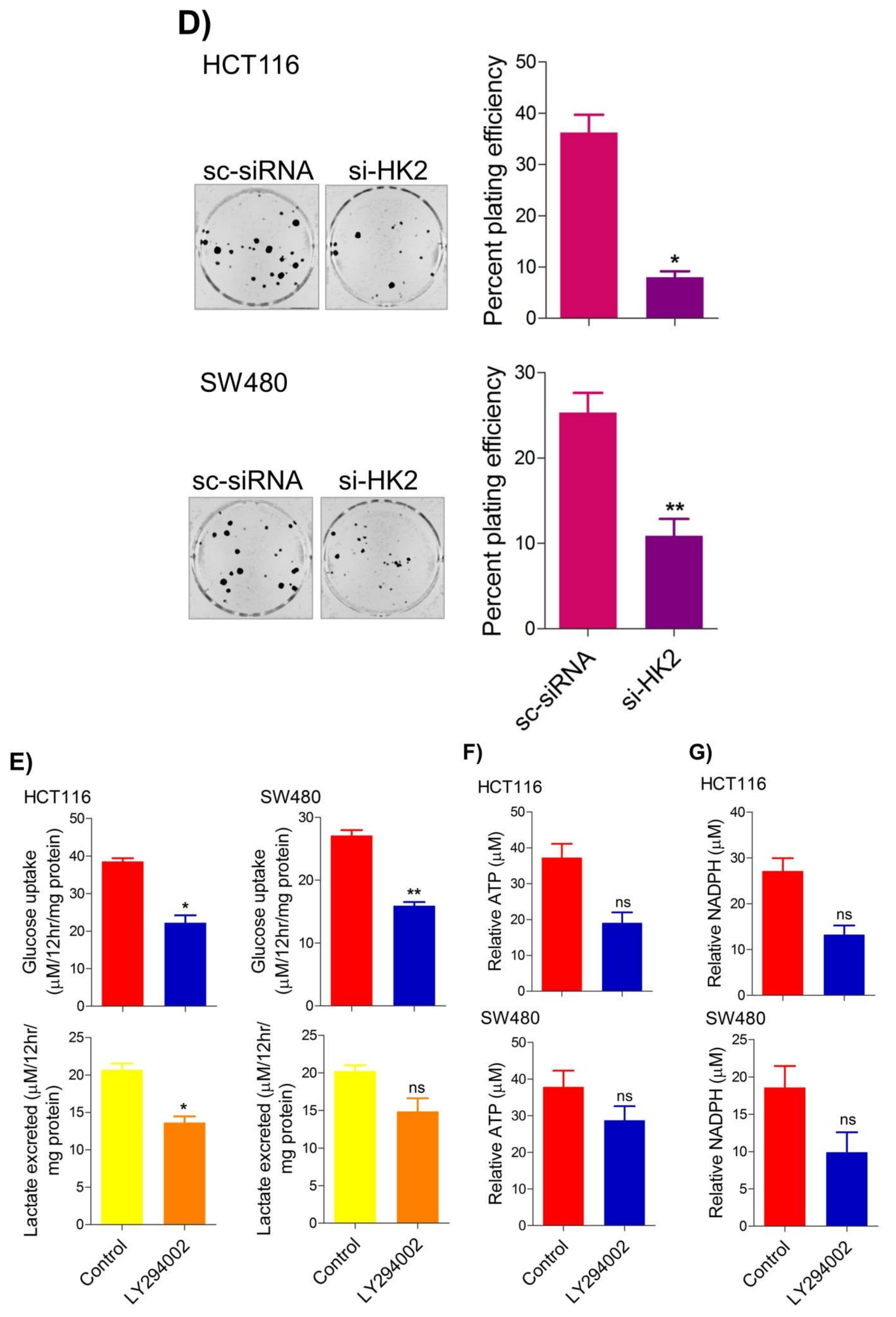
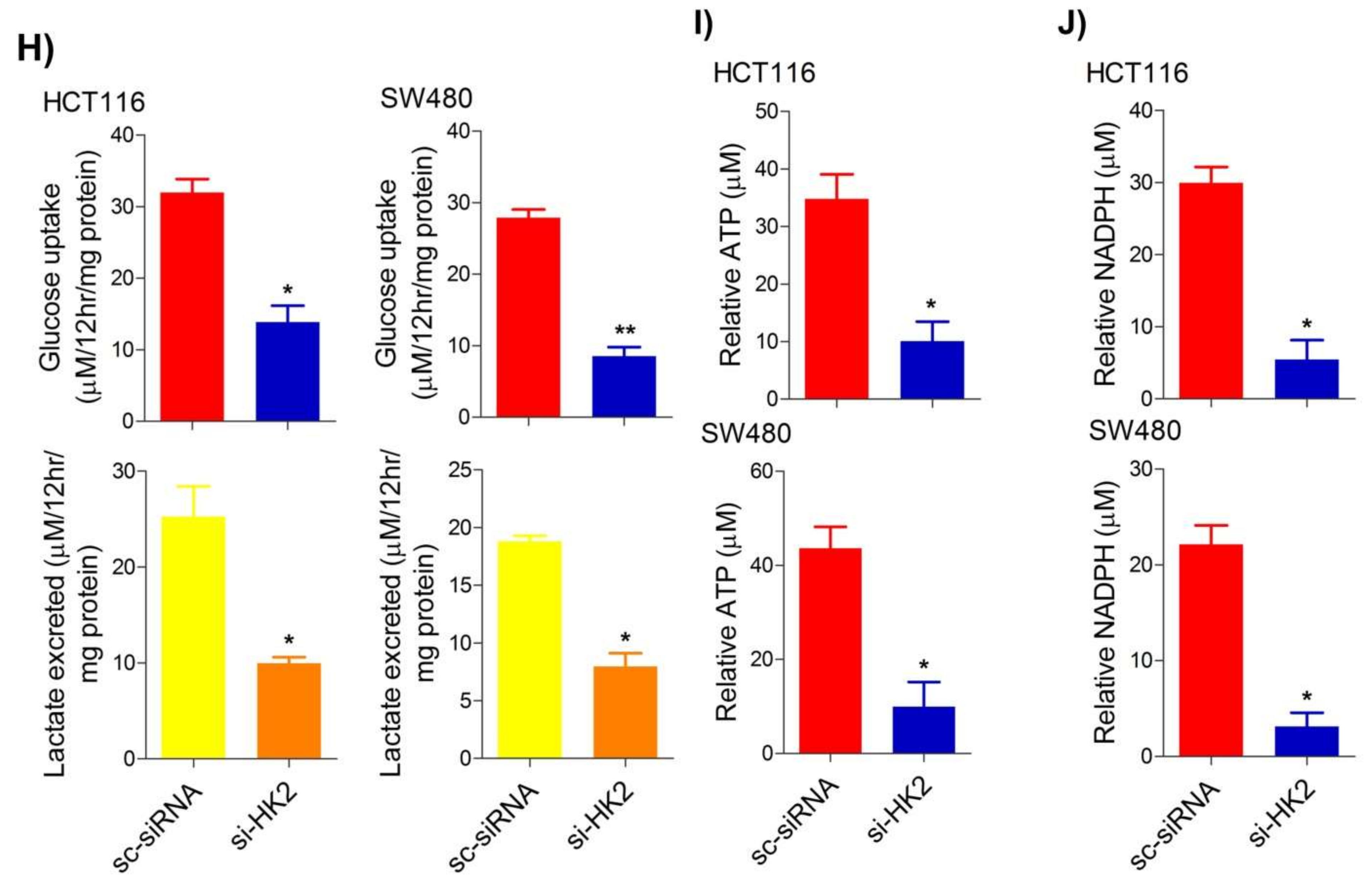
2.4. Inhibition of the PI3K-AKT/ HK2 Pathway Abrogates the Tumorigenic Potential of CRC Cells
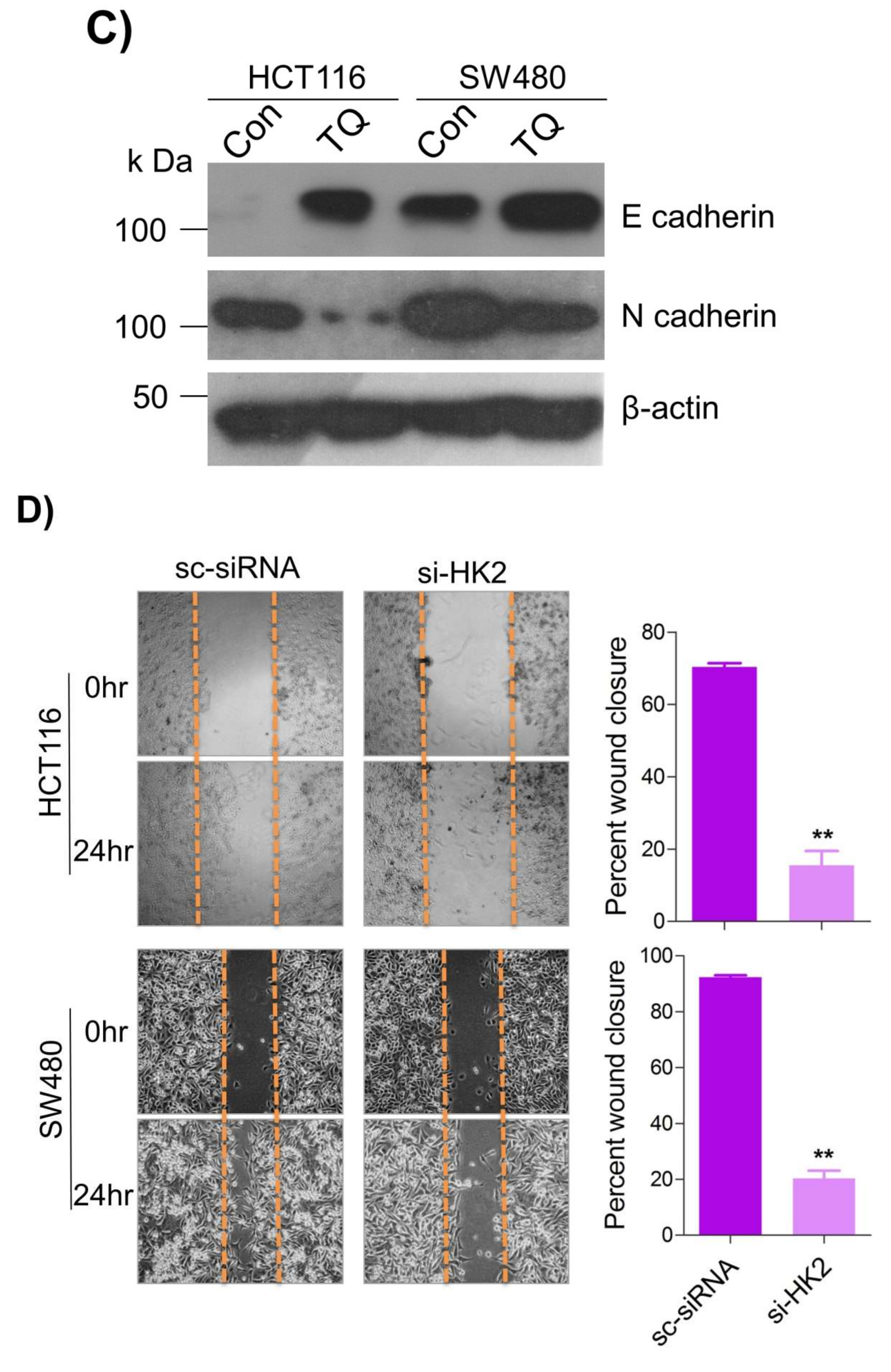
2.5. Thymoquinone Inhibits Cell Migration and Invasion via Modulating Glucose Metabolic Reprogramming
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Cytotoxicity (MTT) and Cell Doubling Assays
4.3. Colony Formation Assay
4.4. Cell Proliferation Assay
4.5. Colony Formation Assay
siRNA Transfection
4.6. Wound Healing and Cell Invasion Assays
4.7. Western Blot
4.8. Glucose Uptake/Lactate Production, ATP, and NADPH Measurement
4.9. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Berardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Budinger, G.R.; Choe, S.H.; Schumacker, P.T. Cellular respiration during hypoxia. Role of cytochrome oxidase as the oxygen sensor in hepatocytes. J. Biol. Chem. 1997, 272, 18808–18816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palm, W.; Thompson, C.B. Nutrient acquisition strategies of mammalian cells. Nature 2017, 546, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, F.A.; Prakasam, G.; Chattopadhyay, S.; Rehman, A.U.; Padder, R.A.; Ansari, M.A.; Irshad, R.; Mangalhara, K.; Bamezai, R.N.K.; Husain, M.; et al. Curcumin decreases Warburg effect in cancer cells by down-regulating pyruvate kinase M2 via mTOR-HIF1α inhibition. Sci. Rep. 2018, 8, 8323. [Google Scholar] [CrossRef] [PubMed]
- Padder, R.A.; Bhat, Z.I.; Ahmad, Z.; Singh, N.; Husain, M. DRP1 Promotes BRAFV600E-Driven Tumor Progression and Metabolic Reprogramming in Colorectal Cancer. Front. Oncol. 2021, 10, 592130. [Google Scholar] [CrossRef] [PubMed]
- Fadaka, A.; Ajiboye, B.; Ojo, O.; Adewale, O.; Olayide, I.; Emuowhochere, R. Biology of glucose metabolization in cancer cells. J. Oncol. Sci. 2017, 3, 45–51. [Google Scholar] [CrossRef]
- Parks, S.K.; Mueller-Klieser, W.; Pouysségur, J. Lactate and Acidity in the Cancer Microenvironment. Annu. Rev. Cancer Biol. 2020, 4, 141–158. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, B.; Li, Y.; Li, Z.; Qin, H.; Sun, Q.; Zhang, F.; Shen, Y.; Shi, Y.; Wang, R. PI3K/Akt signaling mediated Hexokinase-2 expression inhibits cell apoptosis and promotes tumor growth in pediatric osteosarcoma. Biochem. Biophys. Res. Commun. 2015, 464, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.J.; Mejia, E.; Hou, S.; Sher, A.; Hatch, G.; Aliani, M. PI3K-dependent Reprogramming of Hexokinase Isoforms Regulates B Lymphocyte Metabolism. J. Immunol. 2020, 204 (Suppl. S1), 151.30. [Google Scholar]
