
Perspectives on Vascular Regulation of Mechanisms Controlling Selective Immune Cell Function in the Tumor Immune Response


Abstract
:1. Introduction
2. Leukocyte Extravasation
3. EC Cell Adhesion Molecules, Junctions and Leukocyte Extravasation in Tumor Biology
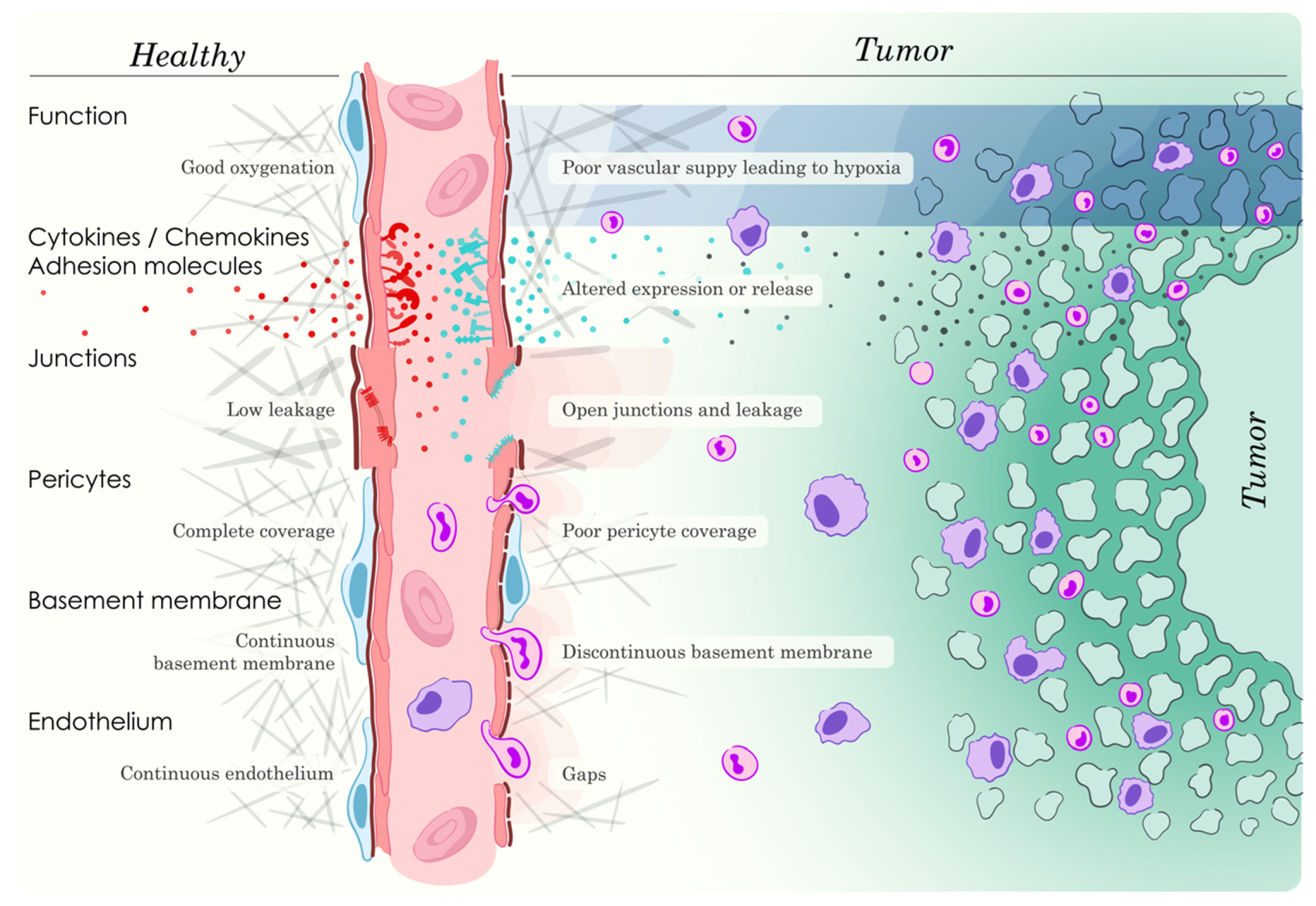
3.1. General Properties of Tumor Angiogenesis and Vasculature
3.2. VEGFA/VEGFR2 Signaling
3.3. Consequences of Alterations in EC Cell Adhesion and Junction Protein Expression/Activities
3.4. Pericytes and IC Infiltration

{kind=link}
| EC Cell Surface Adhesion Molecule/Junction Protein | Condition that Causes Alteration | Consequence | References |
|---|---|---|---|
| P-/E-selectin | TNF-α, IL-1β, IL-4, IFN-γ | Selective effect on neutrophil and memory T cell extravasation | [30,55] |
| ICAM-1 | IL-6, TNF-α, IL-1β, IFN-γ | Selective effect on T cell and monocyte extravasation, deregulated in tumor EC | [31,32,55,56,57] |
| ICAM-2 | IL-6, TNF-α, IL-1β, IFN-γ | General effect on IC extravasation | [31,32,55,56,57] |
| VCAM-1 | IL-1β, TNF-α | Selective effect on T cell and monocyte extravasation, deregulated in tumor EC | [32,58] |
| JAMA | IL-1β, TNF-α, IFN-γ, IL-22, IL-17A | Selective effect on neutrophil extravasation | [10,37,38,40] |
| JAMC | Inflammation | General effect on IC transmigration, monocyte transmigration | [39,59] |
| ESAM | Inflammation | Selective effect on neutrophil transmigration | [35] |
| VE-cadherin (expression and phosphorylation) | VEGFA, inflammation | General effects on IC transmigration, selective effect on T cell transmigration, increased leakage | [8,9,10,23] |
| CD31/PECAM-1 | IL-1β | Selective effect on neutrophil extravasation | [60] |
4. Cytokines/Chemokines and Leukocyte Extravasation in Tumor Biology
5. EC-Produced Immune Checkpoint Inhibitors
6. High Endothelial Venules (HEV) and Leukocyte Extravasation in Tumor Biology
7. Hypoxia and Metabolites in Tumor Biology
8. IC and Intra-/Perivascular Location
9. Platelets and Tumor Vasculature
10. Extracellular Vesicles (EV)
11. Conclusions
12. Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data availability statement
Acknowledgments
Conflicts of Interest
References
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Hegde, S.; Leader, A.M.; Merad, M. MDSC: Markers, development, states, and unaddressed complexity. Immunity 2021, 54, 875–884. [Google Scholar] [CrossRef]
- Paluskievicz, C.M.; Cao, X.; Abdi, R.; Zheng, P.; Liu, Y.; Bromberg, J.S. T Regulatory Cells and Priming the Suppressive Tumor Microenvironment. Front. Immunol. 2019, 10, 2453. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Byrne, K.T.; Yan, F.; Yamazoe, T.; Chen, Z.; Baslan, T.; Richman, L.P.; Lin, J.H.; Sun, Y.H.; Rech, A.J.; et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 2018, 49, 178–193.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Vitale, I.; Shema, E.; Loi, S.; Galluzzi, L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat. Med. 2021, 27, 212–224. [Google Scholar] [CrossRef]
- Bear, A.S.; Vonderheide, R.H.; O’Hara, M.H. Challenges and Opportunities for Pancreatic Cancer Immunotherapy. Cancer Cell 2020, 38, 788–802. [Google Scholar] [CrossRef]
- Claesson-Welsh, L. Vascular permeability—The essentials. Ups. J. Med. Sci. 2015, 120, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Claesson-Welsh, L.; Dejana, E.; McDonald, D.M. Permeability of the Endothelial Barrier: Identifying and Reconciling Controversies. Trends Mol. Med. 2021, 27, 314–331. [Google Scholar] [CrossRef]
