
Role of Distinct Macrophage Populations in the Development of Heart Failure in Macrophage Activation Syndrome

 , , , and
, , , and {kind=link}
Abstract
:1. Introduction
2. Historical and Contemporary Classification of Macrophages
3. Macrophages in the Heart
3.1. Origins of Different Macrophage Groups
3.2. Location, Functions and Replenishment of Distinct Macrophage Groups
3.3. Macrophage Behavior in Response to Myocardial Damage
3.4. Macrophages in Different Types of Heart Failure
4. Role of Various Cytokines in Macrophage Activation Syndrome
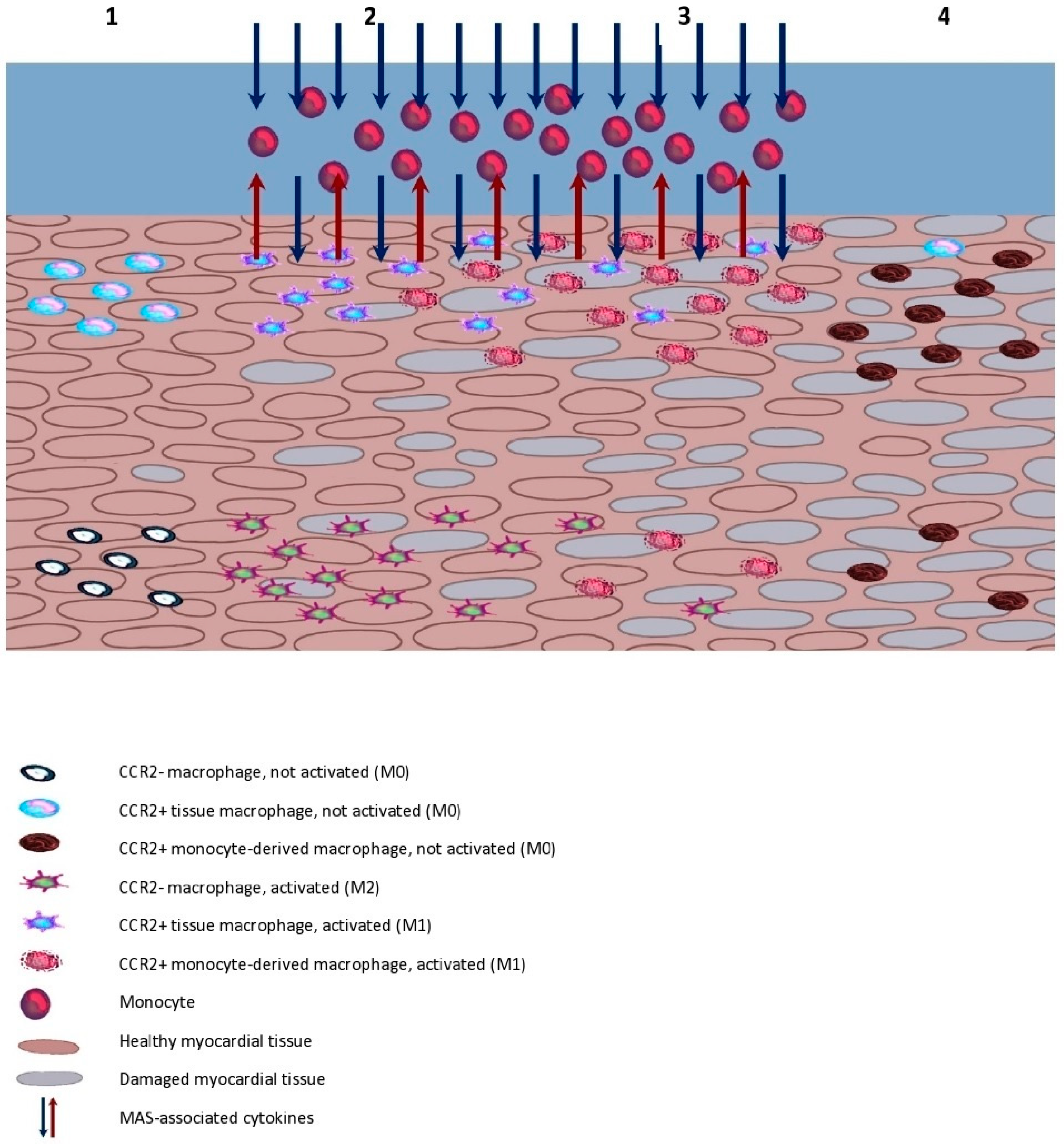
5. Possible Models of Myocarditis Development during MAS
- The heart has not yet been affected by the inflammation and possesses tissue populations of both CCR2− and CCR2+ macrophages. If the CCR2+ subtype is activated by the inflammation, the recruitment of monocytes, depletion of CCR2− and further development of the inflammation may occur with some delay.