- Roberts, D.J.; Miyamoto, S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015, 22, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Nowak, N.; Kulma, A.; Gutowicz, J. Up-regulation of Key Glycolysis Proteins in Cancer Development. Open Life Sci. 2018, 13, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Gandham, S.K.; Talekar, M.; Singh, A.; Amiji, M.M. Inhibition of hexokinase-2 with targeted liposomal 3-bromopyruvate in an ovarian tumor spheroid model of aerobic glycolysis. Int. J. Nanomed. 2015, 10, 4405–4423. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Herschman, H.R. A Tumor Agnostic Therapeutic Strategy for Hexokinase 1-Null/Hexokinase 2-Positive Cancers. Cancer Res. 2019, 79, 5907–5914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Catapang, A.; Braas, D.; Stiles, L.; Doh, H.M.; Lee, J.T.; Graeber, T.G.; Damoiseaux, R.; Shirihai, O.; Herschman, H.R. A precision therapeutic strategy for hexokinase 1-null, hexokinase 2-positive cancers. Cancer Metab. 2018, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Padhye, S.; Banerjee, S.; Ahmad, A.; Mohammad, R.; Sarkar, F.H. From here to eternity—The secret of Pharaohs: Therapeutic potential of black cumin seeds and beyond. Cancer Ther. 2008, 6, 495–510. [Google Scholar] [PubMed]
- Abdelmeguid, N.E.; Fakhoury, R.; Kamal, S.M.; Al Wafai, R.J. Effects of Nigella sativa and thymoquinone on biochemical and subcellular changes in pancreatic β-cells of streptozotocin-induced diabetic rats. J. Diabetes 2010, 2, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Razavi, B.M.; Hosseinzadeh, H. A review of the effects of Nigella sativa L. and its constituent, thymoquinone, in metabolic syndrome. J. Endocrinol. Investig. 2014, 37, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Balbaa, M.; El-Zeftawy, M.; Ghareeb, D.; Taha, N.; Mandour, A.W. Nigella sativa Relieves the Altered Insulin Receptor Signaling in Streptozotocin-Induced Diabetic Rats Fed with a High-Fat Diet. Oxidative Med. Cell. Longev. 2016, 2016, 2492107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imran, M.; Rauf, A.; Khan, I.A.; Shahbaz, M.; Qaisrani, T.B.; Fatmawati, S.; Abu-Izneid, T.; Imran, A.; Rahman, K.U.; Gondal, T.A. Thymoquinone: A novel strategy to combat cancer: A review. Biomed. Pharmacother. 2018, 106, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Kaseb, A.O.; Chinnakannu, K.; Chen, D.; Sivanandam, A.; Tejwani, S.; Menon, M.; Dou, Q.P.; Reddy, G.P. Androgen receptor and E2F-1 targeted thymoquinone therapy for hormone-refractory prostate cancer. Cancer Res. 2007, 67, 7782–7788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badr, G.; Mohany, M.; Abu-Tarboush, F. Thymoquinone decreases F-actin polymerization and the proliferation of human multiple myeloma cells by suppressing STAT3 phosphorylation and Bcl2/Bcl-XL expression. Lipids Health Dis. 2011, 10, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.Q.; Wang, J.; Guo, X.F.; Liu, Z.; Dong, W.G. Thymoquinone inhibits proliferation in gastric cancer via the STAT3 pathway in vivo and in vitro. World J. Gastroenterol. 2016, 22, 4149–4159. [Google Scholar] [CrossRef] [PubMed]
- Kundu, J.; Choi, B.Y.; Jeong, C.H.; Kundu, J.K.; Chun, K.S. Thymoquinone induces apoptosis in human colon cancer HCT116 cells through inactivation of STAT3 by blocking JAK2- and Src-mediated phosphorylation of EGF receptor tyrosine kinase. Oncol. Rep. 2014, 32, 821–828. [Google Scholar] [CrossRef] [Green Version]