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 15, 692–704. [Google Scholar] [CrossRef]
- Chen, X.L.; Nam, J.O.; Jean, C.; Lawson, C.; Walsh, C.T.; Goka, E.; Lim, S.T.; Tomar, A.; Tancioni, I.; Uryu, S.; et al. VEGF-induced vascular permeability is mediated by FAK. Dev. Cell 2012, 22, 146–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooke, V.G.; LeBleu, V.S.; Keskin, D.; Khan, Z.; O’Connell, J.T.; Teng, Y.; Duncan, M.B.; Xie, L.; Maeda, G.; Vong, S.; et al. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell 2012, 21, 66–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xian, X.; Hakansson, J.; Stahlberg, A.; Lindblom, P.; Betsholtz, C.; Gerhardt, H.; Semb, H. Pericytes limit tumor cell metastasis. J. Clin. Investig. 2006, 116, 642–651. [Google Scholar] [CrossRef] [Green Version]
- Richards, M.; Pal, S.; Sjoberg, E.; Martinsson, P.; Venkatraman, L.; Claesson-Welsh, L. Intra-vessel heterogeneity establishes enhanced sites of macromolecular leakage downstream of laminin alpha5. Cell Rep. 2021, 35, 109268. [Google Scholar] [CrossRef]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef]
- McDowell, S.A.C.; Quail, D.F. Immunological Regulation of Vascular Inflammation During Cancer Metastasis. Front. Immunol. 2019, 10, 1984. [Google Scholar] [CrossRef]
- Nourshargh, S.; Alon, R. Leukocyte migration into inflamed tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef] [Green Version]
- Vestweber, D. Relevance of endothelial junctions in leukocyte extravasation and vascular permeability. Ann. N. Y. Acad. Sci. 2012, 1257, 184–192. [Google Scholar] [CrossRef]
- Phillipson, M.; Kaur, J.; Colarusso, P.; Ballantyne, C.M.; Kubes, P. Endothelial domes encapsulate adherent neutrophils and minimize increases in vascular permeability in paracellular and transcellular emigration. PLoS ONE 2008, 3, e1649. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Lanitis, E.; Irving, M.; Coukos, G. Targeting the tumor vasculature to enhance T cell activity. Curr. Opin. Immunol. 2015, 33, 55–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, S.; Wong, A.H.; Jain, R.K. Vascular normalization as a therapeutic strategy for malignant and nonmalignant disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006486. [Google Scholar] [CrossRef] [PubMed]
- Claesson-Welsh, L.; Welsh, M. VEGFA and tumour angiogenesis. J. Intern. Med. 2013, 273, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Padhan, N.; Sjostrom, E.O.; Roche, F.P.; Testini, C.; Honkura, N.; Sainz-Jaspeado, M.; Gordon, E.; Bentley, K.; Philippides, A.; et al. VEGFR2 pY949 signalling regulates adherens junction integrity and metastatic spread. Nat. Commun. 2016, 7, 11017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietila, I.; Van Mourik, D.; Tamelander, A.; Kriz, V.; Claesson-Welsh, L.; Tengholm, A.; Welsh, M. Temporal Dynamics of VEGFA-Induced VEGFR2/FAK Co-Localization Depend on SHB. Cells 2019, 8, 1645. [Google Scholar] [CrossRef] [Green Version]
- Zang, G.; Christoffersson, G.; Tian, G.; Harun-Or-Rashid, M.; Vagesjo, E.; Phillipson, M.; Barg, S.; Tengholm, A.; Welsh, M. Aberrant association between vascular endothelial growth factor receptor-2 and VE-cadherin in response to vascular endothelial growth factor-a in Shb-deficient lung endothelial cells. Cell. Signal. 2013, 25, 85–92. [Google Scholar] [CrossRef] [Green Version]
