
Thiacalixarenes with Sulfur Functionalities at Lower Rim: Heavy Metal Ion Binding in Solution and 2D-Confined Space

 , , , , , and
, , , , , and
Abstract
:
1. Introduction
2. Results
2.1. Synthesis and Characterization of Ligands
2.2. Complexation of Metal Ions at Liquid–Liquid Interface
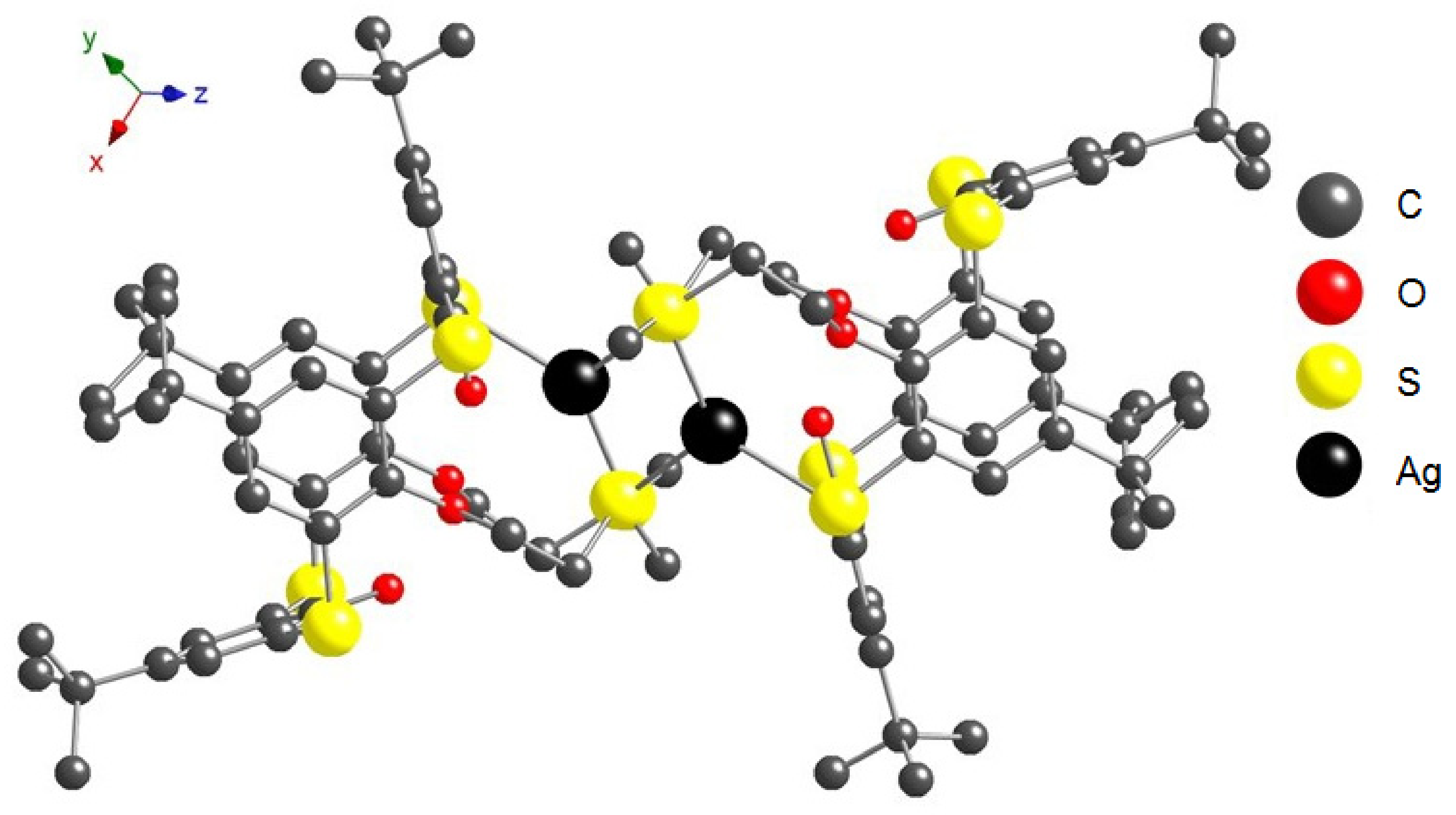
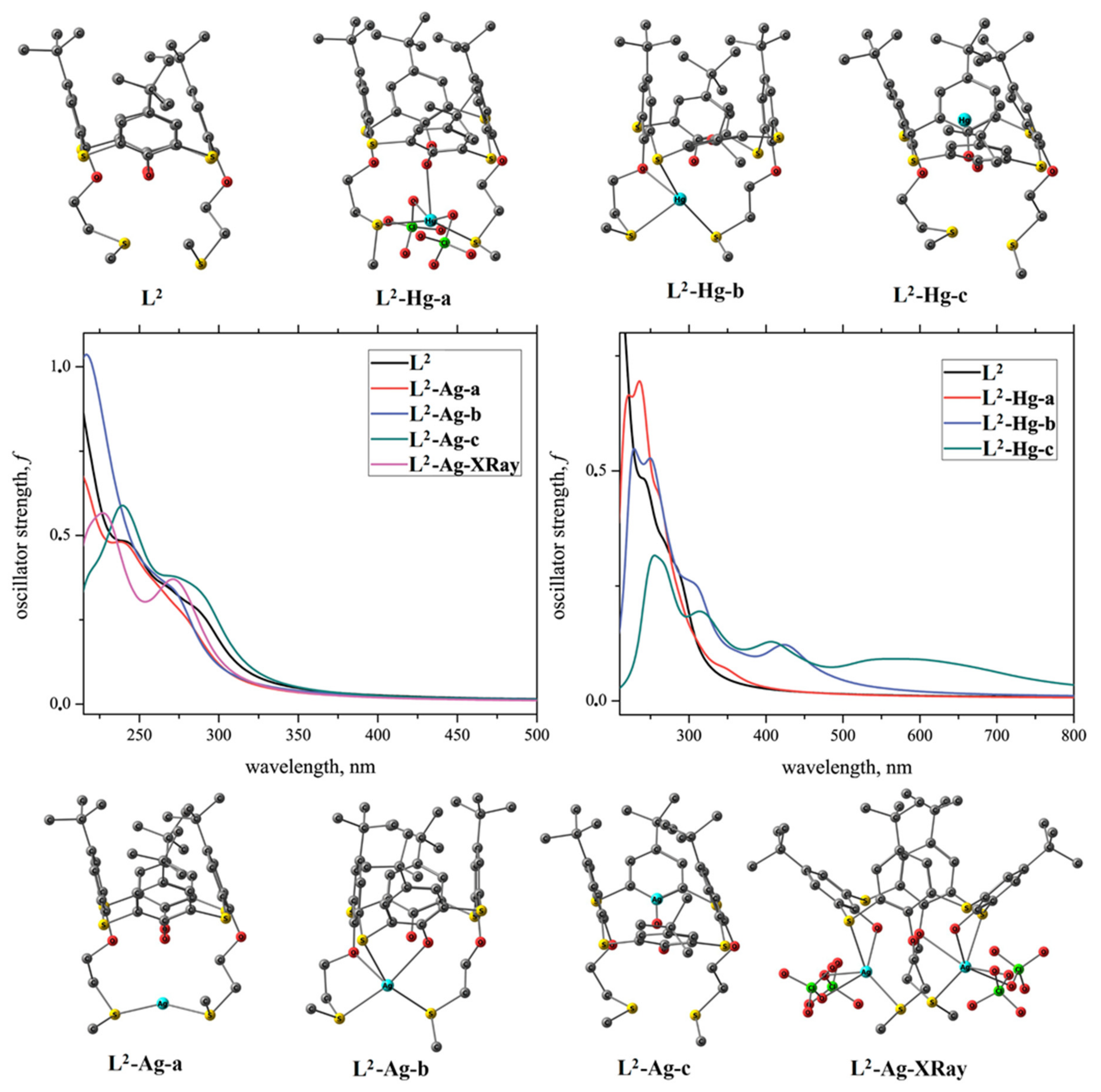
2.3. Solid-State Structures of Ag+ and Hg2+ Complexes
2.4. Metal Ion Binding in Solution
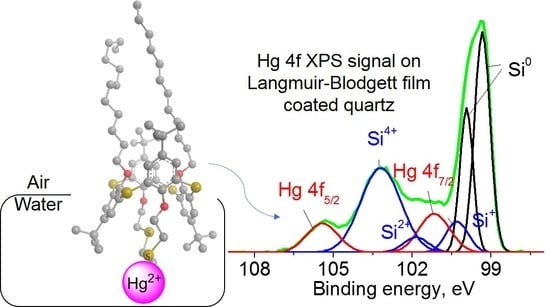
2.5. Aggregation and Interactions with Ions at Air–Water Interface
3. Materials and Methods
Synthesis of Thiacrown-Ether L3
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NMR | Nuclear magnetic resonance |
| LB | Langmuir–Blodgett |
| XPS | X-ray photoelectron spectroscopy |
| IR | Infrared |
| MALDI | Matrix-assisted laser desorption–ionization |
| DLS | Dynamic light scattering |
| DFT | Density functional theory |
| SPOT | Surface potential |
| TZVP | Valence triple-zeta polarization |
| B3LYP | Becke, 3-parameter, Lee–Yang–Parr |
References
- Neri, P.; Sessler, J.L.; Wang, M.-X. Calixarenes and Beyond; Springer: New York, NY, USA, 2016. [Google Scholar] [CrossRef]
- Tan, H.-W.; Liu, Y.-C.; Guo, D.-S. Assembling features of calixarene-based amphiphiles and supra-amphiphiles. Mater. Chem. Front. 2020, 4, 46–98. [Google Scholar] [CrossRef]
- Kumar, R.; Sharma, A.; Singh, H.; Suating, P.; Kim, H.S.; Sunwoo, K.; Shim, I.; Gibb, B.C.; Kim, J.S. Revisiting fluorescent calixarenes: From molecular sensors to smart materials. Chem. Rev. 2019, 119, 9657–9721. [Google Scholar] [CrossRef]
- Iki, N.; Morohashi, N.; Narumi, F.; Miyano, S. High complexation ability of thiacalixarene with transition metal ions. the effects of replacing methylene bridges of tetra(p-t-butyl)calix[4]arenetetrol by epithio groups. Bull. Chem. Soc. Jpn. 1998, 71, 1597–1603. [Google Scholar] [CrossRef]
- Yordanov, A.T.; Whittlesey, B.R.; Roundhill, D.M. Calixarenes derivatized with sulfur-containing functionalities as selective extractants for heavy and precious metal ions. Inorg. Chem. 1998, 37, 3526–3531. [Google Scholar] [CrossRef]
