
HF Formation through Dissociative Electron Attachment—A Combined Experimental and Theoretical Study on Pentafluorothiophenol and 2-Fluorothiophenol

Abstract
:1. Introduction
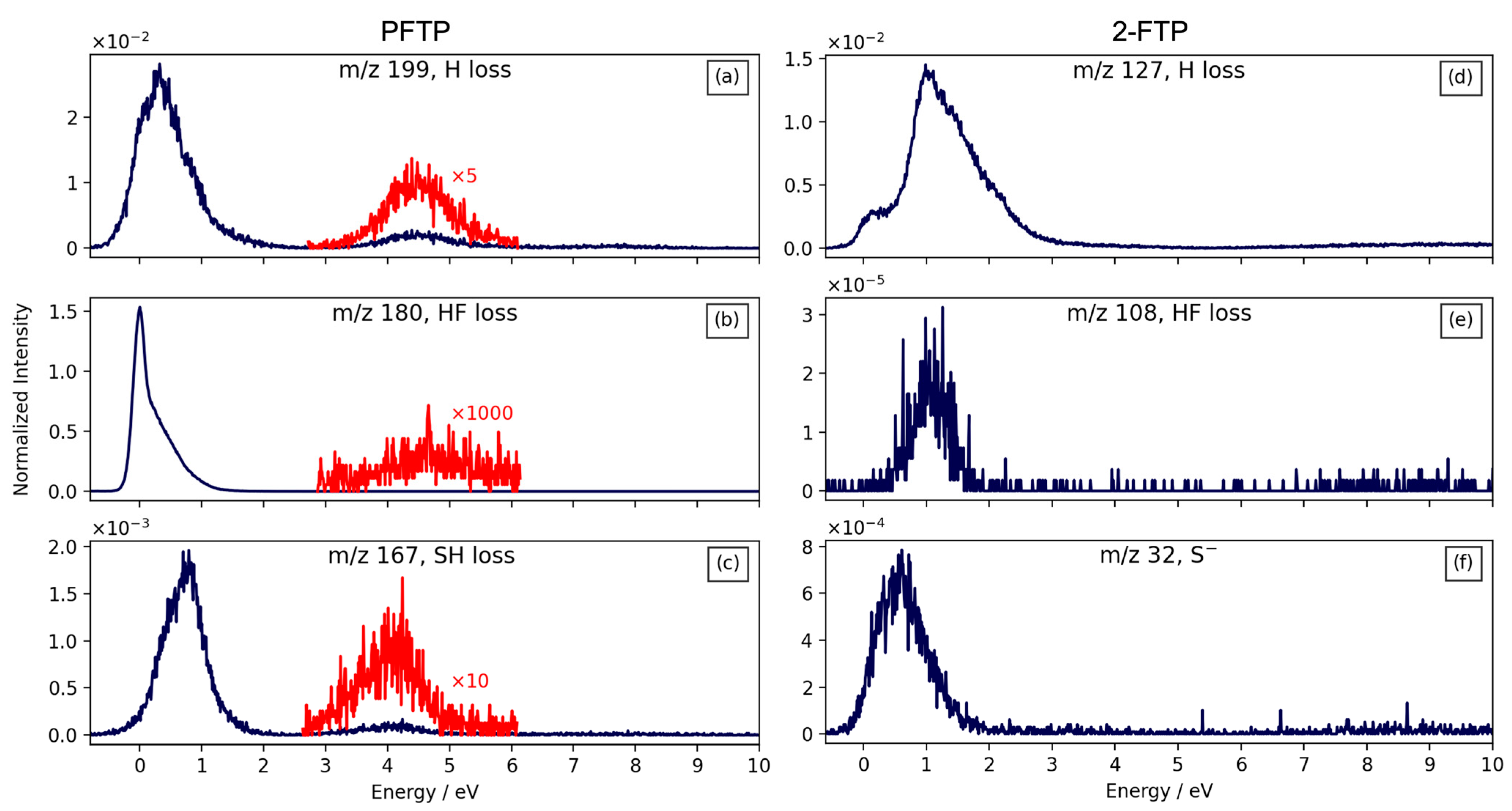
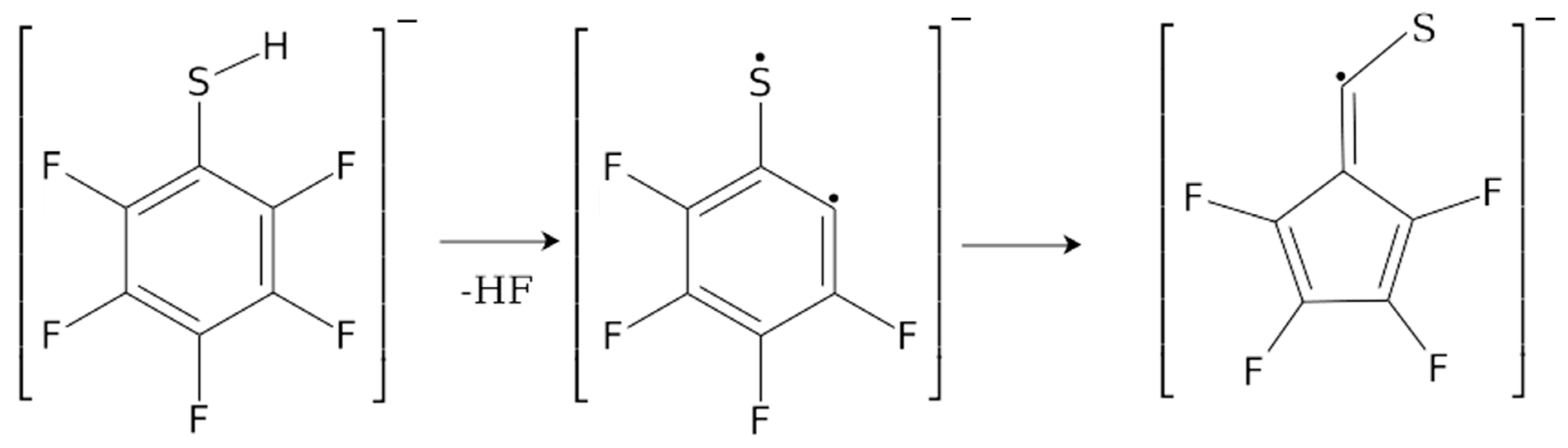
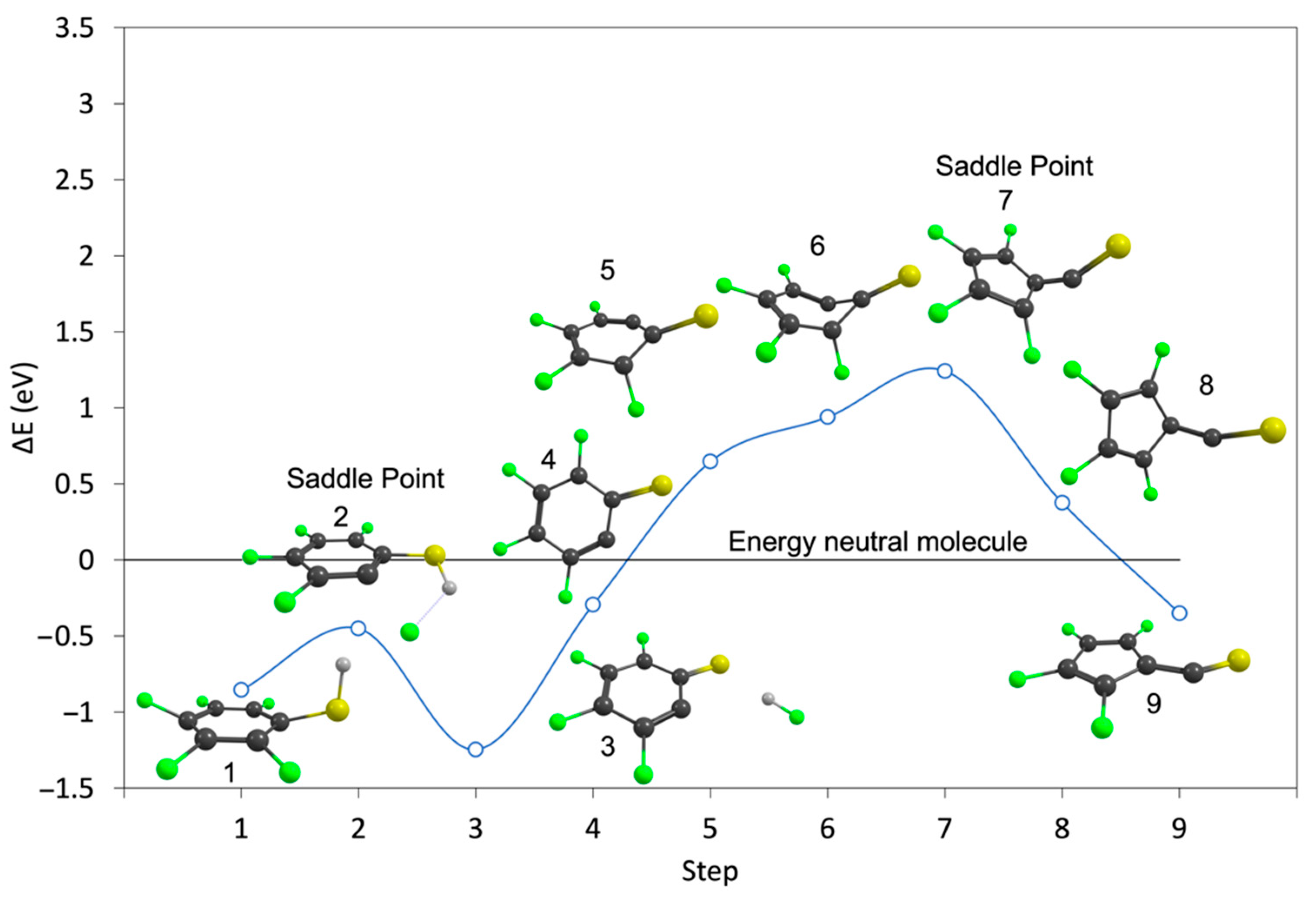
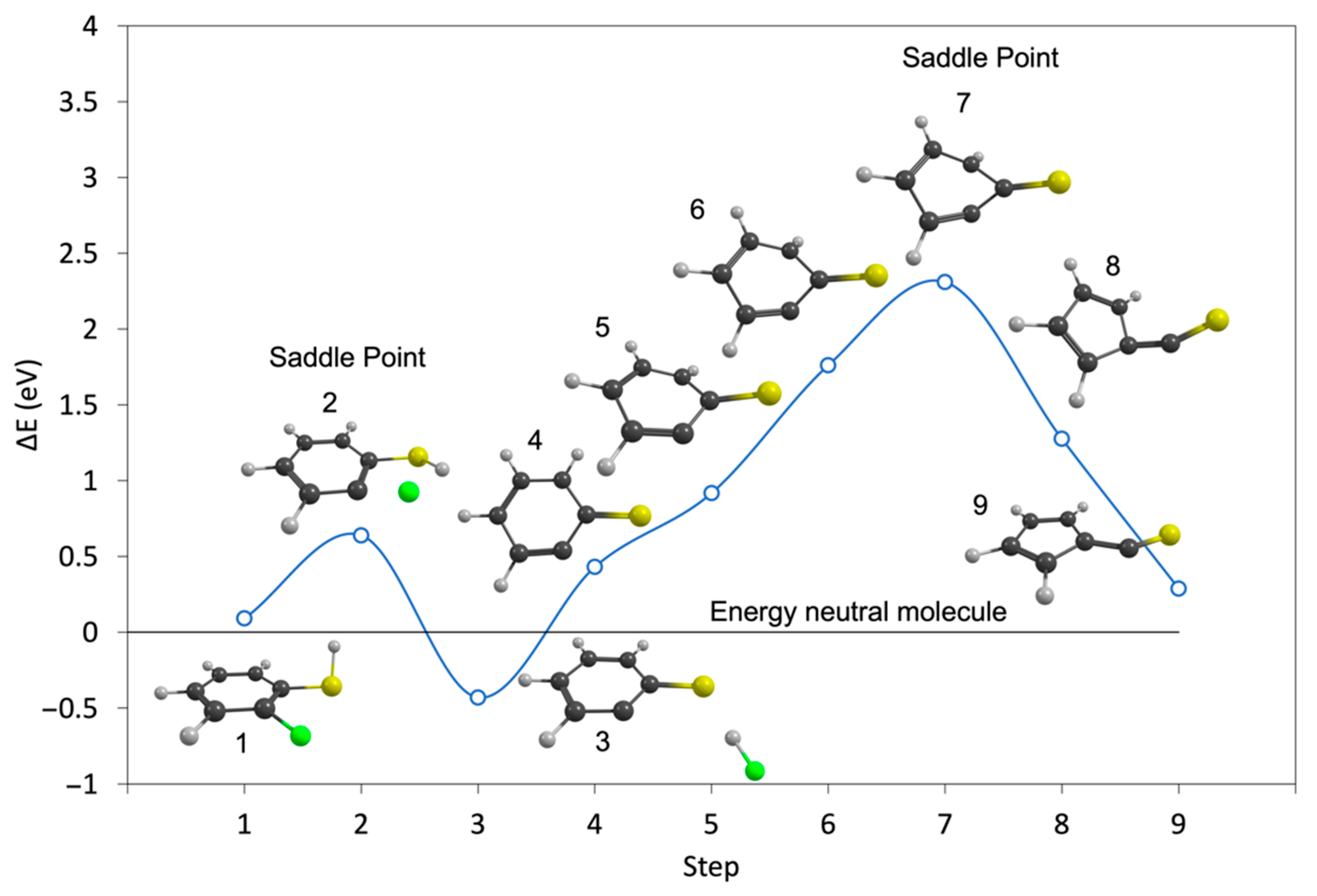
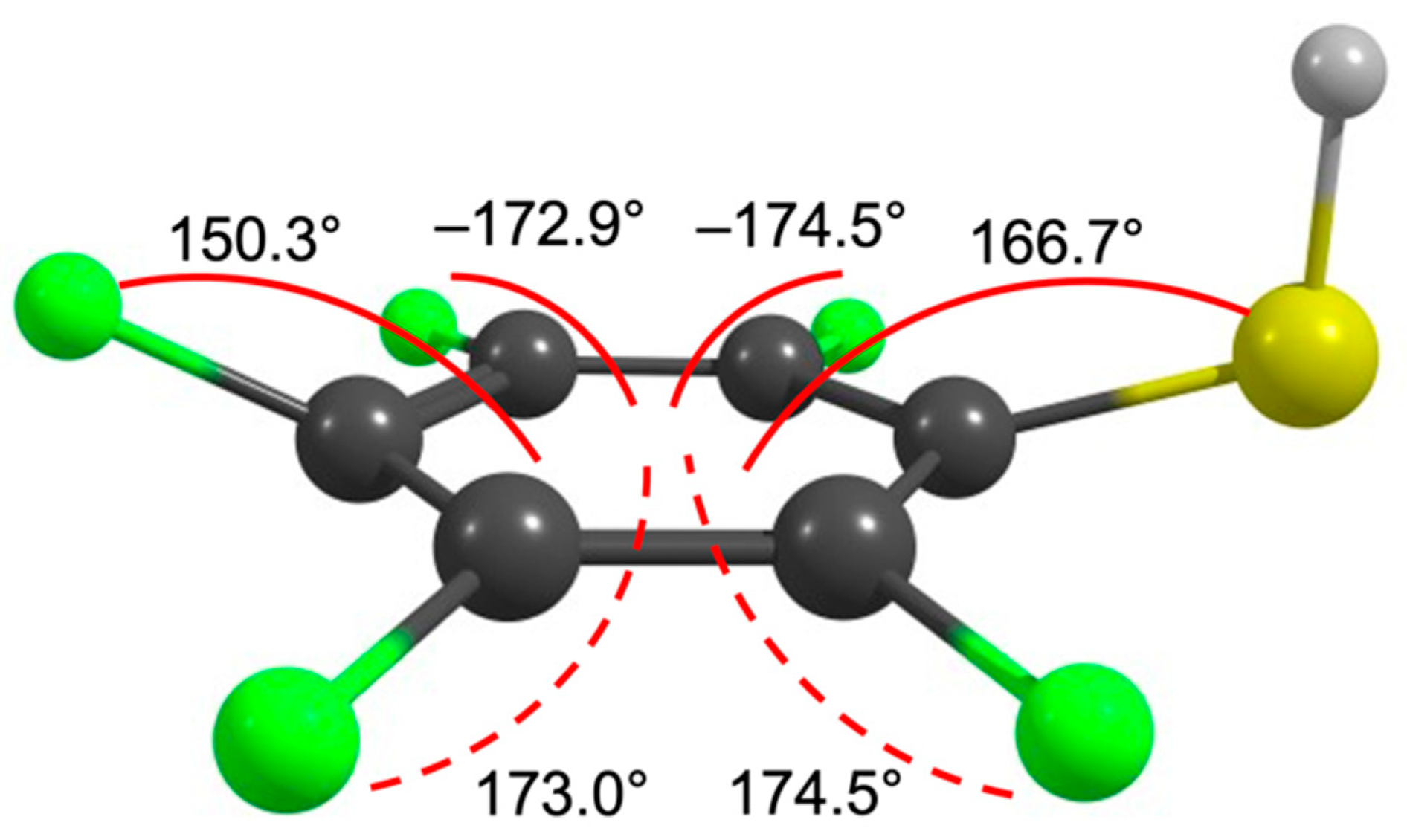
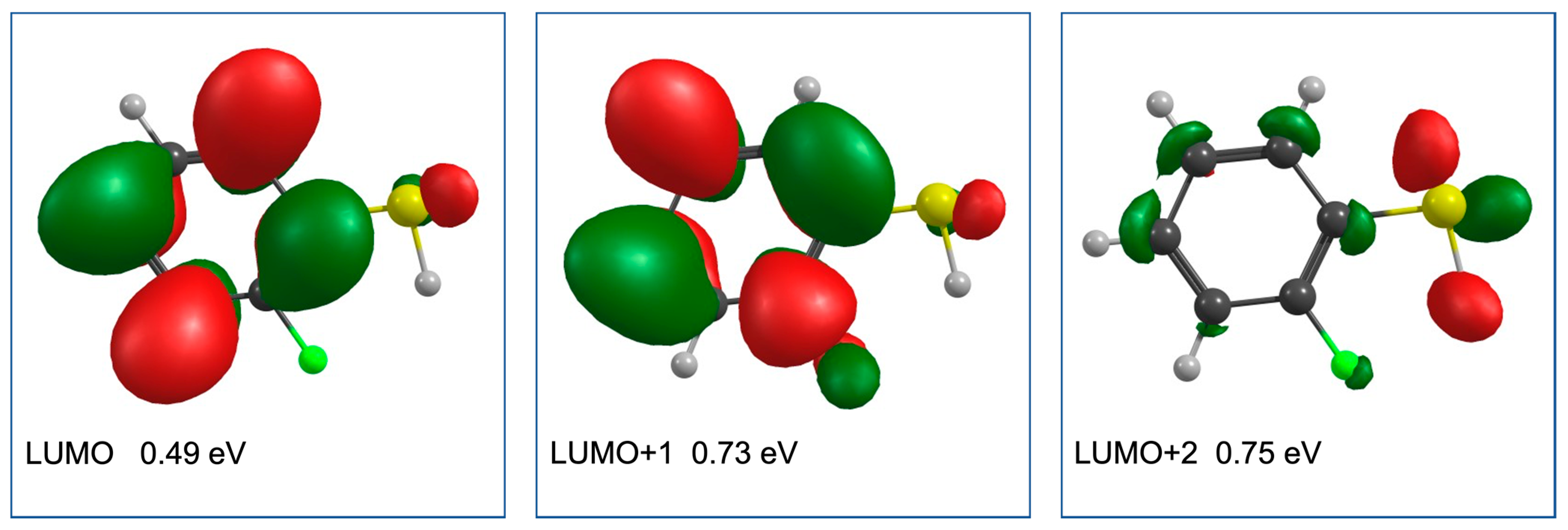
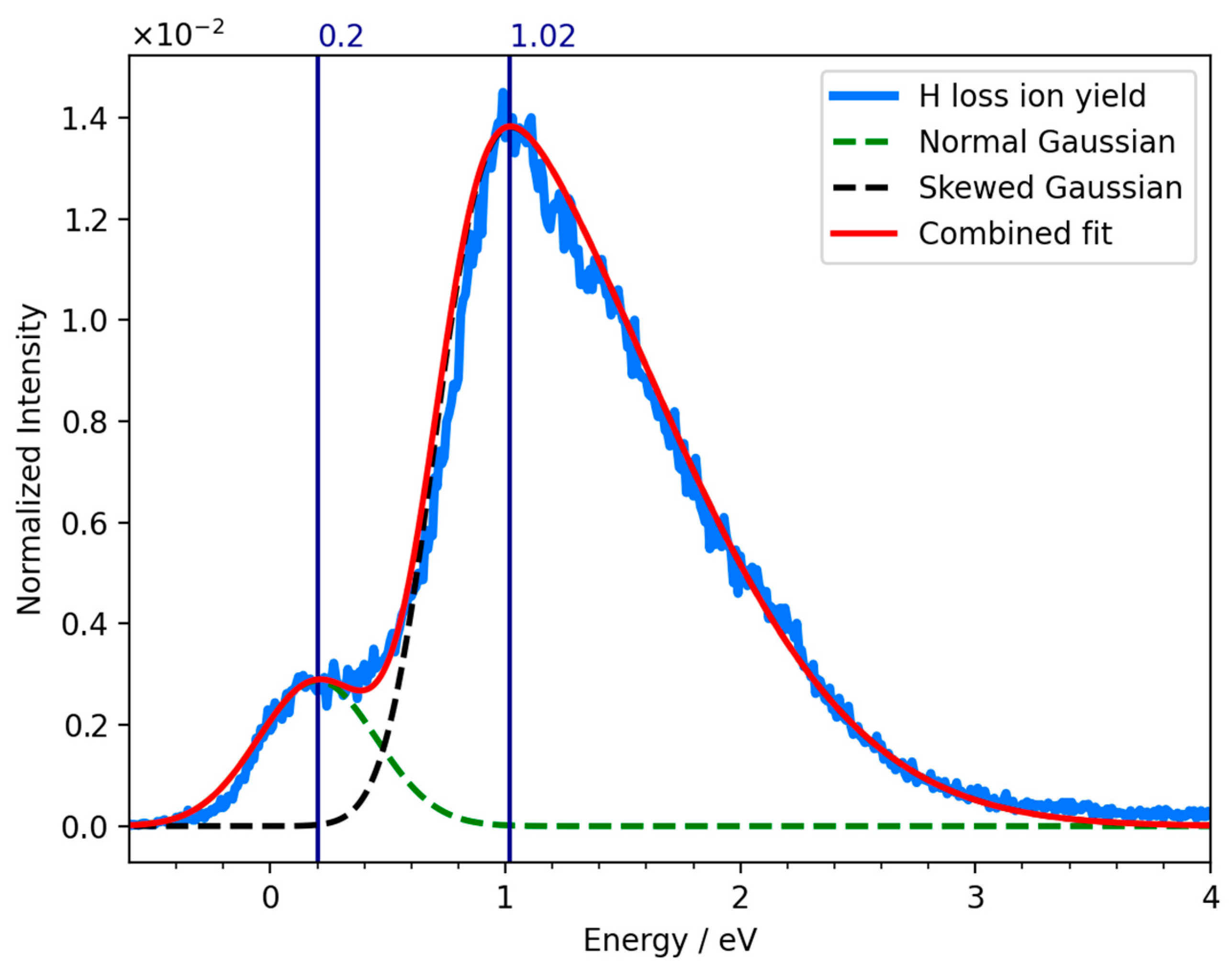
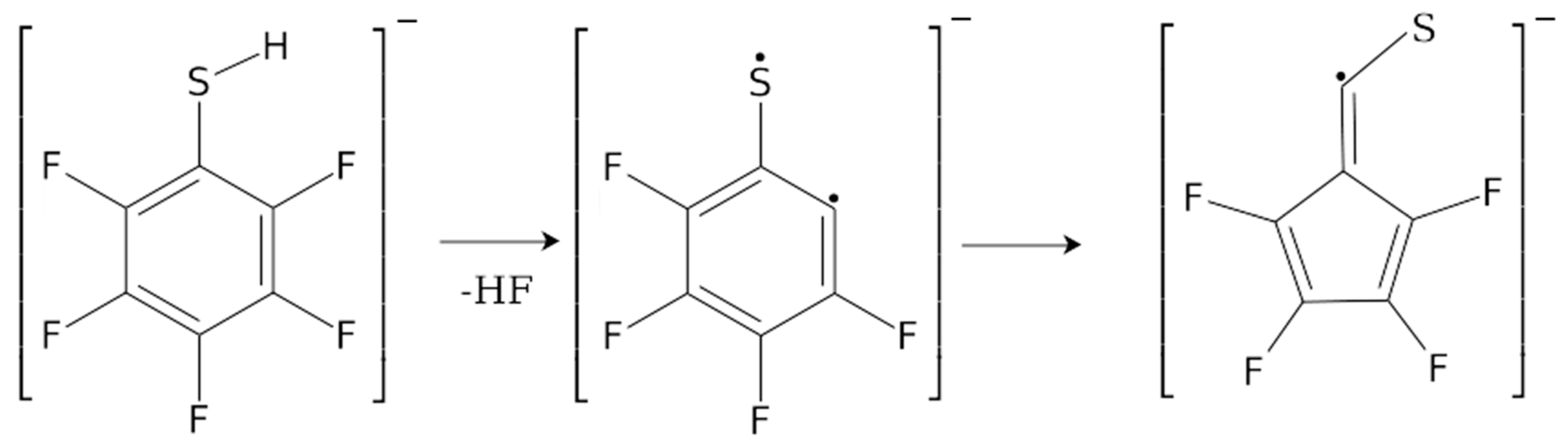
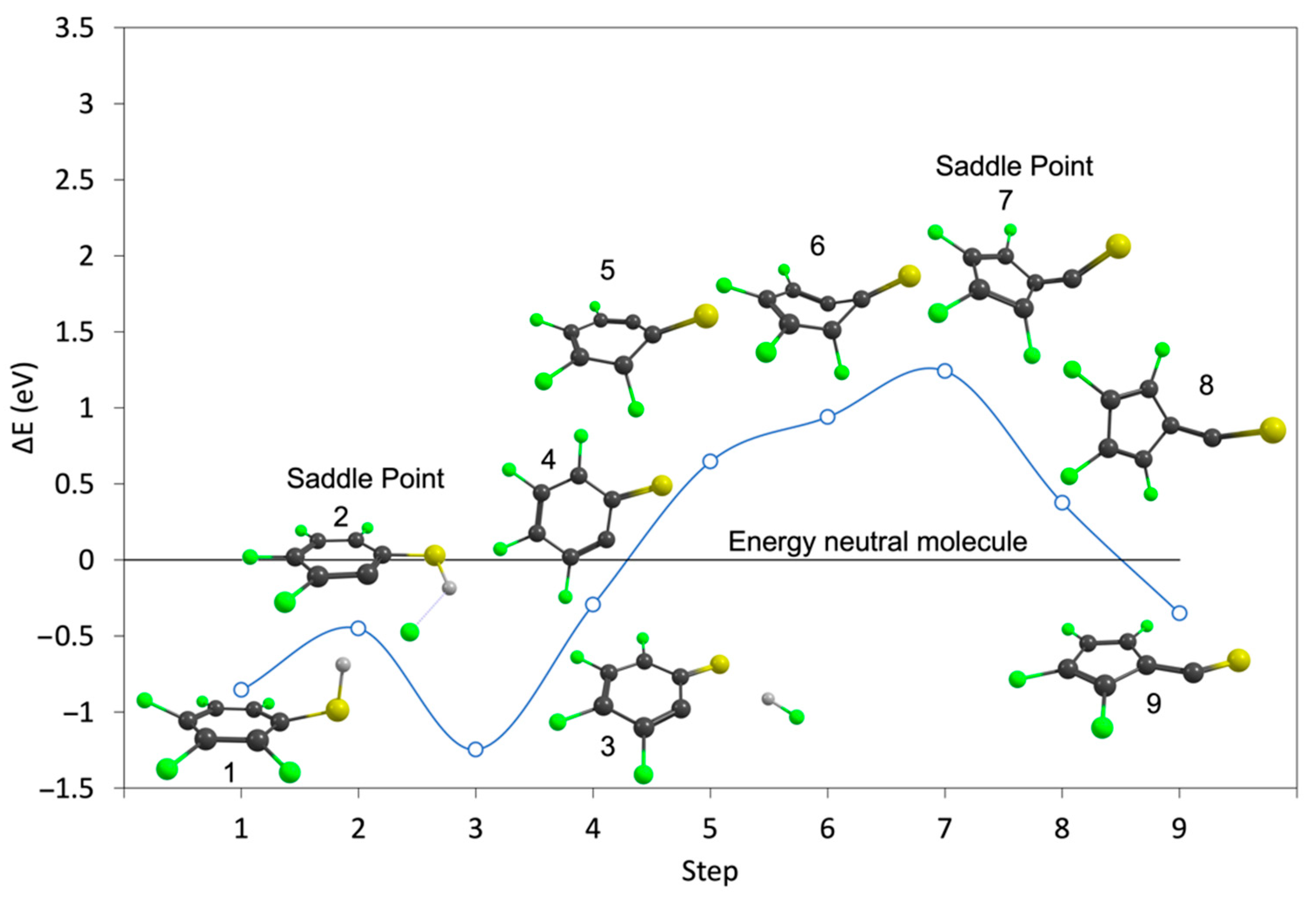
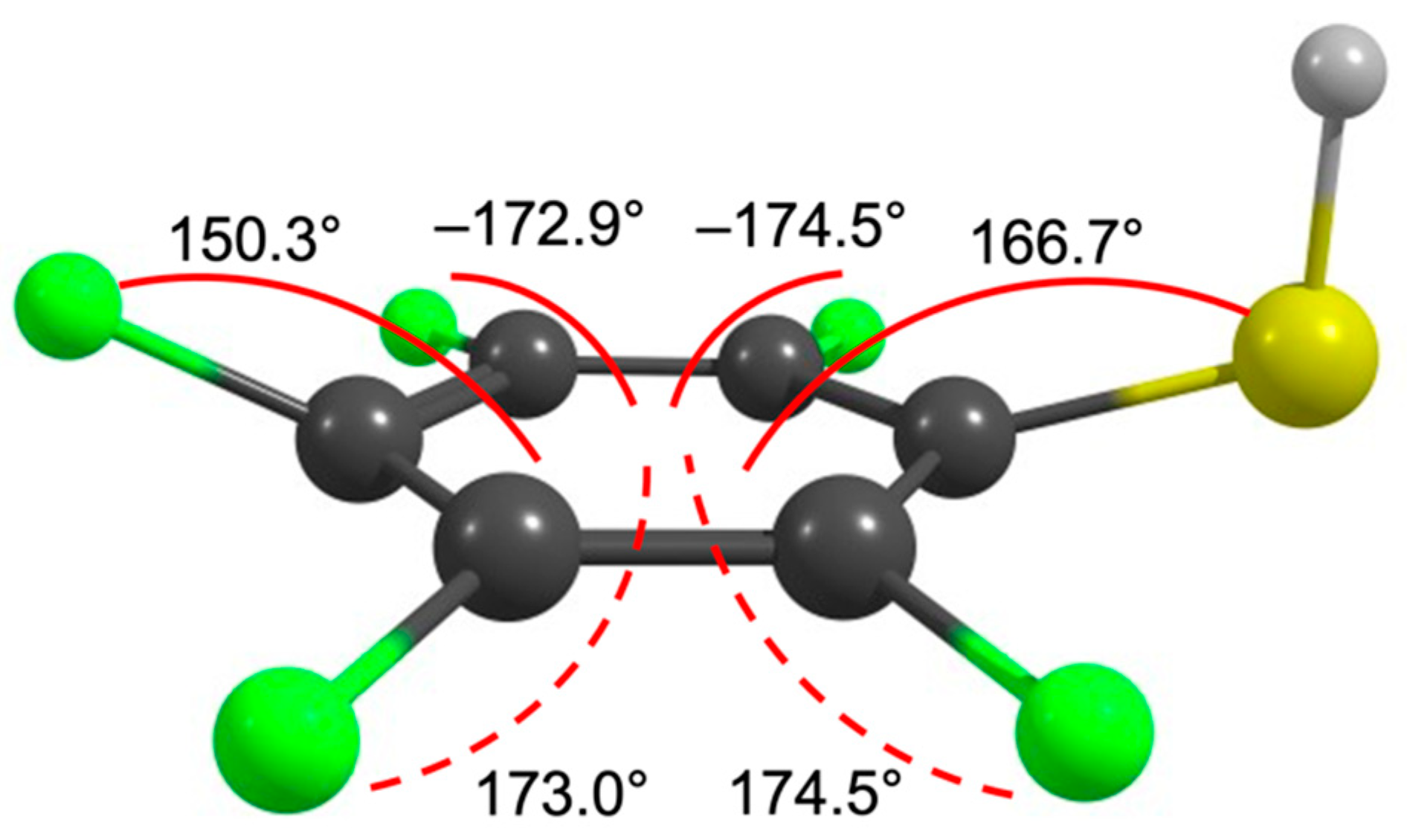
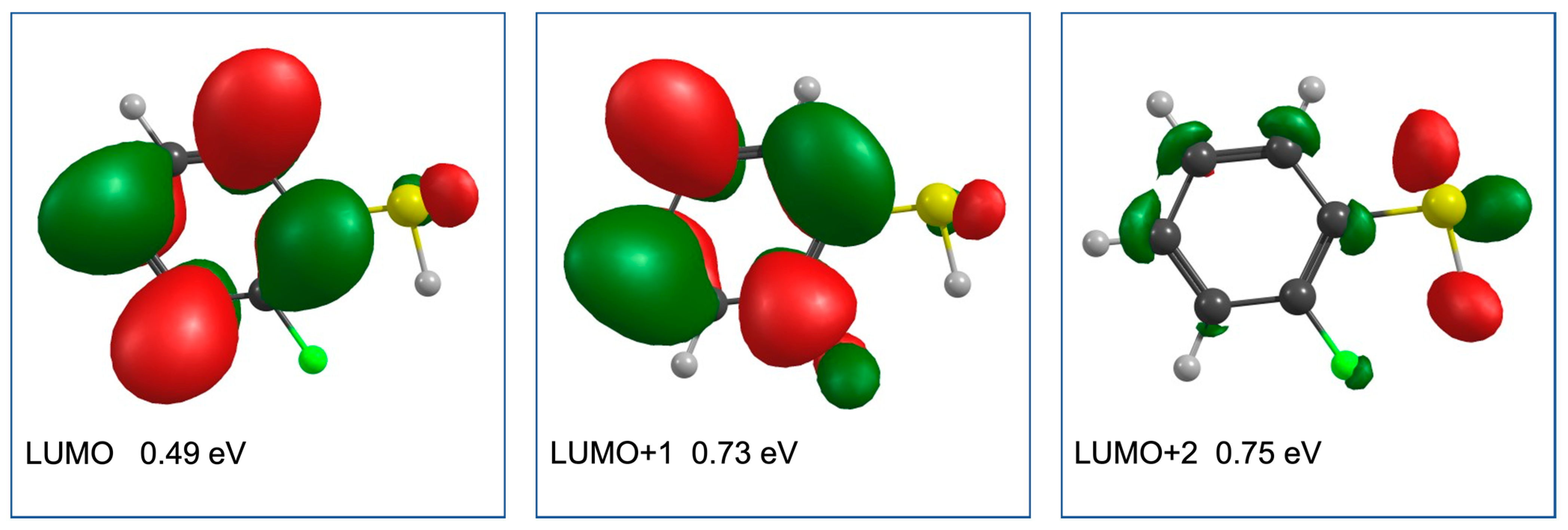
2. Results and Discussion
3. Materials and Methods
3.1. Experimental Setup
3.2. Theoretical Procedures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boudaıffa, B.; Cloutier, P.; Hunting, D.; Huels, M.A.; Sanche, L. Resonant Formation of DNA Strand Breaks by Low-Energy (3 to 20 eV) Electrons. Science 2000, 287, 1658–1660. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Becker, D.; Adhikary, A.; Sevilla, M.D. Reaction of Electrons with DNA: Radiation Damage to Radiosensitization. Int. J. Mol. Sci. 2019, 20, 3998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ameixa, J.; Arthur-Baidoo, E.; Meißner, R.; Makurat, S.; Kozak, W.; Butowska, K.; Ferreira da Silva, F.; Rak, J.; Denifl, S. Low-energy electron-induced decomposition of 5-trifluoromethanesulfonyl-uracil: A potential radiosensitizer. J. Chem. Phys. 2018, 149, 164307. [Google Scholar] [CrossRef] [Green Version]
- Meißner, R.; Feketeová, L.; Bayer, A.; Limão-Vieira, P.; Denifl, S. Formation of negative and positive ions in the radiosensitizer nimorazole upon low-energy electron collisions. J. Chem. Phys. 2021, 154, 074306. [Google Scholar] [CrossRef] [PubMed]
- Tanzer, K.; Feketeová, L.; Puschnigg, B.; Scheier, P.; Illenberger, E.; Denifl, S. Reactions in Nitroimidazole Triggered by Low-Energy (0–2 eV) Electrons: Methylation at N1-H Completely Blocks Reactivity. Angew. Chem. Int. Ed. 2014, 53, 12240–12243. [Google Scholar] [CrossRef] [Green Version]