- Abdelfadil, E.; Cheng, Y.H.; Bau, D.T.; Ting, W.J.; Chen, L.M.; Hsu, H.H.; Lin, Y.M.; Chen, R.J.; Tsai, F.J.; Tsai, C.H.; et al. Thymoquinone induces apoptosis in oral cancer cells through p38β inhibition. Am. J. Chin. Med. 2013, 41, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Alhosin, M.; Abusnina, A.; Achour, M.; Sharif, T.; Muller, C.; Peluso, J.; Chataigneau, T.; Lugnier, C.; Schini-Kerth, V.B.; Bronner, C.; et al. Induction of apoptosis by thymoquinone in lymphoblastic leukemia Jurkat cells is mediated by a p73-dependent pathway which targets the epigenetic integrator UHRF1. Biochem. Pharmacol. 2010, 79, 1251–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abusnina, A.; Alhosin, M.; Keravis, T.; Muller, C.D.; Fuhrmann, G.; Bronner, C.; Lugnier, C. Down-regulation of cyclic nucleotide phosphodiesterase PDE1A is the key event of p73 and UHRF1 deregulation in thymoquinone-induced acute lymphoblastic leukemia cell apoptosis. Cell. Signal. 2011, 23, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.; Zhao, Y.; Lu, X.; Wang, Z.; Liu, Y.; Ren, M.; Lu, G.; Zhang, D.; Sun, Z.; Xu, Z.; et al. TQ inhibits hepatocellular carcinoma growth in vitro and in vivo via repression of Notch signaling. Oncotarget 2015, 6, 32610–32621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Najjar, N.; Chatila, M.; Moukadem, H.; Vuorela, H.; Ocker, M.; Gandesiri, M.; Schneider-Stock, R.; Gali-Muhtasib, H. Reactive oxygen species mediate thymoquinone-induced apoptosis and activate ERK and JNK signaling. Apoptosis 2010, 15, 183–195. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard, M.; Wallace, D.C.; Burelle, Y. The rise of mitochondria in medicine. Mitochondrion 2016, 30, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Chen, Z.; Lu, W.; Garcia-Prieto, C.; Huang, P. The Warburg effect and its cancer therapeutic implications. J. Bioenerg. Biomembr. 2007, 39, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Ma, Y.; Cao, L.; Zhan, S.; Xu, Y.; Fu, F.; Liu, C.; Zhang, G.; Wang, Z.; Wang, R.; et al. B7-H3 promotes aerobic glycolysis and chemoresistance in colorectal cancer cells by regulating HK2. Cell Death Dis. 2019, 10, 308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, A.; Ma, Y.; Yuan, F.; Gong, Y.; Fang, Z.; Mohamed, E.M.; Berrios, E.; Shao, H.; Fang, X. Lysophosphatidic Acid Up-Regulates Hexokinase II and Glycolysis to Promote Proliferation of Ovarian Cancer Cells. Neoplasia 2015, 17, 723–734. [Google Scholar] [CrossRef] [Green Version]
- Coelho, R.G.; Calaça, I.C.; Celestrini, D.M.; Correia-Carneiro, A.H.; Costa, M.M.; Zancan, P.; Sola-Penna, M. Hexokinase and phosphofructokinase activity and intracellular distribution correlate with aggressiveness and invasiveness of human breast carcinoma. Oncotarget 2015, 6, 29375–29387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Li, T.E.; Chen, M.; Xu, D.; Zhu, Y.; Hu, B.Y.; Lin, Z.F.; Pan, J.J.; Wang, X.; Wu, C.; et al. MFN1-dependent alteration of mitochondrial dynamics drives hepatocellular carcinoma metastasis by glucose metabolic reprogramming. Br. J. Cancer 2020, 122, 209–220. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.S.; Barzi, A.; Jemal, A. Colorectal cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 177–193. [Google Scholar] [CrossRef]