- Zhu, S.; Lu, L. Selective chromogenic recognition of copper(ii) ion by thiacalix[4]arene tetrasulfonate and mechanism. Molecules 2020, 25, 612. [Google Scholar] [CrossRef] [Green Version]
- Shetty, D.; Boutros, S.; Eskhan, A.; De Lena, A.M.; Skorjanc, T.; Asfari, Z.; Traboulsi, H.; Mazher, J.; Raya, J.; Banat, F.; et al. Thioether-crown-rich calix[4]arene porous polymer for highly efficient removal of mercury from water. ACS Appl. Mater. Interfaces 2019, 11, 12898–12903. [Google Scholar] [CrossRef]
- Muravev, A.A.; Solovieva, S.E.; Kochetkov, E.N.; Mel’nikova, N.B.; Safiullin, R.A.; Kadirov, M.K.; Latypov, S.K.; Antipin, I.S.; Konovalov, A.I. Thiacalix[4]monocrowns substituted by sulfur-containing anchoring groups: New ligands for gold surface modification. Macroheterocycles 2013, 6, 302–307. [Google Scholar] [CrossRef]
- Jágerszki, G.; Grün, A.; Bitter, I.; Tóth, K.; Gyurcsányi, R.E. Ionophore–gold nanoparticle conjugates for Ag+-selective sensors with nanomolar detection limit. Chem. Commun. 2010, 46, 607–609. [Google Scholar] [CrossRef]
- Lee, J.L.; Kim, H.J.; Jung, J.H.; Sim, W.; Lee, S.S. Networking of calixcrowns: From heteronuclear endo/exocyclic coordination polymers to a photoluminescence switch. J. Am. Chem. Soc. 2008, 130, 13838–13839. [Google Scholar] [CrossRef]
- Shokurov, A.V.; Shcherbina, M.A.; Bakirov, A.V.; Alexandrova, A.V.; Raitman, O.A.; Arslanov, V.V.; Chvalun, S.N.; Selektor, S.L. Rational design of hemicyanine Langmuir monolayers by cation-induced preorganization of their structure for sensory response enhancement. Langmuir 2018, 34, 7690–7697. [Google Scholar] [CrossRef] [PubMed]
- Shokurov, A.V.; Alexandrova, A.V.; Shcherbina, M.A.; Bakirov, A.V.; Rogachev, A.V.; Yakunin, S.N.; Chvalun, S.N.; Arslanov, V.V.; Selektor, S.L. Supramolecular control of the structure and receptor properties of an amphiphilic hemicyanine chromoionophore monolayer at the air/water interface. Soft Matter 2020, 16, 9857–9863. [Google Scholar] [CrossRef] [PubMed]
- Bitter, I.; Csokai, V. An expedient route to p-tert-butylthiacalix[4]arene 1,3-diethers via Mitsunobu reactions. Tetrahedron Lett. 2003, 44, 2261–2265. [Google Scholar] [CrossRef]