- Ribar, A.; Fink, K.; Probst, M.; Huber, S.E.; Feketeová, L.; Denifl, S. Isomer Selectivity in Low-Energy Electron Attachment to Nitroimidazoles. Chem. A Eur. J. 2017, 23, 12892–12899. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef]
- Hockel, M.; Vaupel, P. Tumor Hypoxia: Definitions and Current Clinical, Biologic, and Molecular Aspects. JNCI J. Natl. Cancer Inst. 2001, 93, 266–276. [Google Scholar] [CrossRef] [Green Version]
- Wojtkowiak, J.W.; Verduzco, D.; Schramm, K.J.; Gillies, R.J. Drug Resistance and Cellular Adaptation to Tumor Acidic pH Microenvironment. Mol. Pharm. 2011, 8, 2032–2038. [Google Scholar] [CrossRef]
- Graham, K.; Unger, E. Overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment. Int. J. Nanomed. 2018, 13, 6049–6058. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, M.; Hong, C.R.; Wong, W.W.; Liew, L.P.; Shome, A.; Wang, J.; Gu, Y.; Stevenson, R.J.; Qi, W.; Anderson, R.F.; et al. Next-Generation Hypoxic Cell Radiosensitizers: Nitroimidazole Alkylsulfonamides. J. Med. Chem. 2018, 61, 1241–1254. [Google Scholar] [CrossRef] [PubMed]
- Wardman, P. Chemical Radiosensitizers for Use in Radiotherapy. Clin. Oncol. 2007, 19, 397–417. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Polska, K.; Rak, J.; Wagner, J.R.; Sanche, L. Fundamental Mechanisms of DNA Radiosensitization: Damage Induced by Low-Energy Electrons in Brominated Oligonucleotide Trimers. J. Phys. Chem. B 2012, 116, 9676–9682. [Google Scholar] [CrossRef] [PubMed]
- Pimblott, S.M.; LaVerne, J.A. Production of low-energy electrons by ionizing radiation. Radiat. Phys. Chem. 2007, 76, 1244–1247. [Google Scholar] [CrossRef]
- Farhataziz; Rodgers, M.A.J. Radiation Chemistry-Principles and Applications; VCH: New York, NY, USA, 1987. [Google Scholar]
- Ingólfsson, O. Low Energy Electron–Induced Dissociation. In Low-Energy Electrons; Jenny Stanford Publishing: Delhi, India, 2019; pp. 47–120. [Google Scholar]