- Funa, N.S.; Kriz, V.; Zang, G.; Calounova, G.; Akerblom, B.; Mares, J.; Larsson, E.; Sun, Y.; Betsholtz, C.; Welsh, M. Dysfunctional microvasculature as a consequence of shb gene inactivation causes impaired tumor growth. Cancer Res. 2009, 69, 2141–2148. [Google Scholar] [CrossRef] [Green Version]
- Dirkx, A.E.; Oude Egbrink, M.G.; Kuijpers, M.J.; van der Niet, S.T.; Heijnen, V.V.; Bouma-ter Steege, J.C.; Wagstaff, J.; Griffioen, A.W. Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression. Cancer Res. 2003, 63, 2322–2329. [Google Scholar]
- Tromp, S.C.; oude Egbrink, M.G.; Dings, R.P.; van Velzen, S.; Slaaf, D.W.; Hillen, H.F.; Tangelder, G.J.; Reneman, R.S.; Griffioen, A.W. Tumor angiogenesis factors reduce leukocyte adhesion in vivo. Int. Immunol. 2000, 12, 671–676. [Google Scholar] [CrossRef]
- Doukas, J.; Pober, J.S. IFN-gamma enhances endothelial activation induced by tumor necrosis factor but not IL-1. J. Immunol. 1990, 145, 1727–1733. [Google Scholar]
- Griffioen, A.W.; Damen, C.A.; Martinotti, S.; Blijham, G.H.; Groenewegen, G. Endothelial intercellular adhesion molecule-1 expression is suppressed in human malignancies: The role of angiogenic factors. Cancer Res. 1996, 56, 1111–1117. [Google Scholar] [PubMed]
- Ala, A.; Dhillon, A.P.; Hodgson, H.J. Role of cell adhesion molecules in leukocyte recruitment in the liver and gut. Int. J. Exp. Pathol. 2003, 84, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bujan, J.; Gimeno, M.J.; Prieto, A.; Pascual, G.; Bellon, J.M.; Alvarez-Mon, M. Modulation of PECAM-1 (CD31) expression in human endothelial cells: Effect of IFNgamma and IL-10. J. Vasc. Res. 1999, 36, 106–113. [Google Scholar] [CrossRef]
- Shimizu, Y.; Shaw, S.; Graber, N.; Gopal, T.V.; Horgan, K.J.; Van Seventer, G.A.; Newman, W. Activation-independent binding of human memory T cells to adhesion molecule ELAM-1. Nature 1991, 349, 799–802. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, F.; Petri, B.; Khandoga, A.G.; Moser, C.; Khandoga, A.; Volkery, S.; Li, H.; Nasdala, I.; Brandau, O.; Fassler, R.; et al. ESAM supports neutrophil extravasation, activation of Rho, and VEGF-induced vascular permeability. J. Exp. Med. 2006, 203, 1671–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouzin, C.; Brouet, A.; De Vriese, J.; Dewever, J.; Feron, O. Effects of vascular endothelial growth factor on the lymphocyte-endothelium interactions: Identification of caveolin-1 and nitric oxide as control points of endothelial cell anergy. J. Immunol. 2007, 178, 1505–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, C.; Fraemohs, L.; Dejana, E. The role of junctional adhesion molecules in vascular inflammation. Nat. Rev. Immunol. 2007, 7, 467–477. [Google Scholar] [CrossRef]
- Khandoga, A.; Kessler, J.S.; Meissner, H.; Hanschen, M.; Corada, M.; Motoike, T.; Enders, G.; Dejana, E.; Krombach, F. Junctional adhesion molecule-A deficiency increases hepatic ischemia-reperfusion injury despite reduction of neutrophil transendothelial migration. Blood 2005, 106, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Aurrand-Lions, M.; Lamagna, C.; Dangerfield, J.P.; Wang, S.; Herrera, P.; Nourshargh, S.; Imhof, B.A. Junctional adhesion molecule-C regulates the early influx of leukocytes into tissues during inflammation. J. Immunol. 2005, 174, 6406–6415. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Weight, C.M.; Luissint, A.C.; Hilgarth, R.S.; Brazil, J.C.; Ettel, M.; Nusrat, A.; Parkos, C.A. Role of JAM-A tyrosine phosphorylation in epithelial barrier dysfunction during intestinal inflammation. Mol. Biol. Cell 2019, 30, 566–578. [Google Scholar] [CrossRef]
