
Premarin Reduces Neurodegeneration and Promotes Improvement of Function in an Animal Model of Spinal Cord Injury

 ,
, 
 ,
,  and
and 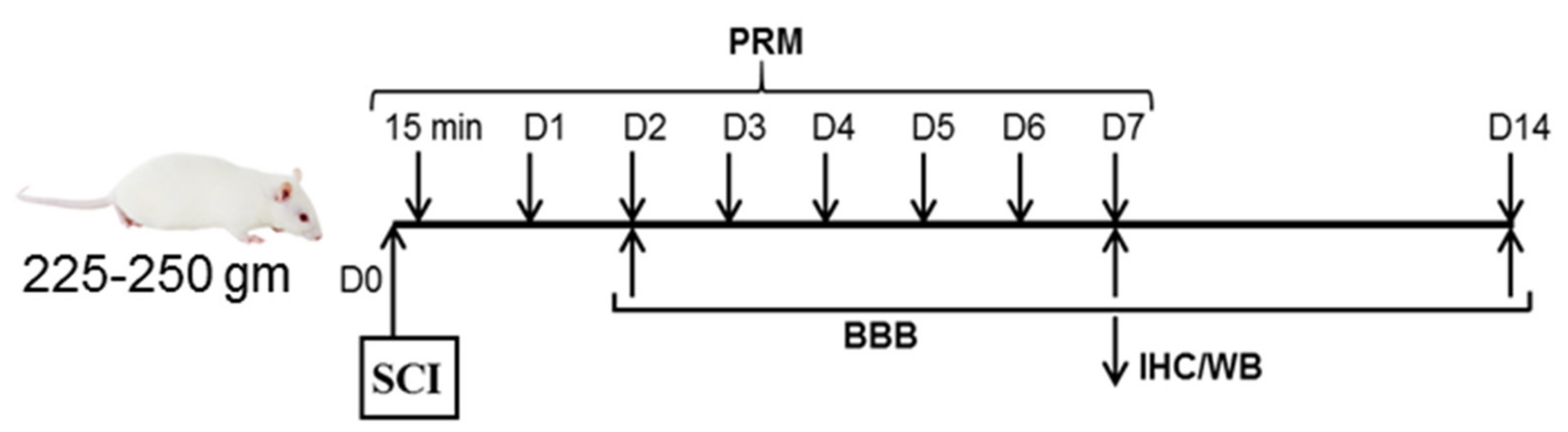{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
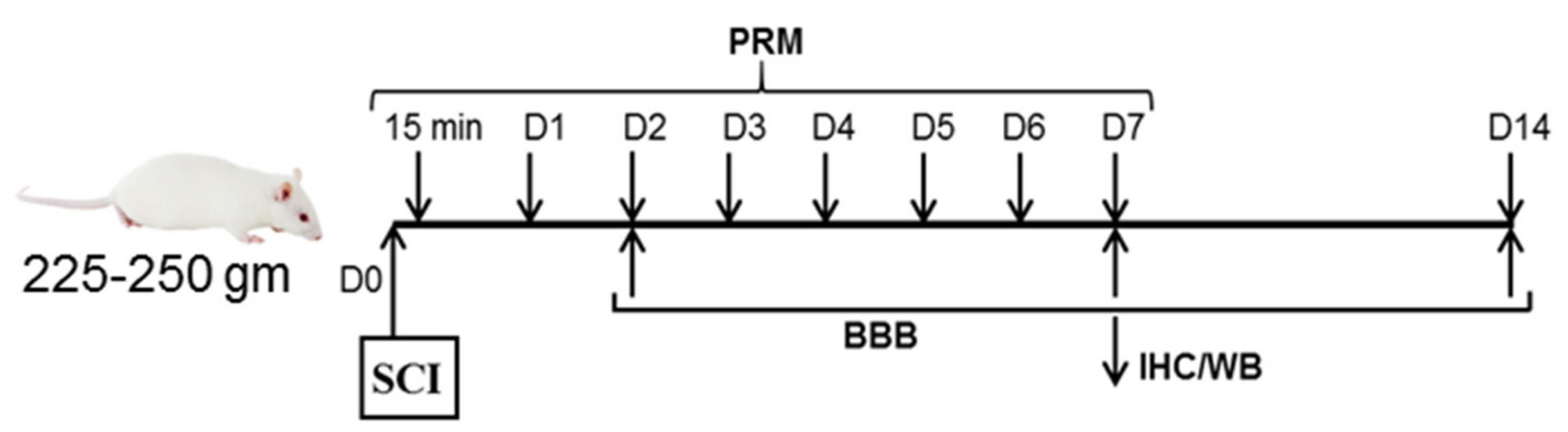
2.1. Induction of SCI and Treatment with PRM
2.2. Magnetic Resonance Imaging (MRI)
2.3. Immunofluorescent Staining
2.4. Luxol Fast Blue (LFB) Staining
2.5. Western Blot Analysis
2.6. Assessment of Locomotor Function (BBB Scoring)
2.7. Statistical Analyses of Data
3. Results
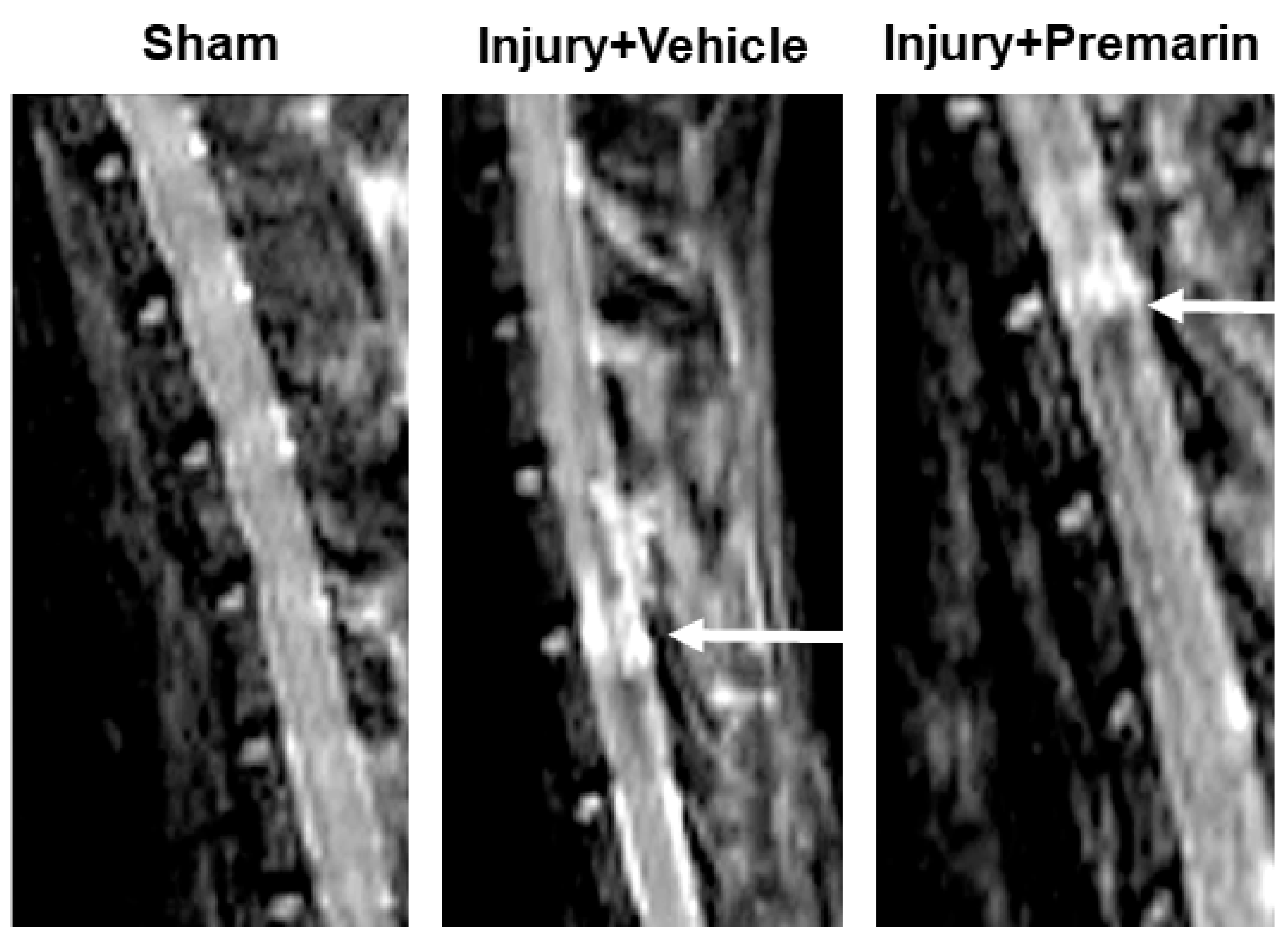
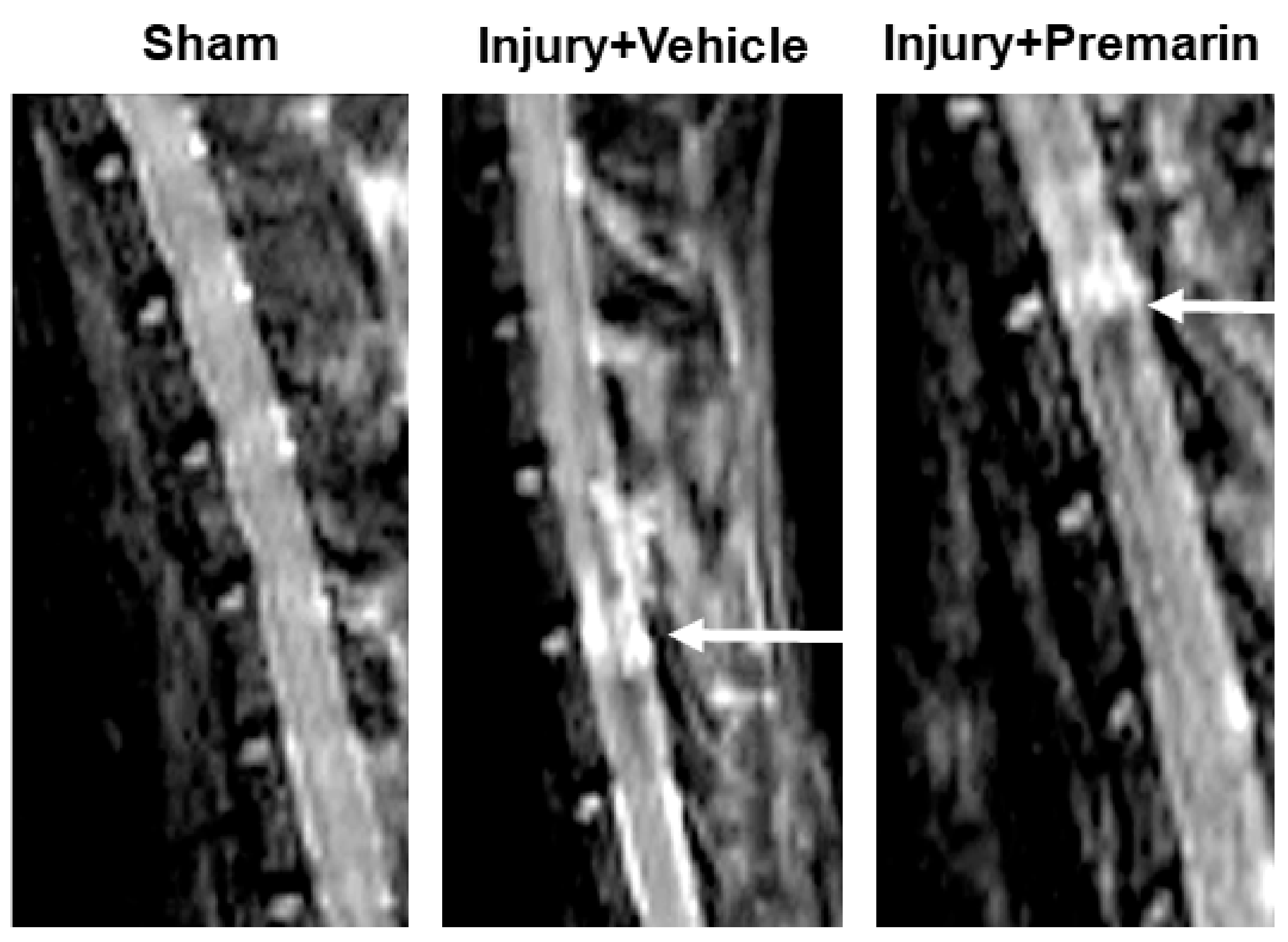
3.1. PRM Decreases Post-SCI Lesion Volume
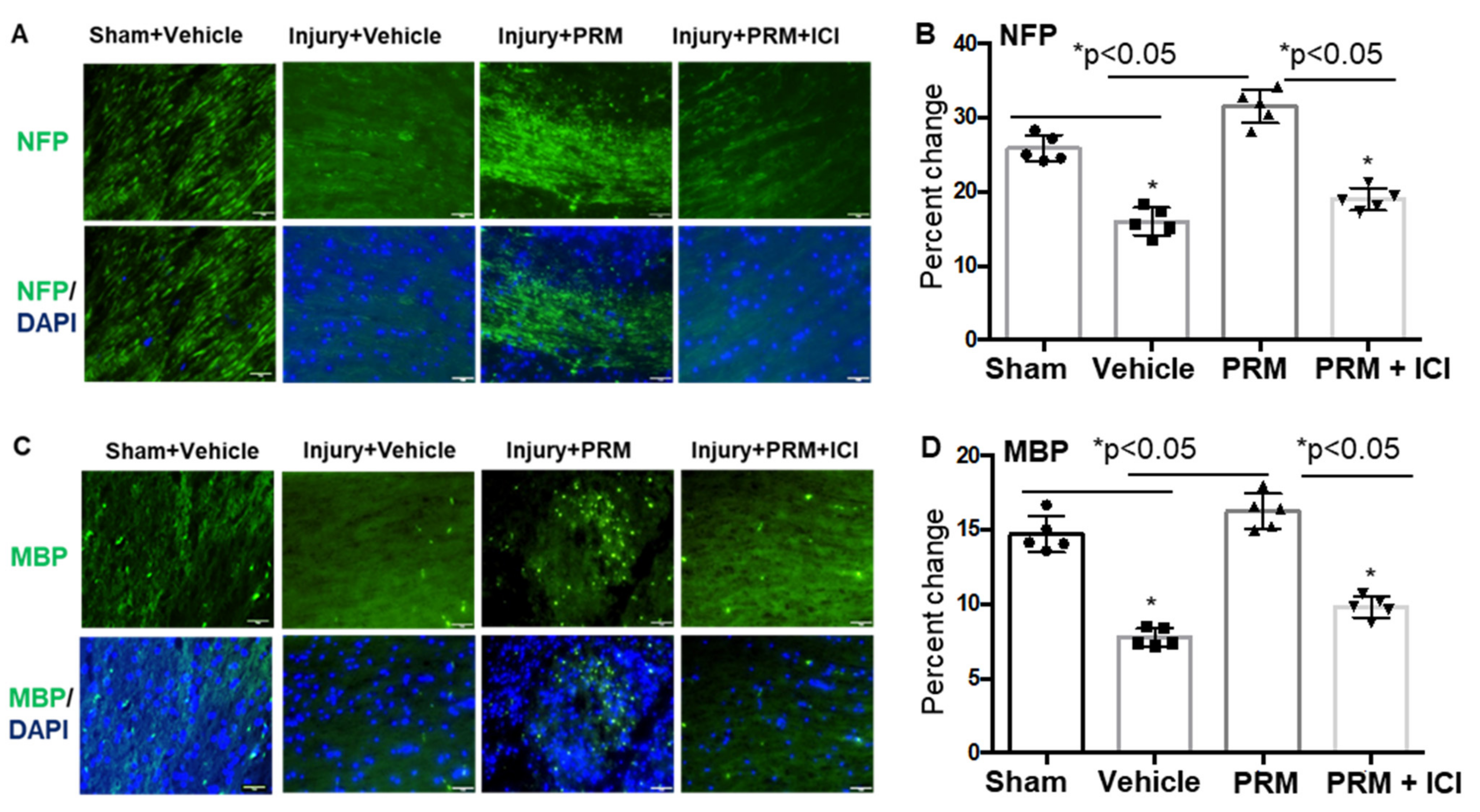
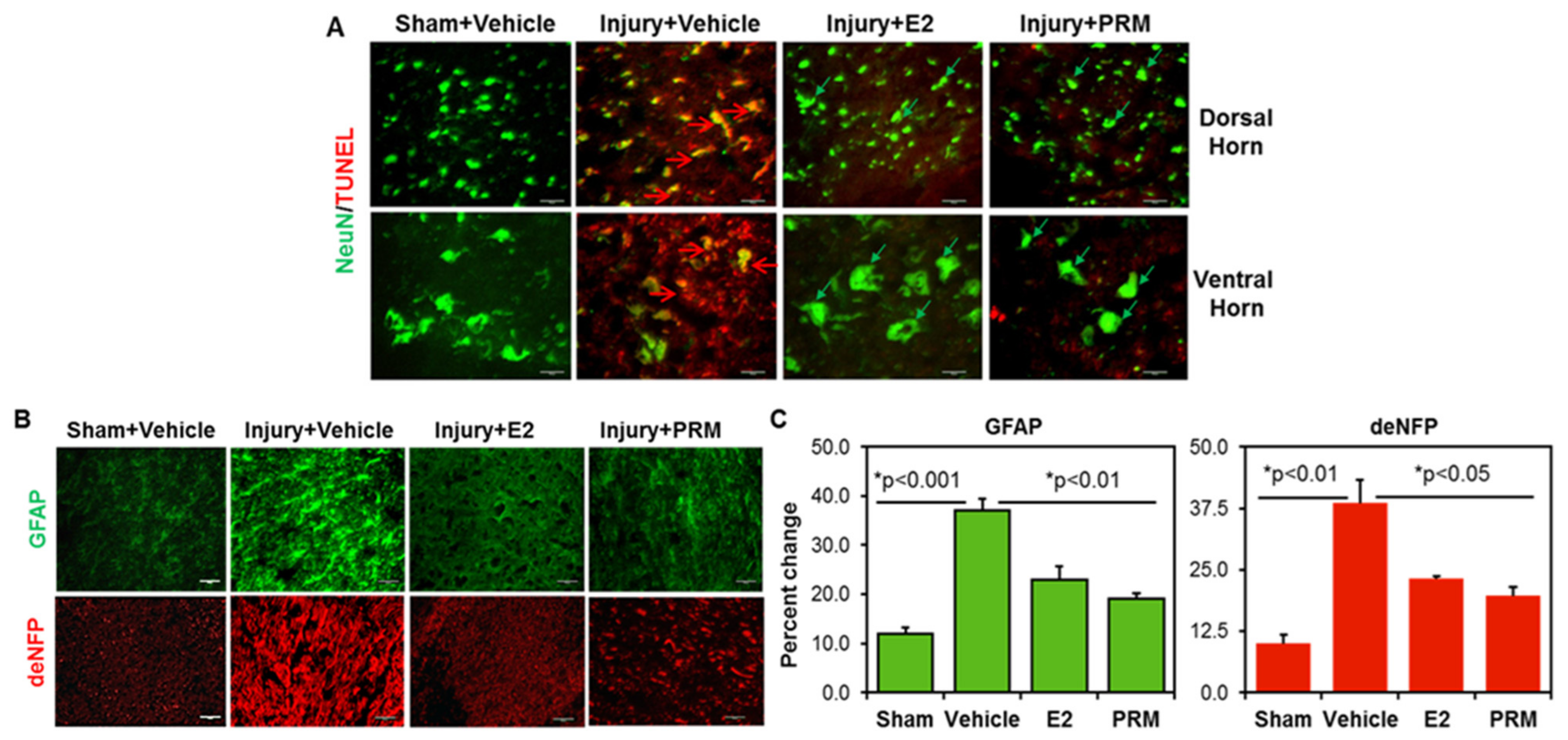
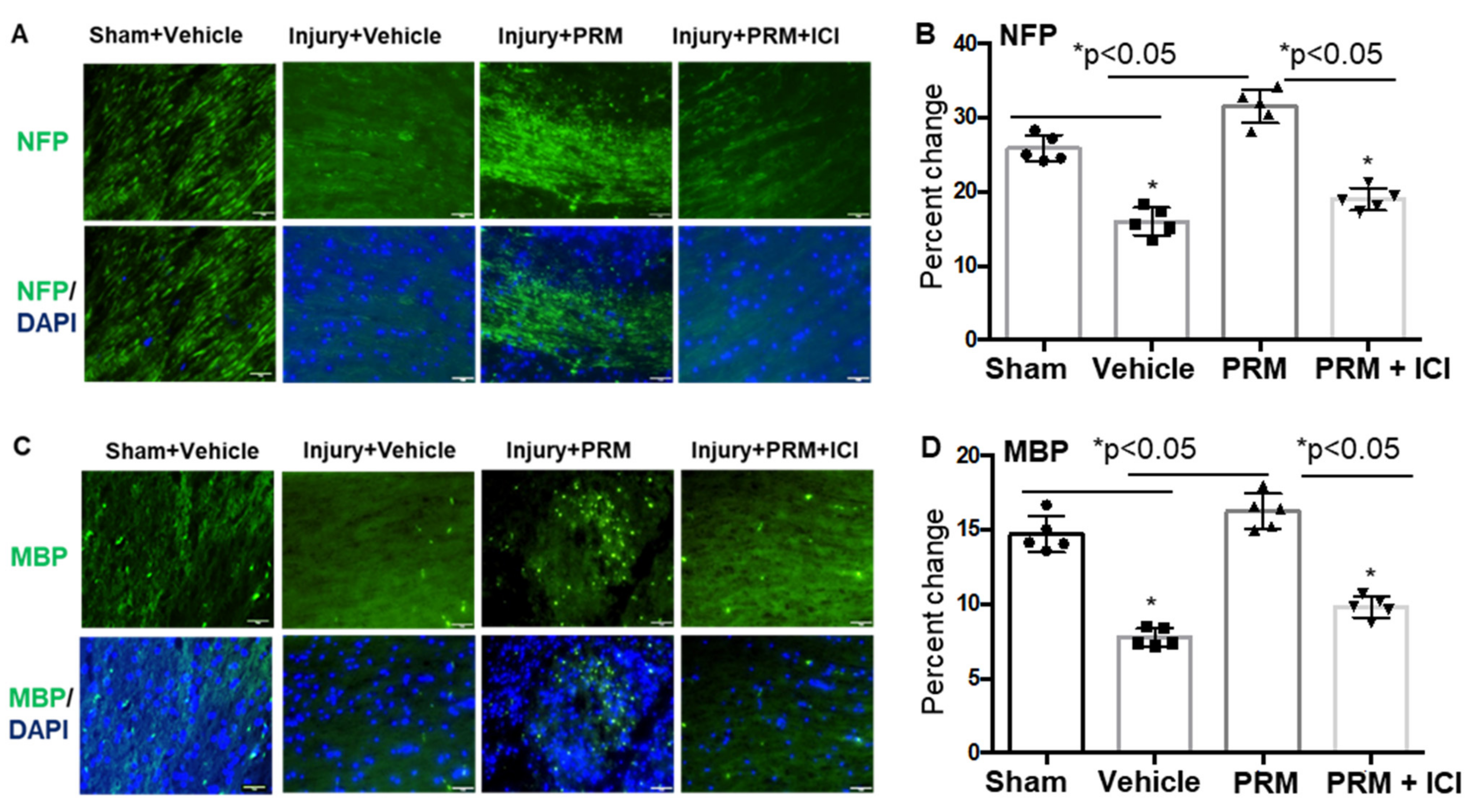
3.2. PRM Attenuates Neuronal Cell Death and Axonal Damage after SCI
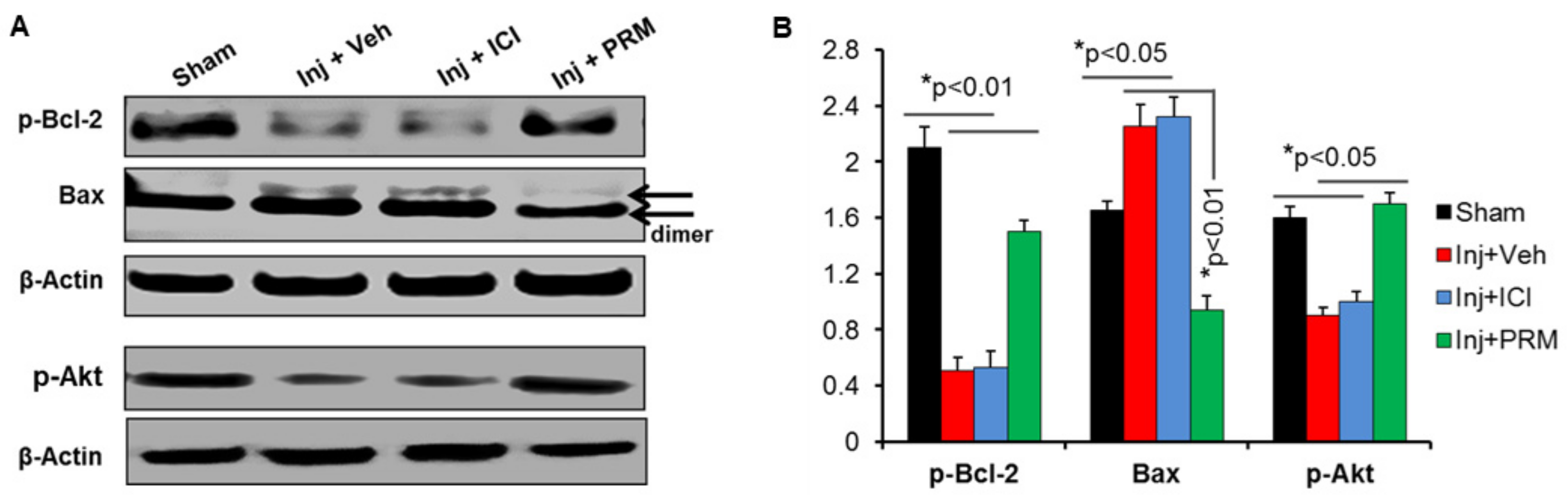
3.3. PRM Alters the Balance of Pro- and Anti-Apoptotic Proteins in Favor of Cell Survival
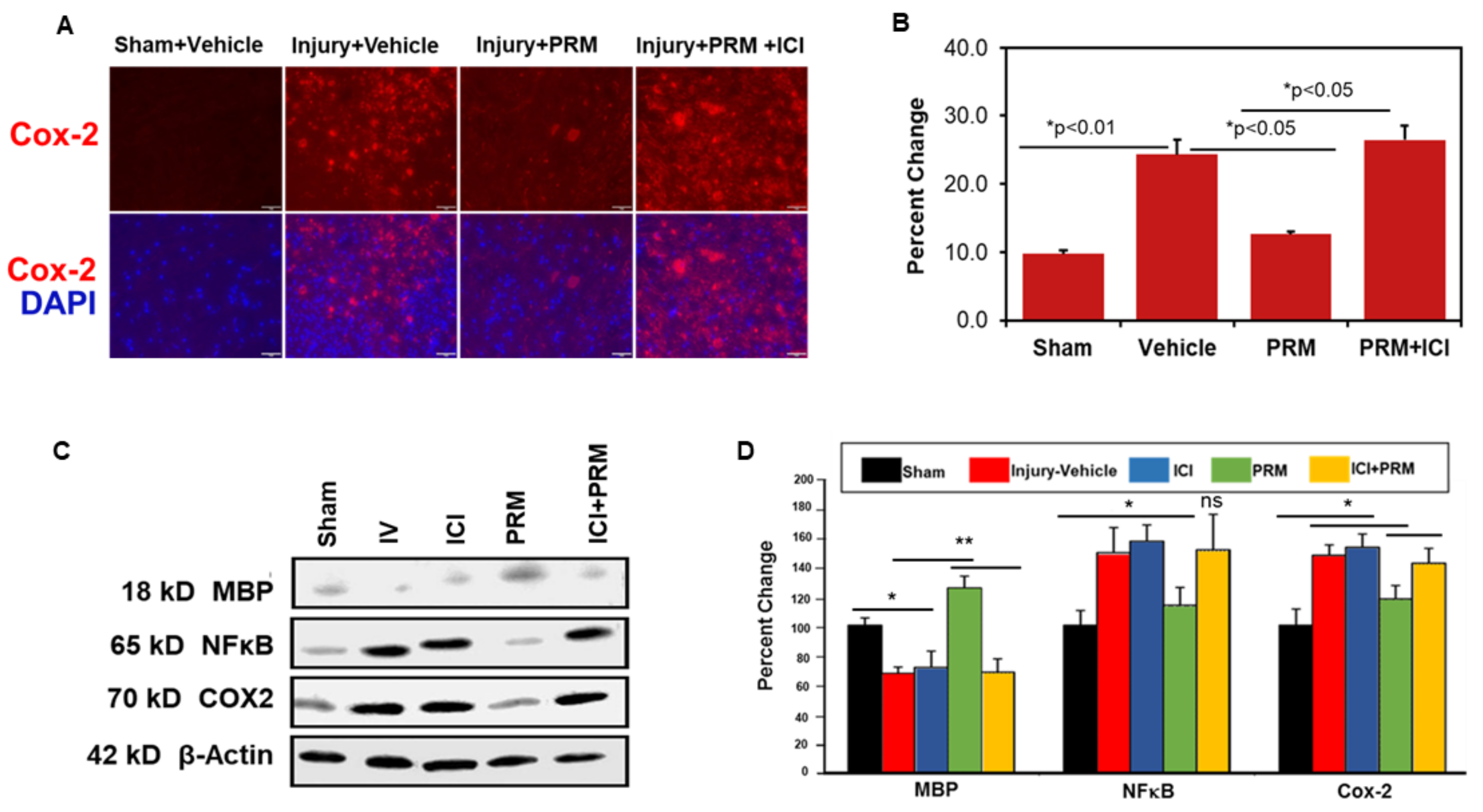
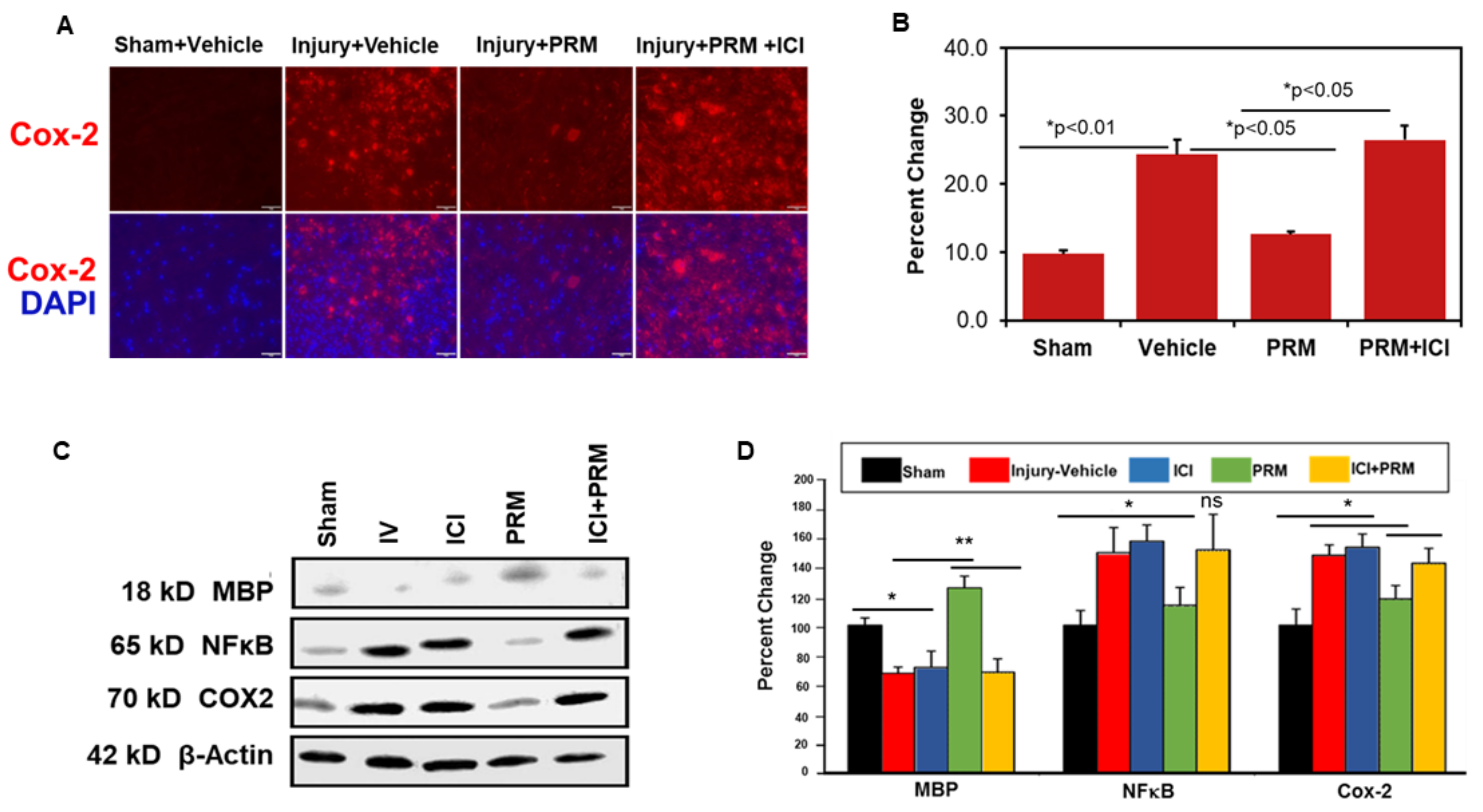
3.4. PRM Attenuates Inflammatory Events following SCI
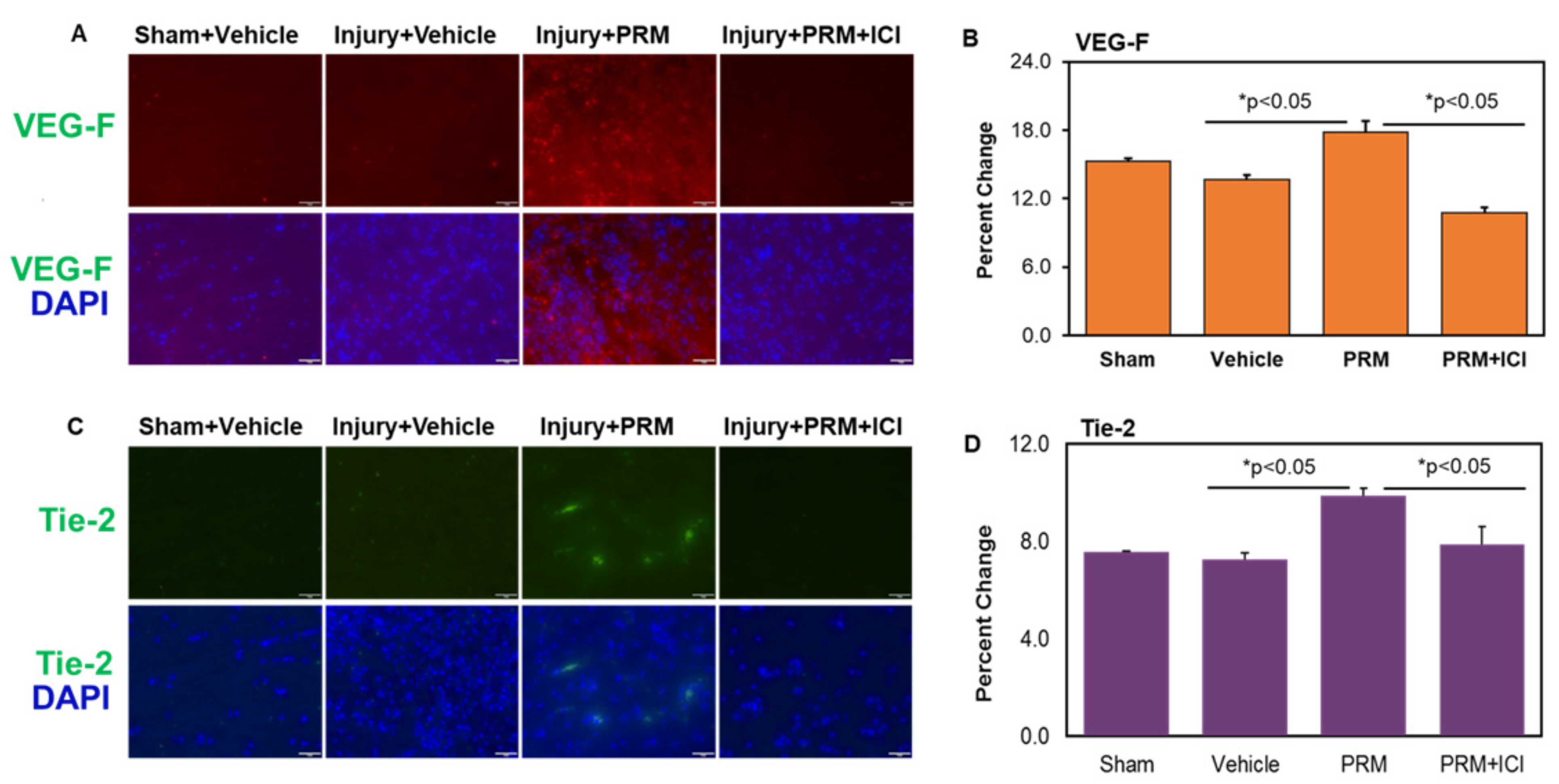
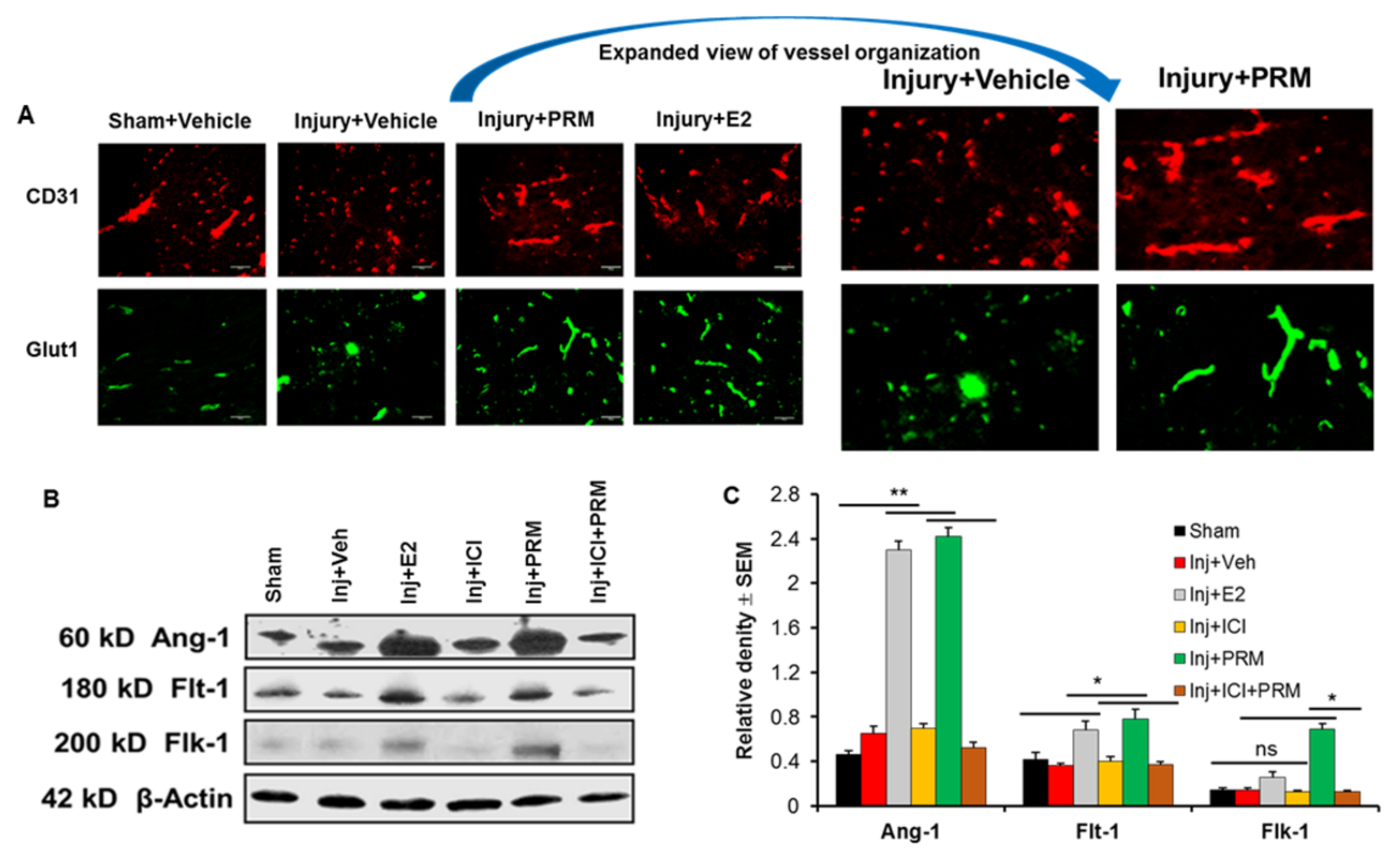
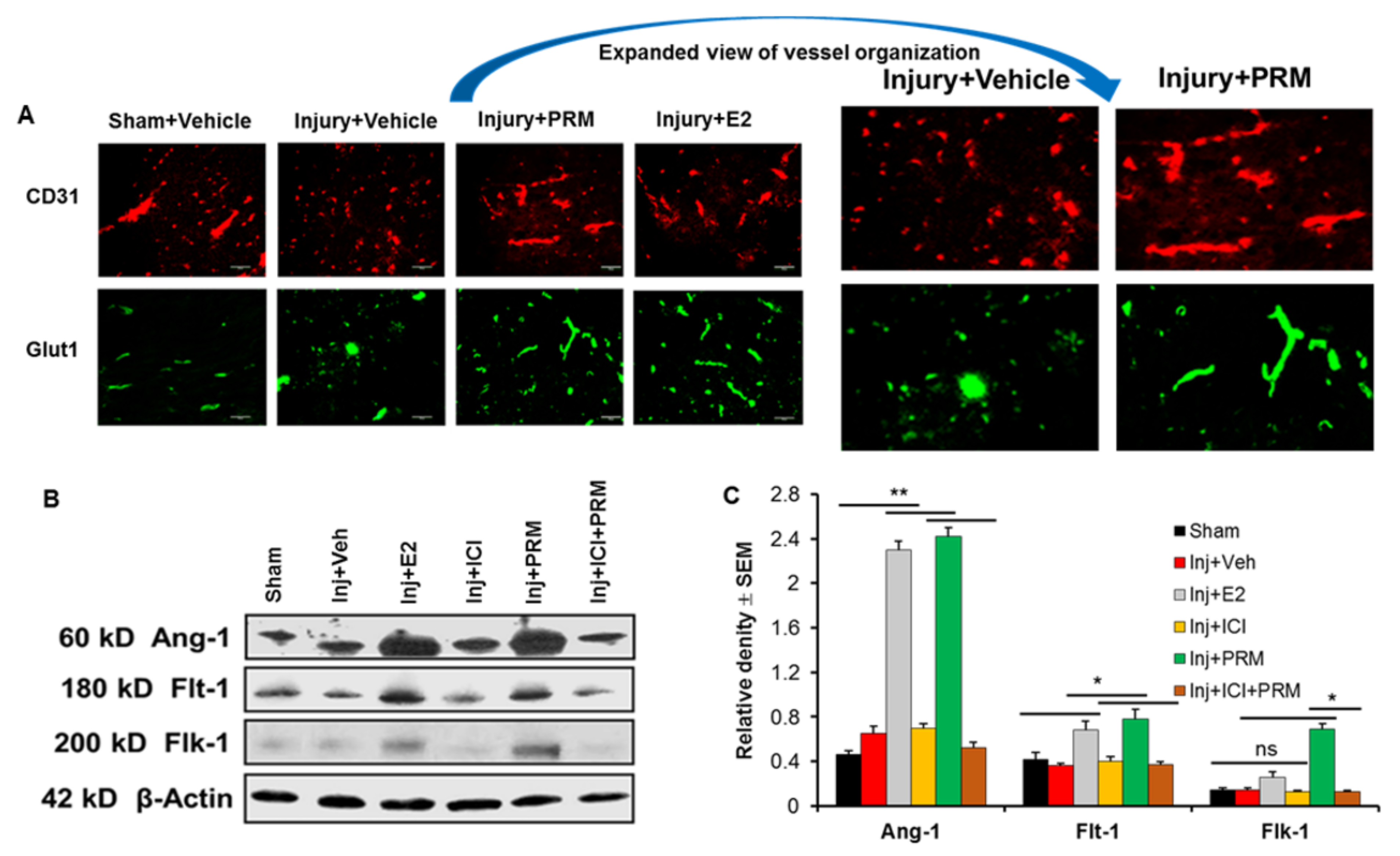
3.5. PRM Improves Microvessel Growth and Increases Expression of Multiple Markers of Angiogenesis
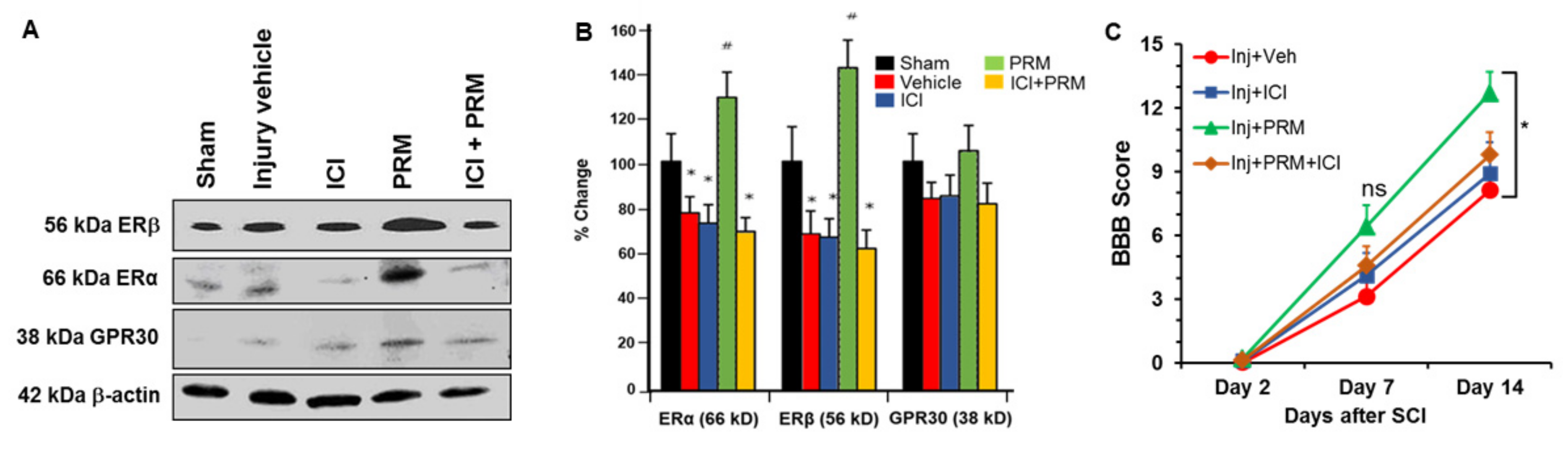
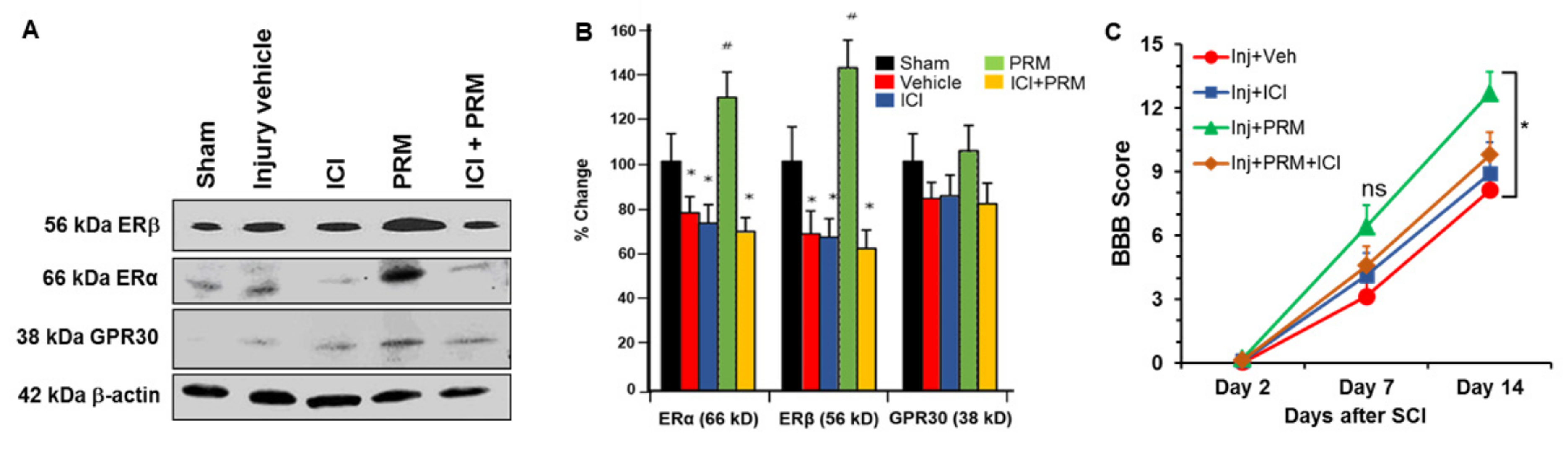
3.6. PRM Increased Expression of Erα and Erβ but Not GPR30
3.7. PRM Improves Motor Function in SCI Rats
3.8. Major Side Effect of PRM Treatment
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matsumoto, T.; Tamaki, T.; Kawakami, M.; Yoshida, M.; Ando, M.; Yamada, H. Early complications of high-dose methylprednisolone sodium succinate treatment in the follow-up of acute cervical spinal cord injury. Spine 2001, 26, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Short, D.J.; El Masry, W.S.; Jones, P.W. High dose methylprednisolone in the management of acute spinal cord injury—A systematic review from a clinical perspective. Spinal Cord 2000, 38, 273–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurlbert, R.J. Methylprednisolone for acute spinal cord injury: An inappropriate standard of care. J. Neurosurg. 2000, 93 (Suppl. 1), 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracken, M.B.; Collins, W.F.; Freeman, D.F.; Shepard, M.J.; Wagner, F.W.; Silten, R.M.; Hellenbrand, K.G.; Ransohoff, J.; Hunt, W.E.; Perot, P.L., Jr.; et al. Efficacy of methylprednisolone in acute spinal cord injury. JAMA 1984, 251, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Sunshine, J.E.; Dagal, A.; Burns, S.P.; Bransford, R.J.; Zhang, F.; Newman, S.F.; Nair, B.G.; Sharar, S.R. Methylprednisolone Therapy in Acute Traumatic Spinal Cord Injury: Analysis of a Regional Spinal Cord Model Systems Database. Anesth. Analg. 2017, 124, 1200–1205. [Google Scholar] [CrossRef]