- Christophorou, L.G.; Olthoff, J.K. Electron interactions with plasma processing gases: Present status and future needs. Appl. Surf. Sci. 2002, 192, 309–326. [Google Scholar] [CrossRef]
- Kohanoff, J.; McAllister, M.; Tribello, G.A.; Gu, B. Interactions between low energy electrons and DNA: A perspective from first-principles simulations. J. Phys. Condens. Matter 2017, 29, 383001. [Google Scholar] [CrossRef]
- Alizadeh, E.; Orlando, T.M.; Sanche, L. Biomolecular Damage Induced by Ionizing Radiation: The Direct and Indirect Effects of Low-Energy Electrons on DNA. Annu. Rev. Phys. Chem. 2015, 66, 379–398. [Google Scholar] [CrossRef]
- Baccarelli, I.; Bald, I.; Gianturco, F.A.; Illenberger, E.; Kopyra, J. Electron-induced damage of DNA and its components: Experiments and theoretical models. Phys. Rep. 2011, 508, 1–44. [Google Scholar] [CrossRef]
- Bald, I.; Denifl, S. The Role of Low-Energy Electrons in DNA Radiation Damage. In Low-Energy Electrons; Jenny Stanford Publishing: Delhi, India, 2019; pp. 285–340. ISBN 9780429058820. [Google Scholar]
- Flosadóttir, H.D.; Ómarsson, B.; Bald, I.; Ingólfsson, O. Metastable decay of DNA components and their compositions—A perspective on the role of reactive electron scattering in radiation damage. Eur. Phys. J. D 2012, 66, 13. [Google Scholar] [CrossRef]
- Scheer, A.M.; Aflatooni, K.; Gallup, G.A.; Burrow, P.D. Bond Breaking and Temporary Anion States in Uracil and Halouracils: Implications for the DNA Bases. Phys. Rev. Lett. 2004, 92, 068102. [Google Scholar] [CrossRef] [Green Version]
- Rackwitz, J.; Kopyra, J.; Dąbkowska, I.; Ebel, K.; Ranković, M.L.; Milosavljević, A.R.; Bald, I. Sensitizing DNA towards Low-Energy Electrons with 2-Fluoroadenine. Angew. Chem. Int. Ed. 2016, 55, 10248–10252. [Google Scholar] [CrossRef] [PubMed]