- Griffioen, A.W.; Damen, C.A.; Mayo, K.H.; Barendsz-Janson, A.F.; Martinotti, S.; Blijham, G.H.; Groenewegen, G. Angiogenesis inhibitors overcome tumor induced endothelial cell anergy. Int. J. Cancer 1999, 80, 315–319. [Google Scholar] [CrossRef]
- Dirkx, A.E.; oude Egbrink, M.G.; Castermans, K.; van der Schaft, D.W.; Thijssen, V.L.; Dings, R.P.; Kwee, L.; Mayo, K.H.; Wagstaff, J.; Bouma-ter Steege, J.C.; et al. Anti-angiogenesis therapy can overcome endothelial cell anergy and promote leukocyte-endothelium interactions and infiltration in tumors. FASEB J. 2006, 20, 621–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Ting, K.K.; Li, J.; Cogger, V.C.; Chen, J.; Johansson-Percival, A.; Ngiow, S.F.; Holst, J.; Grau, G.; Goel, S.; et al. Targeting Vascular Endothelial-Cadherin in Tumor-Associated Blood Vessels Promotes T-cell-Mediated Immunotherapy. Cancer Res. 2017, 77, 4434–4447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, P.J.; Newman, D.K. Signal transduction pathways mediated by PECAM-1: New roles for an old molecule in platelet and vascular cell biology. Arter. Thromb. Vasc. Biol. 2003, 23, 953–964. [Google Scholar] [CrossRef]
- He, Q.; Jamalpour, M.; Bergquist, E.; Anderson, R.L.; Gustafsson, K.; Welsh, M. Mouse Breast Carcinoma Monocytic/Macrophagic Myeloid-Derived Suppressor Cell Infiltration as a Consequence of Endothelial Dysfunction in Shb-Deficient Endothelial Cells Increases Tumor Lung Metastasis. Int. J. Mol. Sci. 2021, 22, 11478. [Google Scholar] [CrossRef]
- Christoffersson, G.; Zang, G.; Zhuang, Z.W.; Vagesjo, E.; Simons, M.; Phillipson, M.; Welsh, M. Vascular adaptation to a dysfunctional endothelium as a consequence of Shb deficiency. Angiogenesis 2012, 15, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Nikpour, M.; Gustafsson, K.; Vagesjo, E.; Seignez, C.; Giraud, A.; Phillipson, M.; Welsh, M. Shb deficiency in endothelium but not in leucocytes is responsible for impaired vascular performance during hindlimb ischaemia. Acta Physiol. 2015, 214, 200–209. [Google Scholar] [CrossRef] [Green Version]
- He, Q.; Li, X.; He, L.; Li, Y.; Betsholtz, C.; Welsh, M. Pericyte dysfunction due to Shb gene deficiency increases B16F10 melanoma lung metastasis. Int. J. Cancer 2020, 147, 2634–2644. [Google Scholar] [CrossRef]
- Hamzah, J.; Jugold, M.; Kiessling, F.; Rigby, P.; Manzur, M.; Marti, H.H.; Rabie, T.; Kaden, S.; Grone, H.J.; Hammerling, G.J.; et al. Vascular normalization in Rgs5-deficient tumours promotes immune destruction. Nature 2008, 453, 410–414. [Google Scholar] [CrossRef]
- Dave, J.M.; Mirabella, T.; Weatherbee, S.D.; Greif, D.M. Pericyte ALK5/TIMP3 Axis Contributes to Endothelial Morphogenesis in the Developing Brain. Dev. Cell 2018, 47, 388–389. [Google Scholar] [CrossRef] [Green Version]
- Lees, D.M.; Reynolds, L.E.; Pedrosa, A.R.; Roy-Luzarraga, M.; Hodivala-Dilke, K.M. Phosphorylation of pericyte FAK-Y861 affects tumour cell apoptosis and tumour blood vessel regression. Angiogenesis 2021, 24, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, S.; Baluk, P.; Kaidoh, T.; Haskell, A.; Jain, R.K.; McDonald, D.M. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am. J. Pathol. 2002, 160, 985–1000. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.; Tobin, N.P.; Rundqvist, H.; Li, T.; Lavergne, M.; Garcia-Ibanez, Y.; Qin, H.; Paulsson, J.; Zeitelhofer, M.; Adzemovic, M.Z.; et al. Role of Tumor Pericytes in the Recruitment of Myeloid-Derived Suppressor Cells. J. Natl. Cancer Inst. 2015, 107, djv209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pico de Coana, Y.; Masucci, G.; Hansson, J.; Kiessling, R. Myeloid-derived suppressor cells and their role in CTLA-4 blockade therapy. Cancer Immunol. Immunother. 2014, 63, 977–983. [Google Scholar] [CrossRef]