- Anemone, A.; Consolino, L.; Conti, L.; Reineri, F.; Cavallo, F.; Aime, S.; Longo, D.L. In vivo evaluation of tumour acidosis for assessing the early metabolic response and onset of resistance to dichloroacetate by using magnetic resonance pH imaging. Int. J. Oncol. 2017, 51, 498–506. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.G.; Rao, S.; Weir, T.L.; O’Malia, J.; Bazan, M.; Brown, R.J.; Ryan, E.P. Metabolomics and metabolic pathway networks from human colorectal cancers, adjacent mucosa, and stool. Cancer Metab. 2016, 4, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kareva, I. Prisoner’s dilemma in cancer metabolism. PLoS ONE 2011, 6, e28576. [Google Scholar] [CrossRef] [Green Version]
- Graziano, F.; Ruzzo, A.; Giacomini, E.; Ricciardi, T.; Aprile, G.; Loupakis, F.; Lorenzini, P.; Ongaro, E.; Zoratto, F.; Catalano, V.; et al. Glycolysis gene expression analysis and selective metabolic advantage in the clinical progression of colorectal cancer. Pharm. J. 2017, 17, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Lu, J. The Warburg metabolism fuels tumor metastasis. Cancer Metastasis Rev. 2019, 38, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zhao, Y.; Ding, Y.; Liu, H.; Liu, Z.; Fodstad, O.; Riker, A.I.; Kamarajugadda, S.; Lu, J.; Owen, L.B.; et al. Warburg effect in chemosensitivity: Targeting lactate dehydrogenase-A re-sensitizes taxol-resistant cancer cells to taxol. Mol. Cancer 2010, 9, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.W.; Lin, S.C.; Chen, K.F.; Lai, Y.Y.; Tsai, S.J. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J. Biol. Chem. 2008, 283, 28106–28114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganapathy-Kanniappan, S.; Geschwind, J.-F.H. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol. Cancer 2013, 12, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundø, K.; Trauelsen, M.; Pedersen, S.F.; Schwartz, T.W. Why Warburg Works: Lactate Controls Immune Evasion through GPR81. Cell Metab. 2020, 31, 666–668. [Google Scholar] [CrossRef] [PubMed]
- Cascone, T.; McKenzie, J.A.; Mbofung, R.M.; Punt, S.; Wang, Z.; Xu, C.; Williams, L.J.; Wang, Z.; Bristow, C.A.; Carugo, A.; et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab. 2018, 27, 977–987.e974. [Google Scholar] [CrossRef]
- Vander Heiden, M.G. Targeting cancer metabolism: A therapeutic window opens. Nat. Rev. Drug Discov. 