- Kopyra, J.; Keller, A.; Bald, I. On the role of fluoro-substituted nucleosides in DNA radiosensitization for tumor radiation therapy. RSC Adv. 2014, 4, 6825. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, T.S.; Davis, M.A.; Maybaum, J.; Stetson, P.L.; Ensminger, W.D. The effect of single versus double-strand substitution on halogenated pyrimidine-induced radiosensitization and DNA strand breakage in human tumor cells. Radiat. Res. 1990, 123, 192–198. [Google Scholar] [CrossRef] [PubMed]
- McGinn, C.J.; Shewach, D.S.; Lawrence, T.S. Radiosensitizing Nucleosides. JNCI J. Natl. Cancer Inst. 1996, 88, 1193–1203. [Google Scholar] [CrossRef] [Green Version]
- Nitsche, M.; Christiansen, H.; Lederer, K.; Griesinger, F.; Schmidberger, H.; Pradier, O. Fludarabine combined with radiotherapy in patients with locally advanced NSCLC lung carcinoma: A phase I study. J. Cancer Res. Clin. Oncol. 2012, 138, 1113–1120. [Google Scholar] [CrossRef] [Green Version]
- Grégoire, V.; Ang, K.K.; Rosier, J.-F.; Beauduin, M.; Garden, A.S.; Hamoir, M.; Hittelman, W.N.; Humblet, Y.; Khuri, F.R.; Milas, L.; et al. A phase I study of fludarabine combined with radiotherapy in patients with intermediate to locally advanced head and neck squamous cell carcinoma. Radiother. Oncol. 2002, 63, 187–193. [Google Scholar] [CrossRef]
- Ómarsson, B.; Bjarnason, E.H.; Ingólfsson, O.; Haughey, S.; Field, T.A. Chemical control through dissociative electron attachment—A study on pentafluorotoluene, pentafluoroaniline and pentafluorophenol. Chem. Phys. Lett. 2012, 539–540, 7–10. [Google Scholar] [CrossRef]
- Ómarsson, B.; Bjarnason, E.H.; Haughey, S.A.; Field, T.A.; Abramov, A.; Klüpfel, P.; Jónsson, H.; Ingólfsson, O. Molecular rearrangement reactions in the gas phase triggered by electron attachment. Phys. Chem. Chem. Phys. 2013, 15, 4754. [Google Scholar] [CrossRef]
- Ómarsson, B.; Bjornsson, R.; Ingólfsson, O. Proton Shuttling and Reaction Paths in Dissociative Electron Attachment to o- and p-Tetrafluorohydroquinone, an Experimental and Theoretical Study. J. Phys. Chem. A 2017, 121, 5580–5585. [Google Scholar] [CrossRef]