- Lidington, E.A.; Moyes, D.L.; McCormack, A.M.; Rose, M.L. A comparison of primary endothelial cells and endothelial cell lines for studies of immune interactions. Transpl. Immunol. 1999, 7, 239–246. [Google Scholar] [CrossRef]
- Fisher, D.T.; Chen, Q.; Skitzki, J.J.; Muhitch, J.B.; Zhou, L.; Appenheimer, M.M.; Vardam, T.D.; Weis, E.L.; Passanese, J.; Wang, W.C.; et al. IL-6 trans-signaling licenses mouse and human tumor microvascular gateways for trafficking of cytotoxic T cells. J. Clin. Investig. 2011, 121, 3846–3859. [Google Scholar] [CrossRef] [Green Version]
- Ayres, R.C.; Neuberger, J.M.; Shaw, J.; Joplin, R.; Adams, D.H. Intercellular adhesion molecule-1 and MHC antigens on human intrahepatic bile duct cells: Effect of pro-inflammatory cytokines. Gut 1993, 34, 1245–1249. [Google Scholar] [CrossRef] [Green Version]
- Haraldsen, G.; Kvale, D.; Lien, B.; Farstad, I.N.; Brandtzaeg, P. Cytokine-regulated expression of E-selectin, intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1) in human microvascular endothelial cells. J. Immunol. 1996, 156, 2558–2565. [Google Scholar]
- Bradfield, P.F.; Menon, A.; Miljkovic-Licina, M.; Lee, B.P.; Fischer, N.; Fish, R.J.; Kwak, B.; Fisher, E.A.; Imhof, B.A. Divergent JAM-C Expression Accelerates Monocyte-Derived Cell Exit from Atherosclerotic Plaques. PLoS ONE 2016, 11, e0159679. [Google Scholar] [CrossRef]
- Thompson, R.D.; Wakelin, M.W.; Larbi, K.Y.; Dewar, A.; Asimakopoulos, G.; Horton, M.A.; Nakada, M.T.; Nourshargh, S. Divergent effects of platelet-endothelial cell adhesion molecule-1 and beta 3 integrin blockade on leukocyte transmigration in vivo. J. Immunol. 2000, 165, 426–434. [Google Scholar] [CrossRef] [Green Version]
- Nourshargh, S.; Krombach, F.; Dejana, E. The role of JAM-A and PECAM-1 in modulating leukocyte infiltration in inflamed and ischemic tissues. J. Leukoc. Biol. 2006, 80, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Casazza, A.; Laoui, D.; Wenes, M.; Rizzolio, S.; Bassani, N.; Mambretti, M.; Deschoemaeker, S.; Van Ginderachter, J.A.; Tamagnone, L.; Mazzone, M. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 2013, 24, 695–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Smith, C.; Shapiro, A.; Monette, R.; Hutchison, J.; Stanimirovic, D. Increased expression of bioactive chemokines in human cerebromicrovascular endothelial cells and astrocytes subjected to simulated ischemia in vitro. J. Neuroimmunol. 1999, 101, 148–160. [Google Scholar] [CrossRef]
- Motz, G.T.; Santoro, S.P.; Wang, L.P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; He, Z.; Huang, M.; Liu, T.; Wang, Y.; Xu, H.; Duan, H.; Ma, P.; Zhang, L.; Zamvil, S.S.; et al. Vascular niche IL-6 induces alternative macrophage activation in glioblastoma through HIF-2alpha. Nat. Commun. 2018, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Han, Y.M.; Song, P.; Liu, Z.; Yuan, Z.; Zou, M.H. Endothelial cell-specific expression of serine/threonine kinase 11 modulates dendritic cell differentiation. Nat. Commun. 2022, 13, 648. [Google Scholar] [CrossRef]