2011, 10, 671–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tennant, D.A.; Durán, R.V.; Gottlieb, E. Targeting metabolic transformation for cancer therapy. Nat. Rev. Cancer 2010, 10, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Papagiannakopoulos, T.; Olenchock, B.A.; Heyman, J.E.; Keibler, M.A.; Luengo, A.; Bauer, M.R.; Jha, A.K.; O'Brien, J.P.; Pierce, K.A.; et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016, 23, 517–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348 Pt 3, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Ben Sahra, I.; Laurent, K.; Giuliano, S.; Larbret, F.; Ponzio, G.; Gounon, P.; Le Marchand-Brustel, Y.; Giorgetti-Peraldi, S.; Cormont, M.; Bertolotto, C.; et al. Targeting cancer cell metabolism: The combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010, 70, 2465–2475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Li, J.; Wang, F.; Hu, J.; Wang, S.; Sun, Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. [Google Scholar] [CrossRef]
- Wang, H.; Wang, L.; Zhang, Y.; Wang, J.; Deng, Y.; Lin, D. Inhibition of glycolytic enzyme hexokinase II (HK2) suppresses lung tumor growth. Cancer Cell Int. 2016, 16, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucantoni, F.; Düssmann, H.; Llorente-Folch, I.; Prehn, J.H.M. BCL2 and BCL(X)L selective inhibitors decrease mitochondrial ATP production in breast cancer cells and are synthetically lethal when combined with 2-deoxy-D-glucose. Oncotarget 2018, 9, 26046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, M.K.; Suk, K. Pyruvate Dehydrogenase Kinase as a Potential Therapeutic Target for Malignant Gliomas. Brain Tumor Res. Treat. 2013, 1, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caino, M.C.; Ghosh, J.C.; Chae, Y.C.; Vaira, V.; Rivadeneira, D.B.; Faversani, A.; Rampini, P.; Kossenkov, A.V.; Aird, K.M.; Zhang, R.; et al. PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proc. Natl. Acad. Sci. USA 2015, 112, 8638–8643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, A.K.; Basu, S.; Sarkar, N.; Ghosh, A.C. Advances in cancer therapy with plant based natural products. Curr. Med. Chem. 2001, 8, 1467–1486. [Google Scholar] [CrossRef] [PubMed]
- Chikara, S.; Nagaprashantha, L.D.; Singhal, J.; Horne, D.; Awasthi, S.; Singhal, S.S. Oxidative stress and dietary phytochemicals: Role in cancer chemoprevention and treatment. Cancer Lett. 2018, 413, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Saunier, E.; Antonio, S.; Regazzetti, A.; Auzeil, N.; Laprévote, O.; Shay, J.W.; Coumoul, X.; Barouki, R.; Benelli, C.; Huc, L.; et al. Resveratrol reverses the Warburg effect by targeting the pyruvate dehydrogenase complex in colon cancer cells. Sci. Rep. 2017, 7, 6945. [Google Scholar] [CrossRef] [Green Version]
- Rajput, S.; Kumar, B.N.P.; Dey, K.K.; Pal, I.; Parekh, A.; Mandal, M. Molecular targeting of Akt by thymoquinone promotes G(1) arrest through translation inhibition of cyclin D1 and induces apoptosis in breast cancer cells. Life Sci. 2013, 93, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Tania, M.; Wei, C.; Mei, Z.; Fu, S.; Cheng, J.; Xu, J.; Fu, J. Thymoquinone inhibits cancer metastasis by downregulating TWIST1 expression to reduce epithelial to mesenchymal transition. Oncotarget 2015, 6, 19580–19591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asaduzzaman Khan, M.; Tania, M.; Fu, S.; Fu, J. Thymoquinone, as an anticancer molecule: From basic research to clinical