- The heart has no CCR2− macrophage population or their number is significantly diminished by prior inflammatory incidents. In this case, the activation of macrophages may prompt a strong and rapid inflammatory response.
- Ongoing infection within the heart, e.g., endocarditis. The inflammatory process may be progressing in the heart even before the onset of a fully symptomatic MAS, in which case, a severe aggravation of the inflammatory response and its extent may occur.
6. Conclusions
Funding
Conflicts of Interest
References
- Crayne, C.B.; Albeituni, S.; Nichols, K.E.; Cron, R.Q. The Immunology of Macrophage Activation Syndrome. Front. Immunol. 2019, 10, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravelli, A.; Minoia, F.; Davì, S.; Horne, A.; Bovis, F.; Pistorio, A.; Aricò, M.; Avcin, T.; Behrens, E.M.; De Benedetti, F.; et al. Classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: A European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Arthritis Rheumatol. 2016, 68, 566–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minoia, F.; Davì, S.; Horne, A.; Bovis, F.; Demirkaya, E.; Akikusa, J.; Ayaz, N.A.; Al-Mayouf, S.M.; Barone, P.; Bica, B.; et al. Dissecting the heterogeneity of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J. Rheumatol. 2015, 42, 994–1001. [Google Scholar] [CrossRef] [PubMed]
- Behrens, E.M.; Beukelman, T.; Paessler, M.; Cron, R.Q. Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J. Rheumatol. 2007, 34, 1133–1138. [Google Scholar] [PubMed]
- Cowley, S.; Ramakrishnan, S. Macrophage activation syndrome in a patient with systemic lupus erythematosus. Pol. Arch. Intern. Med. 2019, 129, 535–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuji, T.; Ohno, S.; Ishigatsubo, Y. Liver manifestations in systemic lupus erythematosus: High incidence of hemophagocytic syndrome. J. Rheumatol. 2002, 29, 1576–1577. [Google Scholar]
- Kim, J.M.; Kwok, S.K.; Ju, J.H.; Kim, H.Y.; Park, S.H. Reactive hemophagocytic syndrome in adult Korean patients with systemic lupus erythematosus: A case-control study and literature review. J. Rheumatol. 2012, 39, 86–93. [Google Scholar] [CrossRef]
- Schulert, G.S.; Grom, A.A. Pathogenesis of macrophage activation syndrome and potential for cytokine-directed therapies. Annu. Rev. Med. 2015, 66, 145–159. [Google Scholar] [CrossRef] [Green Version]
- Stéphan, J.L.; Koné-Paut, I.; Galambrun, C.; Mouy, R.; Bader-Meunier, B.; Prieur, A.M. Reactive haemophagocytic syndrome in children with inflammatory disorders. A retrospective study of 24 patients. Rheumatology 2001, 40, 1285–1292. [Google Scholar] [CrossRef] [Green Version]
- Sawhney, S.; Woo, P.; Murray, K.J. Macrophage activation syndrome: A potentially fatal complication of rheumatic disorders. Arch. Dis. Child. 2001, 85, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Henter, J.I.; Horne, A.; Aricó, M.; Egeler, R.M.; Filipovich, A.H.; Imashuku, S.; Ladisch, S.; McClain, K.; Webb, D.; Winiarski, J.; et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer 2007, 48, 124–131. [Google Scholar] [CrossRef]
- Ravelli, A.; Magni-Manzoni, S.; Pistorio, A.; Besana, C.; Foti, T.; Ruperto, N.; Viola, S.; Martini, A. Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J. Pediatr. 2005, 146, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Davì, S.; Consolaro, A.; Guseinova, D.; Pistorio, A.; Ruperto, N.; Martini, A.; Cron, R.Q.; Ravelli, A. MAS Study Group. An international consensus survey of diagnostic criteria for macrophage activation syndrome in systemic juvenile idiopathic arthritis. J. Rheumatol. 2011, 38, 764–768. [Google Scholar] [CrossRef]