- Dąbkowska, I.; Flosadóttir, H.D.; Orzol, M.; Ptasinska, S.; Bald, I.; Ingólfsson, O.; Illenberger, E. Reactions in gas phase and condensed phase C6F5X (X = NCO, CH2CN) triggered by low energy electrons. Phys. Chem. Chem. Phys. 2009, 11, 5323. [Google Scholar] [CrossRef]
- Marchetti, B.; Karsili, T.N.V.; Cipriani, M.; Hansen, C.S.; Ashfold, M.N.R. The near ultraviolet photodissociation dynamics of 2- and 3-substituted thiophenols: Geometric vs. electronic structure effects. J. Chem. Phys. 2017, 147, 013923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klar, D.; Ruf, M.-W.; Hotop, H. Dissociative electron attachment to CCl4 molecules at low electron energies with meV resolution. Int. J. Mass Spectrom. 2001, 205, 93–110. [Google Scholar] [CrossRef]
- Sanche, L.; Schulz, G.J. Electron transmission spectroscopy: Resonances in triatomic molecules and hydrocarbons. J. Chem. Phys. 1973, 58, 479–493. [Google Scholar] [CrossRef]
- Chowdhury, S.; Grimsrud, E.P.; Heinis, T.; Kebarle, P. Electron affinities of perfluorobenzene and perfluorophenyl compounds. J. Am. Chem. Soc. 1986, 108, 3630–3635. [Google Scholar] [CrossRef]
- Hinde, A.L.; Poppinger, D.; Radom, L. Ab initio study of the benzene radical anion. J. Am. Chem. Soc. 1978, 100, 4681–4685. [Google Scholar] [CrossRef]
- Birch, A.J.; Hinde, A.L.; Radom, L. A theoretical approach to the Birch reduction. Structures and stabilities of the radical anions of substituted benzenes. J. Am. Chem. Soc. 1980, 102, 3370–3376. [Google Scholar] [CrossRef]
- Jordan, K.D.; Michejda, J.A.; Burrow, P.D. Electron transmission studies of the negative ion states of substituted benzenes in the gas phase. J. Am. Chem. Soc. 1976, 98, 7189–7191. [Google Scholar] [CrossRef]
- Christophorou, L.G.; McCorkle, D.L.; Carter, J.G. Compound-negative-ion-resonant states and threshold-electron-excitation spectra of monosubstituted benzene derivatives. J. Chem. Phys. 1974, 60, 3779–3786. [Google Scholar] [CrossRef]