- Elkabes, S.; Nicot, A.B. Sex steroids and neuroprotection in spinal cord injury: A review of preclinical investigations. Exp. Neurol. 2014, 259, 28–37. [Google Scholar] [CrossRef]
- Sribnick, E.A.; Wingrave, J.M.; Matzelle, D.D.; Wilford, G.G.; Ray, S.K.; Banik, N.L. Estrogen attenuated markers of inflammation and decreased lesion volume in acute spinal cord injury in rats. J. Neurosci. Res. 2005, 82, 283–293. [Google Scholar] [CrossRef]
- Chakrabarti, M.; Das, A.; Samantaray, S.; Smith, J.A.; Banik, N.L.; Haque, A.; Ray, S.K. Molecular mechanisms of estrogen for neuroprotection in spinal cord injury and traumatic brain injury. Rev. Neurosci. 2016, 27, 271–281. [Google Scholar] [CrossRef]
- Samantaray, S.; Sribnick, E.A.; Das, A.; Thakore, N.P.; Matzelle, D.; Yu, S.P.; Ray, S.K.; Wei, L.; Banik, N.L. Neuroprotective efficacy of estrogen in experimental spinal cord injury in rats. Ann. N. Y. Acad. Sci. 2010, 1199, 90–94. [Google Scholar] [CrossRef] [Green Version]
- Yune, T.Y.; Kim, S.J.; Lee, S.M.; Lee, Y.K.; Oh, Y.J.; Kim, Y.C.; Markelonis, G.J.; Oh, T.H. Systemic administration of 17beta-estradiol reduces apoptotic cell death and improves functional recovery following traumatic spinal cord injury in rats. J. Neurotrauma 2004, 21, 293–306. [Google Scholar] [CrossRef]
- Sribnick, E.A.; Matzelle, D.D.; Ray, S.K.; Banik, N.L. Estrogen treatment of spinal cord injury attenuates calpain activation and apoptosis. J. Neurosci. Res. 2006, 84, 1064–1075. [Google Scholar] [CrossRef] [PubMed]
- Sribnick, E.A.; Samantaray, S.; Das, A.; Smith, J.; Matzelle, D.D.; Ray, S.K.; Banik, N.L. Postinjury estrogen treatment of chronic spinal cord injury improves locomotor function in rats. J. Neurosci. Res. 2010, 88, 1738–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siriphorn, A.; Dunham, K.A.; Chompoopong, S.; Floyd, C.L. Postinjury administration of 17beta-estradiol induces protection in the gray and white matter with associated functional recovery after cervical spinal cord injury in male rats. J. Comp. Neurol. 2012, 520, 2630–2646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samantaray, S.; Das, A.; Matzelle, D.C.; Yu, S.P.; Wei, L.; Varma, A.; Ray, S.K.; Banik, N.L. Administration of low dose estrogen attenuates persistent inflammation, promotes angiogenesis, and improves locomotor function following chronic spinal cord injury in rats. J. Neurochem. 2016, 137, 604–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samantaray, S.; Das, A.; Matzelle, D.C.; Yu, S.P.; Wei, L.; Varma, A.; Ray, S.K.; Banik, N.L. Administration of low dose estrogen attenuates gliosis and protects neurons in acute spinal cord injury in rats. J. Neurochem. 2016, 136, 1064–1073. [Google Scholar] [CrossRef] [Green Version]