- Bleesing, J.; Prada, A.; Siegel, D.M.; Villanueva, J.; Olson, J.; Ilowite, N.T.; Brunner, H.I.; Griffin, T.; Graham, T.B.; Sherry, D.D.; et al. The diagnostic significance of soluble CD163 and soluble interleukin-2 receptor alpha-chain in macrophage activation syndrome and untreated new-onset systemic juvenile idiopathic arthritis. Arthritis Rheum. 2007, 56, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Minoia, F.; Davì, S.; Horne, A.; Demirkaya, E.; Bovis, F.; Li, C.; Lehmberg, K.; Weitzman, S.; Insalaco, A.; Wouters, C.; et al. Pediatric Rheumatology International Trials Organization; Childhood Arthritis and Rheumatology Research Alliance; Pediatric Rheumatology Collaborative Study Group; Histiocyte Society. Clinical features, treatment, and outcome of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: A multinational, multicenter study of 362 patients. Arthritis Rheumatol. 2014, 66, 3160–3169. [Google Scholar] [CrossRef]
- Sreejit, G.; Fleetwood, A.J.; Murphy, A.J.; Nagareddy, P.R. Origins and diversity of macrophages in health and disease. Clin. Transl. Immunol. 2020, 9, e1222. [Google Scholar] [CrossRef] [PubMed]
- van Furth, R.; Cohn, Z.A.; Hirsch, J.G.; Humphrey, J.H.; Spector, W.G.; Langevoort, H.L. The mononuclear phagocyte system: A new classification of macrophages, monocytes, and their precursor cells. Bull. World Health Organ. 1972, 46, 845–852. [Google Scholar]
- van Furth, R.; Cohn, Z.A. The origin and kinetics of mononuclear phagocytes. J. Exp. Med. 1968, 128, 415–435. [Google Scholar]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [Green Version]
- Ruytinx, P.; Proost, P.; Van Damme, J.; Struyf, S. Chemokine-Induced Macrophage Polarization in Inflammatory Conditions. Front. Immunol. 2018, 9, 1930. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Morris, S.M., Jr. Arginine metabolism: Boundaries of our knowledge. J. Nutr. 2007, 137 (Suppl. S2), 1602S–1609S. [Google Scholar] [CrossRef] [Green Version]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969, Erratum in Nat. Rev. Immunol. 2010, 10, 460. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef] [Green Version]
- Hanna, R.N.; Carlin, L.M.; Hubbeling, H.G.; Nackiewicz, D.; Green, A.M.; Punt, J.A.; Geissmann, F.; Hedrick, C.C. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C- monocytes. Nat. Immunol. 2011, 12, 778–785. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [Green Version]
- Bachelerie, F.; Ben-Baruch, A.; Burkhardt, A.M.; Combadiere, C.; Farber, J.M.; Graham, G.J.; Horuk, R.; Sparre-Ulrich, A.H.; Locati, M.; Luster, A.D.; et al. International Union of Basic and Clinical Pharmacology. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol. Rev. 2013, 66, 1–79, Erratum in Pharmacol. Rev. 2014, 66, 467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soehnlein, O.; Drechsler, M.; Döring, Y.; Lievens, D.; Hartwig, H.; Kemmerich, K.; Ortega-Gómez, A.; Mandl, M.; Vijayan, S.; Projahn, D.; et al. Distinct functions of chemokine receptor axes in the atherogenic mobilization and recruitment of classical monocytes. EMBO Mol. Med. 2013, 5, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Alvarez, D.; Kaplan, T.J.; Jakubzick, C.; Spanbroek, R.; Llodra, J.; Garin, A.; Liu, J.; Mack, M.; van Rooijen, N.; et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J. Clin. Investig. 2007, 117, 185–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafuse, W.P.; Wozniak, D.J.; Rajaram, M.V.S. Role of Cardiac Macrophages on Cardiac Inflammation, Fibrosis and Tissue Repair. Cells 2020, 10, 51. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [Green Version]
- Frangogiannis, N.G. Regulation of the inflammatory response in cardiac repair. Circ. Res. 2012, 110, 159–173. [Google Scholar] [CrossRef]