- Brundle, C.R.; Robin, M.B.; Kuebler, N.A.; Basch, H. Perfluoro effect in photoelectron spectroscopy. I. Nonaromatic molecules. J. Am. Chem. Soc. 1972, 94, 1451–1465. [Google Scholar] [CrossRef]
- Brundle, C.R.; Robin, M.B.; Kuebler, N.A. Perfluoro effect in photoelectron spectroscopy. II. Aromatic molecules. J. Am. Chem. Soc. 1972, 94, 1466–1475. [Google Scholar] [CrossRef]
- Yim, M.B.; Wood, D.E. Free radicals in an adamantane matrix. XII. EPR and INDO study of σ*-π* crossover in fluorinated benzene anions. J. Am. Chem. Soc. 1976, 98, 2053–2059. [Google Scholar] [CrossRef]
- Wang, J.T.; Williams, F. 13C Hyperfine interaction in the hexafluorobenzene radical anion. Chem. Phys. Lett. 1980, 71, 471–475. [Google Scholar] [CrossRef]
- Hitchcock, A.P.; Fischer, P.; Gedanken, A.; Robin, M.B. Antibonding σ* valence MOs in the inner-shell and outer-shell spectra of the fluorobenzenes. J. Phys. Chem. 1987, 91, 531–540. [Google Scholar] [CrossRef]
- Shchegoleva, L.N.; Bilkis, I.I.; Schastnev, P.V. Geometrical and electronic structure of fluoro-substituted benzene radical anions based on quantum chemical analysis of hyperfine interactions. Chem. Phys. 1983, 82, 343–353. [Google Scholar] [CrossRef]
- Shchegoleva, L.N.; Beregovaya, I.V.; Schastnev, P.V. Potential energy surface of C6F6− radical anion. Chem. Phys. Lett. 1999, 312, 325–332. [Google Scholar] [CrossRef]
- Yim, M.B.; DiGregorio, S.; Wood, D.E. Free radicals in an adamantane matrix. 13. Electron paramagnetic resonance study of .sigma.*-.pi.* orbital crossover in fluorinated pyridine anions. J. Am. Chem. Soc. 1977, 99, 4260–4263. [Google Scholar] [CrossRef]
- Skalický, T.; Chollet, C.; Pasquier, N.; Allan, M. Properties of the π* and σ* states of the chlorobenzene anion determined by electron impact spectroscopy. Phys. Chem. Chem. Phys. 2002, 4, 3583–3590. [Google Scholar] [CrossRef]