- Samantaray, S.; Smith, J.A.; Das, A.; Matzelle, D.D.; Varma, A.K.; Ray, S.K.; Banik, N.L. Low dose estrogen prevents neuronal degeneration and microglial reactivity in an acute model of spinal cord injury: Effect of dosing, route of administration, and therapy delay. Neurochem. Res. 2011, 36, 1809–1816. [Google Scholar] [CrossRef] [Green Version]
- Jing, Y.; Bai, F.; Chen, H.; Dong, H. Melatonin prevents blood vessel loss and neurological impairment induced by spinal cord injury in rats. J. Spinal Cord Med. 2017, 40, 222–229. [Google Scholar] [CrossRef] [Green Version]
- Samantaray, S.; Sribnick, E.A.; Das, A.; Knaryan, V.H.; Matzelle, D.D.; Yallapragada, A.V.; Reiter, R.J.; Ray, S.K.; Banik, N.L. Melatonin attenuates calpain upregulation, axonal damage and neuronal death in spinal cord injury in rats. J. Pineal Res. 2008, 44, 348–357. [Google Scholar] [CrossRef]
- Shen, Z.; Zhou, Z.; Gao, S.; Guo, Y.; Gao, K.; Wang, H.; Dang, X. Melatonin Inhibits Neural Cell Apoptosis and Promotes Locomotor Recovery via Activation of the Wnt/beta-Catenin Signaling Pathway After Spinal Cord Injury. Neurochem. Res. 2017, 42, 2336–2343. [Google Scholar] [CrossRef]
- Sribnick, E.A.; Del Re, A.M.; Ray, S.K.; Woodward, J.J.; Banik, N.L. Estrogen attenuates glutamate-induced cell death by inhibiting Ca2+ influx through L-type voltage-gated Ca2+ channels. Brain Res. 2009, 1276, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Sribnick, E.A.; Ray, S.K.; Banik, N.L. Estrogen prevents glutamate-induced apoptosis in C6 glioma cells by a receptor-mediated mechanism. Neuroscience 2006, 137, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; McDowell, M.; Pava, M.J.; Smith, J.A.; Reiter, R.J.; Woodward, J.J.; Varma, A.K.; Ray, S.K.; Banik, N.L. The inhibition of apoptosis by melatonin in VSC4.1 motoneurons exposed to oxidative stress, glutamate excitotoxicity, or TNF-alpha toxicity involves membrane melatonin receptors. J. Pineal Res. 2010, 48, 157–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sur, P.; Sribnick, E.A.; Wingrave, J.M.; Nowak, M.W.; Ray, S.K.; Banik, N.L. Estrogen attenuates oxidative stress-induced apoptosis in C6 glial cells. Brain Res. 2003, 971, 178–188. [Google Scholar] [CrossRef]
- Bruce-Keller, A.J.; Keeling, J.L.; Keller, J.N.; Huang, F.F.; Camondola, S.; Mattson, M.P. Antiinflammatory effects of estrogen on microglial activation. Endocrinology 2000, 141, 3646–3656. [Google Scholar] [CrossRef] [PubMed]
- Beral, V.; Million Women Study, C. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet 2003, 362, 419–427. [Google Scholar] [CrossRef]