- Talvani, A.; Ribeiro, C.S.; Aliberti, J.C.; Michailowsky, V.; Santos, P.V.; Murta, S.M.; Romanha, A.J.; Almeida, I.C.; Farber, J.; Lannes-Vieira, J.; et al. Kinetics of cytokine gene expression in experimental chagasic cardiomyopathy: Tissue parasitism and endogenous IFN-gamma as important determinants of chemokine mRNA expression during infection with Trypanosoma cruzi. Microbes Infect. 2000, 2, 851–866. [Google Scholar] [CrossRef]
- Machado, F.S.; Koyama, N.S.; Carregaro, V.; Ferreira, B.R.; Milanezi, C.M.; Teixeira, M.M.; Rossi, M.A.; Silva, J.S. CCR5 plays a critical role in the development of myocarditis and host protection in mice infected with Trypanosoma cruzi. J. Infect. Dis. 2005, 191, 627–636. [Google Scholar] [CrossRef] [Green Version]
- Ramos, G.C.; van den Berg, A.; Nunes-Silva, V.; Weirather, J.; Peters, L.; Burkard, M.; Friedrich, M.; Pinnecker, J.; Abeßer, M.; Heinze, K.G.; et al. Myocardial aging as a T-cell-mediated phenomenon. Proc. Natl. Acad. Sci. USA 2017, 114, E2420–E2429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zouggari, Y.; Ait-Oufella, H.; Bonnin, P.; Simon, T.; Sage, A.P.; Guérin, C.; Vilar, J.; Caligiuri, G.; Tsiantoulas, D.; Laurans, L.; et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat. Med. 2013, 19, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Dobaczewski, M.; Rai, V.; Haque, Z.; Chen, W.; Li, N.; Frangogiannis, N.G. Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1233–H1242. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Casares, X.; Hosseinzadeh, S.; Barbu, I.; Dick, S.A.; Macklin, J.A.; Wang, Y.; Momen, A.; Kantores, C.; Aronoff, L.; Farno, M.; et al. A CD103+ Conventional Dendritic Cell Surveillance System Prevents Development of Overt Heart Failure during Subclinical Viral Myocarditis. Immunity 2017, 47, 974–989.e8. [Google Scholar] [CrossRef] [Green Version]
- Bracamonte-Baran, W.; Chen, G.; Hou, X.; Talor, M.V.; Choi, H.S.; Davogustto, G.; Taegtmeyer, H.; Sung, J.; Hackam, D.J.; Nauen, D.; et al. Non-cytotoxic Cardiac Innate Lymphoid Cells Are a Resident and Quiescent Type 2-Commited Population. Front. Immunol. 2019, 10, 634. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef] [Green Version]
- Jakubzick, C.; Gautier, E.L.; Gibbings, S.L.; Sojka, D.K.; Schlitzer, A.; Johnson, T.E.; Ivanov, S.; Duan, Q.; Bala, S.; Condon, T.; et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity 2013, 39, 599–610. [Google Scholar] [CrossRef] [Green Version]
- Lavine, K.J.; Epelman, S.; Uchida, K.; Weber, K.J.; Nichols, C.G.; Schilling, J.D.; Ornitz, D.M.; Randolph, G.J.; Mann, D.L. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. USA 2014, 111, 16029–16034. [Google Scholar] [CrossRef] [Green Version]
- Leid, J.; Carrelha, J.; Boukarabila, H.; Epelman, S.; Jacobsen, S.E.; Lavine, K.J. Primitive Embryonic Macrophages are Required for Coronary Development and Maturation. Circ. Res. 2016, 118, 1498–1511. [Google Scholar] [CrossRef]
- Bajpai, G.; Schneider, C.; Wong, N.; Bredemeyer, A.; Hulsmans, M.; Nahrendorf, M.; Epelman, S.; Kreisel, D.; Liu, Y.; Itoh, A.; et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 2018, 24, 1234–1245. [Google Scholar] [CrossRef]
- Palis, J.; Robertson, S.; Kennedy, M.; Wall, C.; Keller, G. Development of erythroid and myeloid progenitors in the yolk sac and embryo proper of the mouse. Development 1999, 126, 5073–5084. [Google Scholar] [CrossRef] [PubMed]
- Tober, J.; Koniski, A.; McGrath, K.E.; Vemishetti, R.; Emerson, R.; de Mesy-Bentley, K.K.; Waugh, R.; Palis, J. The megakaryocyte lineage originates from hemangioblast precursors and is an integral component both of primitive and of definitive hematopoiesis. Blood 2007, 109, 1433–1441. [Google Scholar] [CrossRef] [Green Version]
- Mass, E.; Ballesteros, I.; Farlik, M.; Halbritter, F.; Günther, P.; Crozet, L.; Jacome-Galarza, C.E.; Händler, K.; Klughammer, J.; Kobayashi, Y.; et al. Specification of tissue-resident macrophages during organogenesis. Science 2016, 353, aaf4238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palis, J.; Yoder, M.C. Yolk-sac hematopoiesis: The first blood cells of mouse and man. Exp. Hematol. 2001, 29, 927–936. [Google Scholar] [CrossRef]