investigation. Oncotarget 2017, 8, 51907–51919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, K.S.; Fernandes, P.; O'Donovan, T.R.; McKenna, S.L.; Doddakula, K.K.; Power, D.G.; Soden, D.M.; Forde, P.F. Glycolysis inhibition as a cancer treatment and its role in an anti-tumour immune response. Biochim. Biophys. Acta 2016, 1866, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Hu, L.; Wu, F.; Zou, L.; He, T. Poor prognosis of hexokinase 2 overexpression in solid tumors of digestive system: A meta-analysis. Oncotarget 2017, 8, 32332–32344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Yun, Y.; Wu, B.; Wen, L.; Wen, M.; Yang, H.; Zhao, L.; Liu, W.; Huang, S.; Wen, N.; et al. FOXM1 promotes reprogramming of glucose metabolism in epithelial ovarian cancer cells via activation of GLUT1 and HK2 transcription. Oncotarget 2016, 7, 47985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siveen, K.S.; Mustafa, N.; Li, F.; Kannaiyan, R.; Ahn, K.S.; Kumar, A.P.; Chng, W.J.; Sethi, G. Thymoquinone overcomes chemoresistance and enhances the anticancer effects of bortezomib through abrogation of NF-κB regulated gene products in multiple myeloma xenograft mouse model. Oncotarget 2014, 5, 634–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutton, K.M.; Greenshields, A.L.; Hoskin, D.W. Thymoquinone, a bioactive component of black caraway seeds, causes G1 phase cell cycle arrest and apoptosis in triple-negative breast cancer cells with mutant p53. Nutr. Cancer 2014, 66, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Mu, G.G.; Zhang, L.L.; Li, H.Y.; Liao, Y.; Yu, H.G. Thymoquinone Pretreatment Overcomes the Insensitivity and Potentiates the Antitumor Effect of Gemcitabine Through Abrogation of Notch1, PI3K/Akt/mTOR Regulated Signaling Pathways in Pancreatic Cancer. Dig. Dis. Sci. 2015, 60, 1067–1080. [Google Scholar] [CrossRef] [PubMed]
- El-Baba, C.; Mahadevan, V.; Fahlbusch, F.B.; Mohan, S.S.; Rau, T.T.; Gali-Muhtasib, H.; Schneider-Stock, R. Thymoquinone-induced conformational changes of PAK1 interrupt prosurvival MEK-ERK signaling in colorectal cancer. Mol. Cancer 2014, 13, 201. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Kuang, X.R.; Lv, P.T.; Yan, X.X. Thymoquinone inhibits proliferation and invasion of human nonsmall-cell lung cancer cells via ERK pathway. Tumour Biol. 2015, 36, 259–269. [Google Scholar] [CrossRef]
- Lee, Y.M.; Kim, G.H.; Park, E.J.; Oh, T.I.; Lee, S.; Kan, S.Y.; Kang, H.; Kim, B.M.; Kim, J.H.; Lim, J.H. Thymoquinone Selectively Kills Hypoxic Renal Cancer Cells by Suppressing HIF-1α-Mediated Glycolysis. Int. J. Mol. Sci. 2019, 20, 1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandita, A.; Kumar, B.; Manvati, S.; Vaishnavi, S.; Singh, S.K.; Bamezai, R.N. Synergistic combination of gemcitabine and dietary molecule induces apoptosis in pancreatic cancer cells and down regulates PKM2 expression. PLoS ONE 2014, 9, e107154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yu, X.H.; Yan, Y.G.; Wang, C.; Wang, W.J. PI3K/Akt signaling in osteosarcoma. Clin. Chim. Acta 2015, 444, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Danielsen, S.A.; Eide, P.W.; Nesbakken, A.; Guren, T.; Leithe, E.; Lothe, R.A. Portrait of the PI3K/AKT pathway in colorectal cancer. Biochim. Biophys. Acta 2015, 1855, 104–121. [Google Scholar] [CrossRef] [PubMed]