- Colditz, G.A.; Hankinson, S.E.; Hunter, D.J.; Willett, W.C.; Manson, J.E.; Stampfer, M.J.; Hennekens, C.; Rosner, B.; Speizer, F.E. The use of estrogens and progestins and the risk of breast cancer in postmenopausal women. N. Engl. J. Med. 1995, 332, 1589–1593. [Google Scholar] [CrossRef]
- Li, C.I.; Malone, K.E.; Porter, P.L.; Weiss, N.S.; Tang, M.T.; Cushing-Haugen, K.L.; Daling, J.R. Relationship between long durations and different regimens of hormone therapy and risk of breast cancer. JAMA 2003, 289, 3254–3263. [Google Scholar] [CrossRef] [Green Version]
- Olsson, H.L.; Ingvar, C.; Bladstrom, A. Hormone replacement therapy containing progestins and given continuously increases breast carcinoma risk in Sweden. Cancer 2003, 97, 1387–1392. [Google Scholar] [CrossRef]
- Wysowski, D.K.; Golden, L.; Burke, L. Use of menopausal estrogens and medroxyprogesterone in the United States, 1982–1992. Obstet. Gynecol. 1995, 85, 6–10. [Google Scholar] [CrossRef]
- Rossouw, J.E.; Anderson, G.L.; Prentice, R.L.; LaCroix, A.Z.; Kooperberg, C.; Stefanick, M.L.; Jackson, R.D.; Beresford, S.A.; Howard, B.V.; Johnson, K.C.; et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results From the Women’s Health Initiative randomized controlled trial. JAMA 2002, 288, 321–333. [Google Scholar]
- McCullough, L.D.; Alkayed, N.J.; Traystman, R.J.; Williams, M.J.; Hurn, P.D. Postischemic estrogen reduces hypoperfusion and secondary ischemia after experimental stroke. Stroke 2001, 32, 796–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.H.; Chang, C.Y.; Chang, H.K.; Chen, W.C.; Lin, M.T.; Wang, J.J.; Chen, J.C.; Chang, F.M. Premarin stimulates estrogen receptor-alpha to protect against traumatic brain injury in male rats. Crit. Care Med. 2009, 37, 3097–3106. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Yeh, C.H.; Lin, M.Y.; Kang, C.Y.; Chu, C.C.; Chang, F.M.; Wang, J.J. Premarin improves outcomes of spinal cord injury in male rats through stimulating both angiogenesis and neurogenesis. Crit. Care Med. 2010, 38, 2043–2051. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Smith, J.A.; Gibson, C.; Varma, A.K.; Ray, S.K.; Banik, N.L. Estrogen receptor agonists and estrogen attenuate TNF-alpha-induced apoptosis in VSC4.1 motoneurons. J. Endocrinol. 2011, 208, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Haque, A.; Narang, A.; Das, A.; Matzelle, D.C.; Capone, M.; Banik, N.L. Premarin Treatment Reduces Inflammation and Degeneration and Improves Function in SCI. In Proceedings of the 48th Annual Meeting of the American Society for Neurochemistry, Little Rock, AK, USA, 18–22 March 2017. [Google Scholar]
- Perot, P.L., Jr.; Lee, W.A.; Hsu, C.Y.; Hogan, E.L.; Cox, R.D.; Gross, A.J. Therapeutic model for experimental spinal cord injury in the rat: I. Mortality and motor deficit. Cent. Nerv. Syst. Trauma J. Am. Paralys. Assoc. 1987, 4, 149–159. [Google Scholar] [CrossRef]