- Cross, A.R.; Lion, J.; Poussin, K.; Glotz, D.; Mooney, N. Inflammation Determines the Capacity of Allogenic Endothelial Cells to Regulate Human Treg Expansion. Front. Immunol. 2021, 12, 666531. [Google Scholar] [CrossRef]
- Park, S.; Oh, J.H.; Park, D.J.; Zhang, H.; Noh, M.; Kim, Y.; Kim, Y.S.; Kim, H.; Kim, Y.M.; Ha, S.J.; et al. CU06-1004-Induced Vascular Normalization Improves Immunotherapy by Modulating Tumor Microenvironment via Cytotoxic T Cells. Front. Immunol. 2020, 11, 620166. [Google Scholar] [CrossRef]
- Veluswamy, P.; Wacker, M.; Scherner, M.; Wippermann, J. Delicate Role of PD-L1/PD-1 Axis in Blood Vessel Inflammatory Diseases: Current Insight and Future Significance. Int. J. Mol. Sci. 2020, 21, 8159. [Google Scholar] [CrossRef]
- Majidpoor, J.; Mortezaee, K. Angiogenesis as a hallmark of solid tumors—Clinical perspectives. Cell Oncol. 2021, 44, 715–737. [Google Scholar] [CrossRef]
- Nambiar, D.K.; Aguilera, T.; Cao, H.; Kwok, S.; Kong, C.; Bloomstein, J.; Wang, Z.; Rangan, V.S.; Jiang, D.; von Eyben, R.; et al. Galectin-1-driven T cell exclusion in the tumor endothelium promotes immunotherapy resistance. J. Clin. Investig. 2019, 129, 5553–5567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girard, J.P.; Springer, T.A. High endothelial venules (HEVs): Specialized endothelium for lymphocyte migration. Immunol. Today 1995, 16, 449–457. [Google Scholar] [CrossRef]
- Blanchard, L.; Girard, J.P. High endothelial venules (HEVs) in immunity, inflammation and cancer. Angiogenesis 2021, 24, 719–753. [Google Scholar] [CrossRef] [PubMed]
- Menzel, L.; Zschummel, M.; Crowley, T.; Franke, V.; Grau, M.; Ulbricht, C.; Hauser, A.; Siffrin, V.; Bajenoff, M.; Acton, S.E.; et al. Lymphocyte access to lymphoma is impaired by high endothelial venule regression. Cell Rep. 2021, 37, 109878. [Google Scholar] [CrossRef]
- Qin, M.; Jin, Y.; Pan, L.Y. Tertiary lymphoid structure and B-cell-related pathways: A potential target in tumor immunotherapy. Oncol. Lett. 2021, 22, 836. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.L.; Allaire, N.; Ngam-Ek, A.; Notidis, E.; Hunt, J.; Perrin, S.; Fava, R.A. Lymphotoxin-beta receptor signaling is required for the homeostatic control of HEV differentiation and function. Immunity 2005, 23, 539–550. [Google Scholar] [CrossRef] [Green Version]
- Allen, E.; Jabouille, A.; Rivera, L.B.; Lodewijckx, I.; Missiaen, R.; Steri, V.; Feyen, K.; Tawney, J.; Hanahan, D.; Michael, I.P.; et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci. Transl. Med. 2017, 9, eaak9679. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, J.; Liu, B. Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Front. Immunol. 2018, 9, 978. [Google Scholar] [CrossRef] [Green Version]
- Ramjiawan, R.R.; Griffioen, A.W.; Duda, D.G. Anti-angiogenesis for cancer revisited: Is there a role for combinations with immunotherapy? Angiogenesis 2017, 20, 185–204. [Google Scholar] [CrossRef]
- Chen, Y.; Ramjiawan, R.R.; Reiberger, T.; Ng, M.R.; Hato, T.; Huang, Y.; Ochiai, H.; Kitahara, S.; Unan, E.C.; Reddy, T.P.; et al. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology 2015, 61, 1591–1602. [Google Scholar] [CrossRef]