- Bertrand, J.Y.; Jalil, A.; Klaine, M.; Jung, S.; Cumano, A.; Godin, I. Three pathways to mature macrophages in the early mouse yolk sac. Blood 2005, 106, 3004–3011. [Google Scholar] [CrossRef] [PubMed]
- Boisset, J.C.; van Cappellen, W.; Andrieu-Soler, C.; Galjart, N.; Dzierzak, E.; Robin, C. In vivo imaging of haematopoietic cells emerging from the mouse aortic endothelium. Nature 2010, 464, 116–120. [Google Scholar] [CrossRef]
- Kissa, K.; Herbomel, P. Blood stem cells emerge from aortic endothelium by a novel type of cell transition. Nature 2010, 464, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Lavine, K.J.; Pinto, A.R.; Epelman, S.; Kopecky, B.J.; Clemente-Casares, X.; Godwin, J.; Rosenthal, N.; Kovacic, J.C. The Macrophage in Cardiac Homeostasis and Disease: JACC Macrophage in CVD Series (Part 4). J. Am. Coll. Cardiol. 2018, 72, 2213–2230. [Google Scholar] [CrossRef]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Heidt, T.; Courties, G.; Dutta, P.; Sager, H.B.; Sebas, M.; Iwamoto, Y.; Sun, Y.; Da Silva, N.; Panizzi, P.; van der Laan, A.M.; et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 2014, 115, 284–295, Erratum in Circ. Res. 2014, 115, e95. [Google Scholar] [CrossRef] [Green Version]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Wülfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 2017, 169, 510–522.e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; Gordonov, S.; et al. Immunological Genome Consortium. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosselin, D.; Link, V.M.; Romanoski, C.E.; Fonseca, G.J.; Eichenfield, D.Z.; Spann, N.J.; Stender, J.D.; Chun, H.B.; Garner, H.; Geissmann, F.; et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 2014, 159, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- van de Laar, L.; Saelens, W.; De Prijck, S.; Martens, L.; Scott, C.L.; Van Isterdael, G.; Hoffmann, E.; Beyaert, R.; Saeys, Y.; Lambrecht, B.N.; et al. Yolk Sac Macrophages, Fetal Liver, and Adult Monocytes Can Colonize an Empty Niche and Develop into Functional Tissue-Resident Macrophages. Immunity 2016, 44, 755–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, A.R.; Paolicelli, R.; Salimova, E.; Gospocic, J.; Slonimsky, E.; Bilbao-Cortes, D.; Godwin, J.W.; Rosenthal, N.A. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLoS ONE 2012, 7, e36814. [Google Scholar] [CrossRef] [PubMed]
- DeBerge, M.; Yeap, X.Y.; Dehn, S.; Zhang, S.; Grigoryeva, L.; Misener, S.; Procissi, D.; Zhou, X.; Lee, D.C.; Muller, W.A.; et al. MerTK Cleavage on Resident Cardiac Macrophages Compromises Repair After Myocardial Ischemia Reperfusion Injury. Circ. Res. 2017, 121, 930–940. [Google Scholar] [CrossRef]
- Li, W.; Hsiao, H.M.; Higashikubo, R.; Saunders, B.T.; Bharat, A.; Goldstein, D.R.; Krupnick, A.S.; Gelman, A.E.; Lavine, K.J.; Kreisel, D. Heart-resident CCR2+ macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight 2016, 1, e87315. [Google Scholar] [CrossRef]
- Bajpai, G.; Bredemeyer, A.; Li, W.; Zaitsev, K.; Koenig, A.L.; Lokshina, I.; Mohan, J.; Ivey, B.; Hsiao, H.M.; Weinheimer, C.; et al. Tissue Resident CCR2− and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ. Res. 2019, 124, 263–278. [Google Scholar] [CrossRef]
- Ajami, B.; Bennett, J.L.; Krieger, C.; McNagny, K.M.; Rossi, F.M. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat. Neurosci. 2011, 14, 1142–1149. [Google Scholar] [CrossRef]