- Ray, S.K.; Matzelle, D.C.; Wilford, G.G.; Hogan, E.L.; Banik, N.L. E-64-d prevents both calpain upregulation and apoptosis in the lesion and penumbra following spinal cord injury in rats. Brain Res. 2000, 867, 80–89. [Google Scholar] [CrossRef]
- Sribnick, E.A.; Matzelle, D.D.; Banik, N.L.; Ray, S.K. Direct evidence for calpain involvement in apoptotic death of neurons in spinal cord injury in rats and neuroprotection with calpain inhibitor. Neurochem. Res. 2007, 32, 2210–2216. [Google Scholar] [CrossRef]
- Cox, A.; Capone, M.; Matzelle, D.; Vertegel, A.; Bredikhin, M.; Varma, A.; Haque, A.; Shields, D.C.; Banik, N.L. Nanoparticle-Based Estrogen Delivery to Spinal Cord Injury Site Reduces Local Parenchymal Destruction and Improves Functional Recovery. J. Neurotrauma 2021, 38, 342–352. [Google Scholar] [CrossRef]
- Basso, D.M.; Beattie, M.S.; Bresnahan, J.C. A sensitive and reliable locomotor rating scale for open field testing in rats. J. Neurotrauma 1995, 12, 1–21. [Google Scholar] [CrossRef]
- Polcyn, R.; Capone, M.; Matzelle, D.; Hossain, A.; Chandran, R.; Banik, N.L.; Haque, A. Enolase inhibition alters metabolic hormones and inflammatory factors to promote neuroprotection in spinal cord injury. Neurochem. Int. 2020, 139, 104788. [Google Scholar] [CrossRef]
- Qian, J.; Herrera, J.J.; Narayana, P.A. Neuronal and axonal degeneration in experimental spinal cord injury: In vivo proton magnetic resonance spectroscopy and histology. J. Neurotrauma 2010, 27, 599–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiter, R.J.; Tan, D.X.; Manchester, L.C.; Qi, W. Biochemical reactivity of melatonin with reactive oxygen and nitrogen species: A review of the evidence. Cell. Biochem. Biophys. 2001, 34, 237–256. [Google Scholar] [CrossRef]
- Allegra, M.; Reiter, R.J.; Tan, D.X.; Gentile, C.; Tesoriere, L.; Livrea, M.A. The chemistry of melatonin’s interaction with reactive species. J. Pineal Res. 2003, 34, 1–10. [Google Scholar] [CrossRef]
- Lu, H.; Ma, K.; Jin, L.; Zhu, H.; Cao, R. 17beta-estradiol rescues damages following traumatic brain injury from molecule to behavior in mice. J. Cell. Physiol. 2018, 233, 1712–1722. [Google Scholar] [CrossRef] [PubMed]
- Day, N.L.; Floyd, C.L.; D’Alessandro, T.L.; Hubbard, W.J.; Chaudry, I.H. 17beta-estradiol confers protection after traumatic brain injury in the rat and involves activation of G protein-coupled estrogen receptor 1. J. Neurotrauma 2013, 30, 1531–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, Y.J.; Li, L.Z.; Li, X.G.; Wang, Y.J. 17Beta-estradiol differentially protects cortical pericontusional zone from programmed cell death after traumatic cerebral contusion at distinct stages via non-genomic and genomic pathways. Mol. Cell. Neurosci. 2011, 48, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Li, L.Z.; Bao, Y.J.; Zhao, M. 17beta-estradiol attenuates programmed cell death in cortical pericontusional zone following traumatic brain injury via upregulation of ERalpha and inhibition of caspase-3 activation. Neurochem. Int. 2011, 58, 126–133. [Google Scholar] [CrossRef]