- Iki, N.; Kabuto, C.; Fukushima, T.; Kumagai, H.; Takeya, H.; Miyanari, S.; Miyashi, T.; Miyano, S. Synthesis of p-tert-butylthiacalix[4]arene and its inclusion property. Tetrahedron 2000, 56, 1437–1443. [Google Scholar] [CrossRef]
- Muravev, A.A.; Solovieva, S.E.; Latypov, S.K.; Antipin, I.S.; Konovalov, A.I. Synthesis and characterization of thiacalix[4]monocrowns modified by thioether groups on the lower rim. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 499–502. [Google Scholar] [CrossRef]
- Muravev, A.A.; Trushina, E.A.; Yakupov, A.T.; Solovieva, S.E.; Antipin, I.S. Ag-selective nanotubes based on bisthiacalix[4]arene with ethylene sulfide bridges. Dokl. Chem. 2019, 487, 212–214. [Google Scholar] [CrossRef]
- Muravev, A.A.; Yakupov, A.T.; Solovieva, S.E.; Antipin, I.S. A new approach to the synthesis of thiacrowns on a thiacalix[4]arene scaffold. Dokl. Chem. 2019, 487, 188–191. [Google Scholar] [CrossRef]
- Dvořáková, H.; Lang, J.; Vlach, J.; Sýkora, J.; Čajan, M.; Himl, M.; Pojarová, M.; Stibor, I.; Lhoták, P. Partially O-alkylated thiacalix[4]arenes: Synthesis, molecular and crystal structures, conformational behavior. J. Org. Chem. 2007, 72, 7157–7166. [Google Scholar] [CrossRef]
- Latypov, S.K.; Kharlamov, S.V.; Muravev, A.A.; Balandina, A.A.; Solovieva, S.E.; Antipin, I.S.; Konovalov, A.I. Conformational diversity and dynamics of distally disubstituted calix and thiacalix[4]arenes in solution. J. Phys. Org. Chem. 2013, 26, 407–414. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Yang, W.-P.; Chen, Y.-Y.; Gong, S.-L. Different metal-ion-induced dimeric self-assembling cavities based on thiacalix[4]benzocrown-4 isomers. Dalton Trans. 2011, 40, 367–376. [Google Scholar] [CrossRef]
- Ju, H.; Park, I.-H.; Lee, E.; Kim, C.; Kim, S.; Kuwahara, S.; Habata, Y.; Lee, S.S. A thiacalix basket and its anion-dependent 2-D and 3-D silver(I) coordination polymers via exo-coordination. Eur. J. Inorg. Chem. 2020, 2020, 356–360. [Google Scholar] [CrossRef]
- Ju, H.; Kim, C.; Choi, K.-S.; Lee, E.; Kim, S.; Jung, J.H.; Habata, Y.; Lindoy, L.F.; Lee, S.S. Thiacalix[4]-bis-crown with hard cavities and soft bridges exhibiting endocyclic potassium(I) complexes and exocyclic silver(I) coordination polymers. Eur. J. Inorg. Chem. 2018, 2018, 3587–3594. [Google Scholar] [CrossRef]