- Wang, X.; Shen, Y.; Li, S.; Lv, M.; Zhang, X.; Yang, J.; Wang, F.; Yang, J. Importance of the interaction between immune cells and tumor vasculature mediated by thalidomide in cancer treatment. Int. J. Mol. Med. 2016, 38, 1021–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.K. Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity 2015, 43, 435–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Massena, S.; Christoffersson, G.; Vagesjo, E.; Seignez, C.; Gustafsson, K.; Binet, F.; Herrera Hidalgo, C.; Giraud, A.; Lomei, J.; Westrom, S.; et al. Identification and characterization of VEGF-A-responsive neutrophils expressing CD49d, VEGFR1, and CXCR4 in mice and humans. Blood 2015, 126, 2016–2026. [Google Scholar] [CrossRef]
- Shrimali, R.K.; Yu, Z.; Theoret, M.R.; Chinnasamy, D.; Restifo, N.P.; Rosenberg, S.A. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010, 70, 6171–6180. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Georganaki, M.; Conze, L.L.; Lavina, B.; van Hooren, L.; Vemuri, K.; van de Walle, T.; Ramachandran, M.; Zhang, L.; Ponten, F.; et al. ELTD1-deletion reduces vascular abnormality and improves T-cell recruitment after PD-1 blockade in glioma. Neuro Oncol. 2021, noab181. [Google Scholar] [CrossRef]
- Georganaki, M.; Ramachandran, M.; Tuit, S.; Nunez, N.G.; Karampatzakis, A.; Fotaki, G.; van Hooren, L.; Huang, H.; Lugano, R.; Ulas, T.; et al. Tumor endothelial cell up-regulation of IDO1 is an immunosuppressive feed-back mechanism that reduces the response to CD40-stimulating immunotherapy. Oncoimmunology 2020, 9, 1730538. [Google Scholar] [CrossRef] [Green Version]
- Hodi, F.S.; Lawrence, D.; Lezcano, C.; Wu, X.; Zhou, J.; Sasada, T.; Zeng, W.; Giobbie-Hurder, A.; Atkins, M.B.; Ibrahim, N.; et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol. Res. 2014, 2, 632–642. [Google Scholar] [CrossRef] [Green Version]
- Zang, G.; Gustafsson, K.; Jamalpour, M.; Hong, J.; Genove, G.; Welsh, M. Vascular dysfunction and increased metastasis of B16F10 melanomas in Shb deficient mice as compared with their wild type counterparts. BMC Cancer 2015, 15, 234. [Google Scholar] [CrossRef] [Green Version]
- Maehara, T.; Kaneko, N.; Perugino, C.A.; Mattoo, H.; Kers, J.; Allard-Chamard, H.; Mahajan, V.S.; Liu, H.; Murphy, S.J.; Ghebremichael, M.; et al. Cytotoxic CD4+ T lymphocytes may induce endothelial cell apoptosis in systemic sclerosis. J. Clin. Investig. 2020, 130, 2451–2464. [Google Scholar] [CrossRef] [Green Version]
- Vollmann, E.H.; Rattay, K.; Barreiro, O.; Thiriot, A.; Fuhlbrigge, R.A.; Vrbanac, V.; Kim, K.W.; Jung, S.; Tager, A.M.; von Andrian, U.H. Specialized transendothelial dendritic cells mediate thymic T-cell selection against blood-borne macromolecules. Nat. Commun. 2021, 12, 6230. [Google Scholar] [CrossRef] [PubMed]
- Dib, P.R.B.; Quirino-Teixeira, A.C.; Merij, L.B.; Pinheiro, M.B.M.; Rozini, S.V.; Andrade, F.B.; Hottz, E.D. Innate immune receptors in platelets and platelet-leukocyte interactions. J. Leukoc. Biol. 2020, 108, 1157–1182. [Google Scholar] [CrossRef] [PubMed]
- Wojtukiewicz, M.Z.; Sierko, E.; Hempel, D.; Tucker, S.C.; Honn, K.V. Platelets and cancer angiogenesis nexus. Cancer Metastasis Rev. 2017, 36, 249–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Ren, M.; Chen, N.; Luo, M.; Deng, X.; Xia, J.; Yu, G.; Liu, J.; He, B.; Zhang, X.; et al. Presence of intratumoral platelets is associated with tumor vessel structure and metastasis. BMC Cancer 2014, 14, 167. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Cedervall, J.; Hamidi, A.; Herre, M.; Viitaniemi, K.; D’Amico, G.; Miao, Z.; Unnithan, R.V.M.; Vaccaro, A.; van Hooren, L.; et al. Platelet-Specific PDGFB Ablation Impairs Tumor Vessel Integrity and Promotes Metastasis. Cancer Res. 2020, 80, 3345–3358. [Google Scholar] [CrossRef]