- Lapanantasin, S.; Chongthammakun, S.; Floyd, C.L.; Berman, R.F. Effects of 17beta-estradiol on intracellular calcium changes and neuronal survival after mechanical strain injury in neuronal-glial cultures. Synapse 2006, 60, 406–410. [Google Scholar] [CrossRef]
- Merchenthaler, I.; Dellovade, T.L.; Shughrue, P.J. Neuroprotection by estrogen in animal models of global and focal ischemia. Ann. N. Y. Acad. Sci. 2003, 1007, 89–100. [Google Scholar] [CrossRef]
- Benedek, G.; Zhang, J.; Bodhankar, S.; Nguyen, H.; Kent, G.; Jordan, K.; Manning, D.; Vandenbark, A.A.; Offner, H. Estrogen induces multiple regulatory B cell subtypes and promotes M2 microglia and neuroprotection during experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2016, 293, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Zhang, P.; Li, X.; Lei, S.; Li, W.; He, X.; Zhang, J.; Wang, N.; Qi, C.; Chen, X.; et al. Post-stroke estradiol treatment enhances neurogenesis in the subventricular zone of rats after permanent focal cerebral ischemia. Neuroscience 2013, 231, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Ardelt, A.A.; Carpenter, R.S.; Lobo, M.R.; Zeng, H.; Solanki, R.B.; Zhang, A.; Kulesza, P.; Pike, M.M. Estradiol modulates post-ischemic cerebral vascular remodeling and improves long-term functional outcome in a rat model of stroke. Brain Res. 2012, 1461, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, K.; Yang, Y.; Takayasu, Y.; Gertner, M.; Hwang, J.Y.; Aromolaran, K.; Bennett, M.V.; Zukin, R.S. Estradiol pretreatment ameliorates impaired synaptic plasticity at synapses of insulted CA1 neurons after transient global ischemia. Brain Res. 2015, 1621, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakkar, R.; Wang, R.; Wang, J.; Vadlamudi, R.K.; Brann, D.W. 17beta-Estradiol Regulates Microglia Activation and Polarization in the Hippocampus Following Global Cerebral Ischemia. Oxidative Med. Cell. Longev. 2018, 2018, 4248526. [Google Scholar] [CrossRef] [Green Version]
- Losordo, D.W.; Isner, J.M. Estrogen and angiogenesis: A review. Arter. Thromb. Vasc. Biol. 2001, 21, 6–12. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.G.; Zhang, L.; Jiang, Q.; Zhang, R.; Davies, K.; Powers, C.; Bruggen, N.; Chopp, M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J. Clin. Investig. 2000, 106, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Augustin, H.G. Angiogenesis in the female reproductive system. Mech. Angiogenesis 2005, 35–52. [Google Scholar]