- Wang, Z.; Su, H.-F.; Gong, Y.-W.; Qu, Q.-P.; Bi, Y.-F.; Tung, C.-H.; Sun, D.; Zheng, L.-S. A hierarchically assembled 88-nuclei silver-thiacalix[4]arene nanocluster. Nat. Commun. 2020, 11, 308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamsieva, A.V.; Musina, E.I.; Gerasimova, T.P.; Fayzullin, R.R.; Kolesnikov, I.E.; Samigullina, A.I.; Katsyuba, S.A.; Karasik, A.A.; Sinyashin, O.G. Intriguing near-infrared solid-state luminescence of binuclear silver(I) complexes based on pyridylphospholane scaffolds. Inorg. Chem. 2019, 58, 7698–7704. [Google Scholar] [CrossRef] [PubMed]
- Yam, V.W.W.; Cheng, E.C.C.; Zhu, N. A novel polynuclear gold–sulfur cube with an unusually large Stokes shift. Angew. Chem. Int. Ed. 2001, 113, 1813–1815. [Google Scholar] [CrossRef]
- Danil de Namor, A.F.; Chahine, S.; Castellano, E.E.; Piroc, O.E.; Jenkins, H.D.B. A preliminary observation of additive thermodynamic contribution of pendant arms to the complexation of calixarene derivatives with mercury(II). Chem. Commun. 2005, 41, 3844–3846. [Google Scholar] [CrossRef] [PubMed]
- Praveen, L.; Ganga, V.B.; Thirumalai, R.; Sreeja, T.; Reddy, M.L.P.; Luxmi Varma, R. A new Hg2+ selective fluorescent sensor based on a 1,3-alternate thiacalixarene anchored with four 8-quinolinoloxy groups. Inorg. Chem. 2007, 46, 6277–6282. [Google Scholar] [CrossRef]
- Ju, H.; Lee, J.Y.; Lee, S.S. Influence of anions and mole ratio on the formation of 2-D coordination networks of thiacalix[4]-bis-monothiacrown-5. CrystEngComm 2020, 22, 7617–7622. [Google Scholar] [CrossRef]
- Colleran, J.J.; Creaven, B.S.; Donlon, D.F.; McGinley, J. Non-trivial solution chemistry between amido-pyridylcalix[4]arenes and some metal salts. Dalton Trans. 2010, 39, 10928–10936. [Google Scholar] [CrossRef] [Green Version]
- Muravev, A.; Yakupov, A.; Gerasimova, T.; Nugmanov, R.; Trushina, E.; Babaeva, O.; Nizameeva, G.; Syakaev, V.; Katsyuba, S.; Selektor, S.; et al. Switching ion binding selectivity of thiacalix[4]arene monocrowns at liquid–liquid and 2D-confined interfaces. Int. J. Mol. Sci. 2021, 22, 3535. [Google Scholar] [CrossRef] [PubMed]