- Schulz, G.J.; Asundi, R.K. Isotope Effect in the Dissociative Attachment in H2 at Low Energy. Phys. Rev. 1967, 158, 25–29. [Google Scholar] [CrossRef]
- Krishnakumar, E.; Denifl, S.; Čadež, I.; Markelj, S.; Mason, N.J. Dissociative Electron Attachment Cross Sections for H2 and D2. Phys. Rev. Lett. 2011, 106, 243201. [Google Scholar] [CrossRef] [Green Version]
- Newville, M.; Stensitzki, T.; Allen, D.B.; Ingargiola, A. LMFIT: Non-Linear Least-Square Minimization and Curve-Fitting for Python (0.8.0). Zenodo 2014. [Google Scholar] [CrossRef]
- Bjarnason, E.H.; Ómarsson, B.; Engmann, S.; Ómarsson, F.H.; Ingólfsson, O. Dissociative electron attachment to titatinum tetrachloride and titanium tetraisopropoxide. Eur. Phys. J. D 2014, 68, 121. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615. [Google Scholar] [CrossRef] [Green Version]
- Riplinger, C.; Neese, F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. J. Chem. Phys. 2013, 138, 034106. [Google Scholar] [CrossRef]
- Riplinger, C.; Pinski, P.; Becker, U.; Valeev, E.F.; Neese, F. Sparse maps—A systematic infrastructure for reduced-scaling electronic structure methods. II. Linear scaling domain based pair natural orbital coupled cluster theory. J. Chem. Phys. 2016, 144, 024109. [Google Scholar] [CrossRef]
- Riplinger, C.; Sandhoefer, B.; Hansen, A.; Neese, F. Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J. Chem. Phys. 2013, 139, 134101. [Google Scholar] [CrossRef]
- Saitow, M.; Becker, U.; Riplinger, C.; Valeev, E.F.; Neese, F. A new near-linear scaling, efficient and accurate, open-shell domain-based local pair natural orbital coupled cluster singles and doubles theory. J. Chem. Phys. 2017, 146, 164105. [Google Scholar] [CrossRef]
- Neese, F. Importance of Direct Spin−Spin Coupling and Spin-Flip Excitations for the Zero-Field Splittings of Transition Metal Complexes: A Case Study. J. Am. Chem. Soc. 2006, 128, 10213–10222. [Google Scholar] [CrossRef]