- Augustin, H.G. Antiangiogenic tumour therapy: Will it work? Trends Pharm. Sci. 1998, 19, 216–222. [Google Scholar] [CrossRef]
- Skold, M.; Cullheim, S.; Hammarberg, H.; Piehl, F.; Suneson, A.; Lake, S.; Sjogren, A.; Walum, E.; Risling, M. Induction of VEGF and VEGF receptors in the spinal cord after mechanical spinal injury and prostaglandin administration. Eur. J. Neurosci. 2000, 12, 3675–3686. [Google Scholar] [CrossRef]
- Mocchetti, I.; Rabin, S.J.; Colangelo, A.M.; Whittemore, S.R.; Wrathall, J.R. Increased basic fibroblast growth factor expression following contusive spinal cord injury. Exp. Neurol. 1996, 141, 154–164. [Google Scholar] [CrossRef]
- Follesa, P.; Wrathall, J.R.; Mocchetti, I. Increased basic fibroblast growth factor mRNA following contusive spinal cord injury. Brain Res. Mol. Brain Res. 1994, 22, 1–8. [Google Scholar] [CrossRef]
- Bartholdi, D.; Rubin, B.P.; Schwab, M.E. VEGF mRNA induction correlates with changes in the vascular architecture upon spinal cord damage in the rat. Eur. J. Neurosci. 1997, 9, 2549–2560. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Keogh, C.L.; Whitaker, V.R.; Theus, M.H.; Yu, S.P. Angiogenesis and stem cell transplantation as potential treatments of cerebral ischemic stroke. Pathophysiology 2005, 12, 47–62. [Google Scholar] [CrossRef]
- Arai, K.; Jin, G.; Navaratna, D.; Lo, E.H. Brain angiogenesis in developmental and pathological processes: Neurovascular injury and angiogenic recovery after stroke. FEBS J. 2009, 276, 4644–4652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santagati, S.; Melcangi, R.C.; Celotti, F.; Martini, L.; Maggi, A. Estrogen receptor is expressed in different types of glial cells in culture. J. Neurochem. 1994, 63, 2058–2064. [Google Scholar] [CrossRef]
- Mor, G.; Nilsen, J.; Horvath, T.; Bechmann, I.; Brown, S.; Garcia-Segura, L.M.; Naftolin, F. Estrogen and microglia: A regulatory system that affects the brain. J. Neurobiol. 1999, 40, 484–496. [Google Scholar] [CrossRef]
- McEwen, B.S. Clinical review 108: The molecular and neuroanatomical basis for estrogen effects in the central nervous system. J. Clin. Endocrinol. Metab. 1999, 84, 1790–1797. [Google Scholar] [CrossRef]
- Raghava, N.; Das, B.C.; Ray, S.K. Neuroprotective effects of estrogen in CNS injuries: Insights from animal models. Neurosci. Neuroecon 2017, 6, 15–29. [Google Scholar] [CrossRef] [Green Version]
- McEwen, B.S.; Alves, S.E. Estrogen actions in the central nervous system. Endocr. Rev. 1999, 20, 279–307. [Google Scholar] [CrossRef]
- Weaver, C.E., Jr.; Park-Chung, M.; Gibbs, T.T.; Farb, D.H. 17beta-Estradiol protects against NMDA-induced excitotoxicity by direct inhibition of NMDA receptors. Brain Res. 1997, 761, 338–341. [Google Scholar] [CrossRef]