- Narayanankutty, A. PI3K/ Akt/ mTOR Pathway as a Therapeutic Target for Colorectal Cancer: A Review of Preclinical and Clinical Evidence. Curr. Drug Targets 2019, 20, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Rathmell, J.C.; Fox, C.J.; Plas, D.R.; Hammerman, P.S.; Cinalli, R.M.; Thompson, C.B. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol. Cell. Biol. 2003, 23, 7315–7328. [Google Scholar] [CrossRef] [Green Version]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlob, K.; Majewski, N.; Kennedy, S.; Kandel, E.; Robey, R.B.; Hay, N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001, 15, 1406–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plas, D.R.; Talapatra, S.; Edinger, A.L.; Rathmell, J.C.; Thompson, C.B. Akt and Bcl-xL promote growth factor-independent survival through distinct effects on mitochondrial physiology. J. Biol. Chem. 2001, 276, 12041–12048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edinger, A.L.; Thompson, C.B. Antigen-presenting cells control T cell proliferation by regulating amino acid availability. Proc. Natl. Acad. Sci. USA 2002, 99, 1107–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzzai, M.; Bauer, D.E.; Jones, R.G.; DeBerardinis, R.J.; Hatzivassiliou, G.; Elstrom, R.L.; Thompson, C.B. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid β-oxidation. Oncogene 2005, 24, 4165–4173. [Google Scholar] [CrossRef] [Green Version]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg's contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, R.; Jain, M.; Madhusudhan, N.; Sheppard, N.G.; Strittmatter, L.; Kampf, C.; Huang, J.; Asplund, A.; Mootha, V.K. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat. Commun. 2014, 5, 3128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, G.P.; Massagué, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [Green Version]
- Vaquero, J.; Guedj, N.; Clapéron, A.; Nguyen Ho-Bouldoires, T.H.; Paradis, V.; Fouassier, L. Epithelial-mesenchymal transition in cholangiocarcinoma: From clinical evidence to regulatory networks. J. Hepatol. 2017, 66, 424–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.J.; Yoon, C.; Lee, J.H.; Chang, K.K.; Lin, J.X.; Kim, Y.H.; Kook, M.C.; Aksoy, B.A.; Park, D.J.; Ashktorab, H.; et al. KMT2C Mutations in Diffuse-Type Gastric Adenocarcinoma Promote Epithelial-to-Mesenchymal Transition. Clin. Cancer Res. 2018, 24, 6556–6569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjöberg, E.; Meyrath, M.; Milde, L.; Herrera, M.; Lövrot, J.; Hägerstrand, D.; Frings, O.; Bartish, M.; Rolny, C.; Sonnhammer, E.; et al. A Novel ACKR2-Dependent Role of Fibroblast-Derived CXCL14 in Epithelial-to-Mesenchymal Transition and Metastasis of Breast Cancer. Clin. Cancer Res. 2019, 25, 3702–3717. [Google Scholar] [CrossRef] [Green Version]