- Leontidis, E. Chaotropic salts interacting with soft matter: Beyond the lyotropic series. Curr. Opin. Colloid Interface Sci. 2016, 23, 100–109. [Google Scholar] [CrossRef]
- Xie, J.; Zheng, Q.-Y.; Zheng, Y.-S.; Chen, C.-F.; Huang, Z.-T. Syntheses and metal-ion binding properties of calix[4]arene derivatives containing soft donor atoms: Highly selective extraction reagents for Ag+. J. Incl. Phenom. Macrocycl. Chem. 2001, 40, 125–130. [Google Scholar] [CrossRef]
- Tabakci, M.; Memon, S.; Yilmaz, M.; Roundhill, D.M. Oligomeric calix[4]arene-thiacrown ether for toxic heavy metals. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 186–193. [Google Scholar] [CrossRef]
- Armarego, W.L.F. Purification of Laboratory Chemicals; Armarego, W.L.F., Chai, C.L.L., Eds.; Butterworth-Heinemann: Oxford, UK, 2009. [Google Scholar]
- Naumkin, A.V.; Kraut-Vass, A.; Gaarenstroom, S.W.; Powell, C.J. NIST X-Ray Photoelectron Spectroscopy Database, Version 4.1; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2012. Available online: http://srdata.nist.gov/xps/ (accessed on 1 September 2021).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, UK, 2016. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Häser, M.; Treutler, O.; Ahlrichs, R. Calculation of excitation energies within time-dependent density functional theory using auxiliary basis set expansions. Chem. Phys. Lett. 1997, 264, 573–578. [Google Scholar] [CrossRef]
- Furche, F. On the density matrix based approach to time-dependent density functional response theory. J. Chem. Phys. 2001, 114, 5982–5992. [Google Scholar] [CrossRef]
- Rudberg, E.; Sałek, P. Calculations of two-photon charge-transfer excitations using Coulomb-attenuated density-functional theory. J. Chem. Phys. 2005, 123, 184108. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, R.; Amos, R.D. The application of CAM-B3LYP to the charge-transfer band problem of the zincbacteriochlorin-bacteriochlorin complex. Chem. Phys. Lett. 2006, 420, 106–109. [Google Scholar] [CrossRef]