- Honjo, H.; Tamura, T.; Matsumoto, Y.; Kawata, M.; Ogino, Y.; Tanaka, K.; Yamamoto, T.; Ueda, S.; Okada, H. Estrogen as a growth factor to central nervous cells. Estrogen treatment promotes development of acetylcholinesterase-positive basal forebrain neurons transplanted in the anterior eye chamber. J. Steroid. Biochem. Mol. Biol. 1992, 41, 633–635. [Google Scholar] [CrossRef]
- Pinkerton, J.V. Tissue-selective Estrogen Complex for Menopausal Hormone Therapy. Clin. Obstet. Gynecol. 2018, 61, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Hiroi, R.; Weyrich, G.; Koebele, S.V.; Mennenga, S.E.; Talboom, J.S.; Hewitt, L.T.; Lavery, C.N.; Mendoza, P.; Jordan, A.; Bimonte-Nelson, H.A. Benefits of Hormone Therapy Estrogens Depend on Estrogen Type: 17beta-Estradiol and Conjugated Equine Estrogens Have Differential Effects on Cognitive, Anxiety-Like, and Depressive-Like Behaviors and Increase Tryptophan Hydroxylase-2 mRNA Levels in Dorsal Raphe Nucleus Subregions. Front. Neurosci. 2016, 10, 517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagan, R.; Abreu, P.; Andrews, E. Vaginal bleeding/spotting with conjugated estrogens/bazedoxifene, conjugated estrogens/medroxyprogesterone acetate, and placebo. Postgrad. Med. 2018, 130, 687–693. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haque, A.; Das, A.; Samantaray, S.; Matzelle, D.; Capone, M.; Wallace, G.; Husarik, A.N.; Taheri, S.; Reiter, R.J.; Varma, A.; et al. Premarin Reduces Neurodegeneration and Promotes Improvement of Function in an Animal Model of Spinal Cord Injury. Int. J. Mol. Sci. 2022, 23, 2384. https://doi.org/10.3390/ijms23042384
Haque A, Das A, Samantaray S, Matzelle D, Capone M, Wallace G, Husarik AN, Taheri S, Reiter RJ, Varma A, et al. Premarin Reduces Neurodegeneration and Promotes Improvement of Function in an Animal Model of Spinal Cord Injury. International Journal of Molecular Sciences. 2022; 23(4):2384. https://doi.org/10.3390/ijms23042384
Chicago/Turabian StyleHaque, Azizul, Arabinda Das, Supriti Samantaray, Denise Matzelle, Mollie Capone, Gerald Wallace, Aarti N. Husarik, Saied Taheri, Russel J. Reiter, Abhay Varma, and et al. 2022. "Premarin Reduces Neurodegeneration and Promotes Improvement of Function in an Animal Model of Spinal Cord Injury" International Journal of Molecular Sciences 23, no. 4: 2384. https://doi.org/10.3390/ijms23042384
APA StyleHaque, A., Das, A., Samantaray, S., Matzelle, D., Capone, M., Wallace, G., Husarik, A. N., Taheri, S., Reiter, R. J., Varma, A., Ray, S. K., & Banik, N. L. (2022). Premarin Reduces Neurodegeneration and Promotes Improvement of Function in an Animal Model of Spinal Cord Injury. International Journal of Molecular Sciences, 23(4), 2384. https://doi.org/10.3390/ijms23042384