- Bylsma, L.C.; Gillezeau, C.; Garawin, T.A.; Kelsh, M.A.; Fryzek, J.P.; Sangaré, L.; Lowe, K.A. Prevalence of RAS and BRAF mutations in metastatic colorectal cancer patients by tumor sidedness: A systematic review and meta-analysis. Cancer Med. 2020, 9, 1044–1057. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Fan, Y.; Huang, S.; Wang, G.; Han, R.; Lei, F.; Luo, A.; Jing, X.; Zhao, L.; Gu, S.; et al. Thymoquinone inhibits the metastasis of renal cell cancer cells by inducing autophagy via AMPK/mTOR signaling pathway. Cancer Sci. 2018, 109, 3865–3873. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Ahn, K.S.; Hsu, A.; Woo, C.C.; Yuan, Y.; Tan, K.H.B.; Chinnathambi, A.; Alahmadi, T.A.; Alharbi, S.A.; Koh, A.P.F.; et al. Thymoquinone Inhibits Bone Metastasis of Breast Cancer Cells Through Abrogation of the CXCR4 Signaling Axis. Front. Pharmacol. 2018, 9, 1294. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenes, M.; Shang, M.; Di Matteo, M.; Goveia, J.; Martín-Pérez, R.; Serneels, J.; Prenen, H.; Ghesquière, B.; Carmeliet, P.; Mazzone, M. Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis. Cell Metab. 2016, 24, 701–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, D.; Scimè, A. Metabolic Regulation of Epithelial to Mesenchymal Transition: Implications for Endocrine Cancer. Front. Endocrinol. 2019, 10, 773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gui, D.Y.; Sullivan, L.B.; Luengo, A.; Hosios, A.M.; Bush, L.N.; Gitego, N.; Davidson, S.M.; Freinkman, E.; Thomas, C.J.; Vander Heiden, M.G. Environment dictates dependence on mitochondrial complex I for NAD+ and aspartate production and determines cancer cell sensitivity to metformin. Cell Metab. 2016, 24, 716–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, F.; Wang, J.H.; Fan, W.J.; Meng, Y.T.; Li, M.M.; Li, T.T.; Cui, B.; Wang, H.F.; Zhao, Y.; An, F.; et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene 2018, 37, 1062–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karim, S.; Burzangi, A.S.; Ahmad, A.; Siddiqui, N.A.; Ibrahim, I.M.; Sharma, P.; Abualsunun, W.A.; Gabr, G.A. PI3K-AKT Pathway Modulation by Thymoquinone Limits Tumor Growth and Glycolytic Metabolism in Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 2305. https://doi.org/10.3390/ijms23042305
Karim S, Burzangi AS, Ahmad A, Siddiqui NA, Ibrahim IM, Sharma P, Abualsunun WA, Gabr GA. PI3K-AKT Pathway Modulation by Thymoquinone Limits Tumor Growth and Glycolytic Metabolism in Colorectal Cancer. International Journal of Molecular Sciences. 2022; 23(4):2305. https://doi.org/10.3390/ijms23042305
Chicago/Turabian StyleKarim, Shahid, Abdulhadi S. Burzangi, Aftab Ahmad, Nasir Ali Siddiqui, Ibrahim M. Ibrahim, Priyanka Sharma, Walaa A. Abualsunun, and Gamal A. Gabr. 2022. "PI3K-AKT Pathway Modulation by Thymoquinone Limits Tumor Growth and Glycolytic Metabolism in Colorectal Cancer" International Journal of Molecular Sciences 23, no. 4: 2305. https://doi.org/10.3390/ijms23042305