- Chatterjee, M.; Gawaz, M. Platelet-derived CXCL12 (SDF-1alpha): Basic mechanisms and clinical implications. J. Thromb. Haemost. 2013, 11, 1954–1967. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, J.S.; Talmage, K.E.; Massari, J.V.; La Jeunesse, C.M.; Flick, M.J.; Kombrinck, K.W.; Jirouskova, M.; Degen, J.L. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood 2005, 105, 178–185. [Google Scholar] [CrossRef] [Green Version]
- Shirai, T.; Inoue, O.; Tamura, S.; Tsukiji, N.; Sasaki, T.; Endo, H.; Satoh, K.; Osada, M.; Sato-Uchida, H.; Fujii, H.; et al. C-type lectin-like receptor 2 promotes hematogenous tumor metastasis and prothrombotic state in tumor-bearing mice. J. Thromb. Haemost. 2017, 15, 513–525. [Google Scholar] [CrossRef]
- Ringvall, M.; Thulin, A.; Zhang, L.; Cedervall, J.; Tsuchida-Straeten, N.; Jahnen-Dechent, W.; Siegbahn, A.; Olsson, A.K. Enhanced platelet activation mediates the accelerated angiogenic switch in mice lacking histidine-rich glycoprotein. PLoS ONE 2011, 6, e14526. [Google Scholar] [CrossRef]
- Mathiesen, A.; Hamilton, T.; Carter, N.; Brown, M.; McPheat, W.; Dobrian, A. Endothelial Extracellular Vesicles: From Keepers of Health to Messengers of Disease. Int. J. Mol. Sci. 2021, 22, 4640. [Google Scholar] [CrossRef]
- Grigoryeva, E.S.; Savelieva, O.E.; Popova, N.O.; Cherdyntseva, N.V.; Perelmuter, V.M. Do tumor exosome integrins alone determine organotropic metastasis? Mol. Biol. Rep. 2020, 47, 8145–8157. [Google Scholar] [CrossRef] [PubMed]
- Sabra, M.; Karbasiafshar, C.; Aboulgheit, A.; Raj, S.; Abid, M.R.; Sellke, F.W. Clinical Application of Novel Therapies for Coronary Angiogenesis: Overview, Challenges, and Prospects. Int. J. Mol. Sci. 2021, 22, 3722. [Google Scholar] [CrossRef] [PubMed]
- Wadey, R.M.; Connolly, K.D.; Mathew, D.; Walters, G.; Rees, D.A.; James, P.E. Inflammatory adipocyte-derived extracellular vesicles promote leukocyte attachment to vascular endothelial cells. Atherosclerosis 2019, 283, 19–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kugeratski, F.G.; McAndrews, K.M.; Kalluri, R. Multifunctional Applications of Engineered Extracellular Vesicles in the Treatment of Cancer. Endocrinology 2021, 162, bqaa250. [Google Scholar] [CrossRef]
- Arwert, E.N.; Harney, A.S.; Entenberg, D.; Wang, Y.; Sahai, E.; Pollard, J.W.; Condeelis, J.S. A Unidirectional Transition from Migratory to Perivascular Macrophage Is Required for Tumor Cell Intravasation. Cell Rep. 2018, 23, 1239–1248. [Google Scholar] [CrossRef] [Green Version]
- Honkura, N.; Richards, M.; Lavina, B.; Sainz-Jaspeado, M.; Betsholtz, C.; Claesson-Welsh, L. Intravital imaging-based analysis tools for vessel identification and assessment of concurrent dynamic vascular events. Nat. Commun. 2018, 9, 2746. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Welsh, M. Perspectives on Vascular Regulation of Mechanisms Controlling Selective Immune Cell Function in the Tumor Immune Response. Int. J. Mol. Sci. 2022, 23, 2313. https://doi.org/10.3390/ijms23042313
Welsh M. Perspectives on Vascular Regulation of Mechanisms Controlling Selective Immune Cell Function in the Tumor Immune Response. International Journal of Molecular Sciences. 2022; 23(4):2313. https://doi.org/10.3390/ijms23042313
Chicago/Turabian StyleWelsh, Michael. 2022. "Perspectives on Vascular Regulation of Mechanisms Controlling Selective Immune Cell Function in the Tumor Immune Response" International Journal of Molecular Sciences 23, no. 4: 2313. https://doi.org/10.3390/ijms23042313