- Cai, Z.L.; Crossley, M.J.; Reimers, J.R.; Kobayashi, R.; Amos, R.D. Density functional theory for charge transfer: The nature of the N-Bands of porphyrins and chlorophylls revealed through CAM-B3LYP, CASPT2, and SAC-CI calculations. J. Phys. Chem. B 2006, 110, 15624–15632. [Google Scholar] [CrossRef] [PubMed]
- Battye, T.G.; Kontogiannis, L.; Johnson, O.H.; Powell, R.; Leslie, A.W. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, P.R. Scaling and assessment of data quality. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Adv. 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | ||
|---|---|---|
| L2 | 8.65 (1:1) | 11.36 (1:2) |
| L3 | 7.51 (1:1) | 9.81 (1:3) |
| L4 | 8.33 (1:1) | 12.90 (1:1) |
| Ligand | Technique | Ag+ | Hg2+ |
|---|---|---|---|
| L2 | X-ray | epithio and alkyl sulfide | n/a |
| UV/Vis | epithio and/or alkyl sulfide | alkyl sulfide | |
| NMR | alkyl sulfide | alkyl sulfide | |
| L3 | NMR | alkyl sulfide | alkyl sulfide |
| L4 | X-ray | n/a | alkyl sulfide |
| UV/Vis | epithio and/or alkyl sulfide | alkyl sulfide |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muravev, A.; Yakupov, A.; Gerasimova, T.; Islamov, D.; Lazarenko, V.; Shokurov, A.; Ovsyannikov, A.; Dorovatovskii, P.; Zubavichus, Y.; Naumkin, A.; et al. Thiacalixarenes with Sulfur Functionalities at Lower Rim: Heavy Metal Ion Binding in Solution and 2D-Confined Space. Int. J. Mol. Sci. 2022, 23, 2341. https://doi.org/10.3390/ijms23042341
Muravev A, Yakupov A, Gerasimova T, Islamov D, Lazarenko V, Shokurov A, Ovsyannikov A, Dorovatovskii P, Zubavichus Y, Naumkin A, et al. Thiacalixarenes with Sulfur Functionalities at Lower Rim: Heavy Metal Ion Binding in Solution and 2D-Confined Space. International Journal of Molecular Sciences. 2022; 23(4):2341. https://doi.org/10.3390/ijms23042341
Chicago/Turabian StyleMuravev, Anton, Ayrat Yakupov, Tatiana Gerasimova, Daut Islamov, Vladimir Lazarenko, Alexander Shokurov, Alexander Ovsyannikov, Pavel Dorovatovskii, Yan Zubavichus, Alexander Naumkin, and et al. 2022. "Thiacalixarenes with Sulfur Functionalities at Lower Rim: Heavy Metal Ion Binding in Solution and 2D-Confined Space" International Journal of Molecular Sciences 23, no. 4: 2341. https://doi.org/10.3390/ijms23042341