- Ásgeirsson, V.; Birgisson, B.O.; Bjornsson, R.; Becker, U.; Neese, F.; Riplinger, C.; Jónsson, H. Nudged Elastic Band Method for Molecular Reactions Using Energy-Weighted Springs Combined with Eigenvector Following. J. Chem. Theory Comput. 2021, 17, 4929–4945. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| m/z | Fragment | ΔH0K | ΔEth |
|---|---|---|---|
| PFTP | |||
| 199 | [M-H]−/C6F5S− | 0.42 | 0.13 |
| 180 | [M-HF]−/C6F4S− | −0.0084 | −0.29 |
| 180 | * [M-HF]−/C5F4CS− | −0.066 | −0.35 |
| 167 | [M-SH]−/C6F5− | 0.61 | 0.32 |
| 2-FTP | |||
| 127 | [M-H]−/C6H4FS− | 1.03 | 0.84 |
| 108 | [M-HF]−/C6H4S− | 0.62 | 0.42 |
| 108 | * [M-HF]−/C5H4CS− | 0.47 | 0.28 |
| 32 | S− | 0.33 | 0.14 |
| PFP | |||
| 164 | [M-HF]−/C6F4O− | −0.066 | −0.33 |
| 164 | * [M-HF]−/C5F4CO− | −0.27 | −0.54 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cipriani, M.; Ingólfsson, O. HF Formation through Dissociative Electron Attachment—A Combined Experimental and Theoretical Study on Pentafluorothiophenol and 2-Fluorothiophenol. Int. J. Mol. Sci. 2022, 23, 2430. https://doi.org/10.3390/ijms23052430
Cipriani M, Ingólfsson O. HF Formation through Dissociative Electron Attachment—A Combined Experimental and Theoretical Study on Pentafluorothiophenol and 2-Fluorothiophenol. International Journal of Molecular Sciences. 2022; 23(5):2430. https://doi.org/10.3390/ijms23052430
Chicago/Turabian StyleCipriani, Maicol, and Oddur Ingólfsson. 2022. "HF Formation through Dissociative Electron Attachment—A Combined Experimental and Theoretical Study on Pentafluorothiophenol and 2-Fluorothiophenol" International Journal of Molecular Sciences 23, no. 5: 2430. https://doi.org/10.3390/ijms23052430
APA StyleCipriani, M., & Ingólfsson, O. (2022). HF Formation through Dissociative Electron Attachment—A Combined Experimental and Theoretical Study on Pentafluorothiophenol and 2-Fluorothiophenol. International Journal of Molecular Sciences, 23(5), 2430. https://doi.org/10.3390/ijms23052430