- Molawi, K.; Wolf, Y.; Kandalla, P.K.; Favret, J.; Hagemeyer, N.; Frenzel, K.; Pinto, A.R.; Klapproth, K.; Henri, S.; Malissen, B.; et al. Progressive replacement of embryo-derived cardiac macrophages with age. J. Exp. Med. 2014, 211, 2151–2158. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.L.; Zheng, F.; De Baetselier, P.; Martens, L.; Saeys, Y.; De Prijck, S.; Lippens, S.; Abels, C.; Schoonooghe, S.; Raes, G.; et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat. Commun. 2016, 7, 10321. [Google Scholar] [CrossRef]
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; Althagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 2019, 20, 29–39, Erratum in Nat. Immunol. 2019, 20, 664. [Google Scholar] [CrossRef] [PubMed]
- Miyanishi, M.; Tada, K.; Koike, M.; Uchiyama, Y.; Kitamura, T.; Nagata, S. Identification of Tim4 as a phosphatidylserine receptor. Nature 2007, 450, 435–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, E.; Yeap, X.Y.; Dehn, S.; Terry, R.; Novak, M.; Zhang, S.; Iwata, S.; Han, X.; Homma, S.; Drosatos, K.; et al. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ. Res. 2013, 113, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.Y.; Lim, S.Y.; Tan, C.K.; Thiam, C.H.; Goh, C.C.; Carbajo, D.; Chew, S.H.S.; See, P.; Chakarov, S.; Wang, X.N.; et al. Hyaluronan Receptor LYVE-1-Expressing Macrophages Maintain Arterial Tone through Hyaluronan-Mediated Regulation of Smooth Muscle Cell Collagen. Immunity 2018, 49, 326–341.e7, Erratum in Immunity 2018, 49, 1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagar, S.; Liu, P.P.; Cooper, L.T., Jr. Myocarditis. Lancet 2012, 379, 738–747. [Google Scholar] [CrossRef] [Green Version]
- Coura, J.R.; Borges-Pereira, J. Chagas disease: 100 years after its discovery. A systemic review. Acta Trop. 2010, 115, 5–13. [Google Scholar] [CrossRef]
- Kindermann, I.; Barth, C.; Mahfoud, F.; Ukena, C.; Lenski, M.; Yilmaz, A.; Klingel, K.; Kandolf, R.; Sechtem, U.; Cooper, L.T.; et al. Update on myocarditis. J. Am. Coll. Cardiol. 2012, 59, 779–792. [Google Scholar] [CrossRef]
- Westermann, D.; Savvatis, K.; Schultheiss, H.P.; Tschöpe, C. Immunomodulation and matrix metalloproteinases in viral myocarditis. J. Mol. Cell. Cardiol. 2010, 48, 468–473. [Google Scholar] [CrossRef]
- Esfandiarei, M.; McManus, B.M. Molecular biology and pathogenesis of viral myocarditis. Annu. Rev. Pathol. 2008, 3, 127–155. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.M.; Fairweather, D.; Huber, S.A.; Cunningham, M.W. Autoimmune myocarditis, valvulitis, and cardiomyopathy. Curr. Protoc. Immunol. 2013, 101, 15.14.1-15.14.51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Ong, S.; Talor, M.V.; Barin, J.G.; Baldeviano, G.C.; Kass, D.A.; Bedja, D.; Zhang, H.; Sheikh, A.; Margolick, J.B.; et al. Cardiac fibroblasts mediate IL-17A-driven inflammatory dilated cardiomyopathy. J. Exp. Med. 2014, 211, 1449–1464. [Google Scholar] [CrossRef] [PubMed]
- Leuschner, F.; Courties, G.; Dutta, P.; Mortensen, L.J.; Gorbatov, R.; Sena, B.; Novobrantseva, T.I.; Borodovsky, A.; Fitzgerald, K.; Koteliansky, V.; et al. Silencing of CCR2 in myocarditis. Eur. Heart J. 2015, 36, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Deci, M.B.; Ferguson, S.W.; Scatigno, S.L.; Nguyen, J. Modulating Macrophage Polarization through CCR2 Inhibition and Multivalent Engagement. Mol. Pharm. 2018, 15, 2721–2731. [Google Scholar] [CrossRef]
- Moskalik, A.; Niderla-Bielińska, J.; Ratajska, A. Multiple roles of cardiac macrophages in heart homeostasis and failure. Heart Fail. Rev. 2021. Online ahead of print. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.; Coats, A.J.; Falk, V.; González-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef]
- Mishra, S.; Kass, D.A. Cellular and molecular pathobiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2021, 18, 400–423. [Google Scholar] [CrossRef]
- Tromp, J.; Westenbrink, B.D.; Ouwerkerk, W.; van Veldhuisen, D.J.; Samani, N.J.; Ponikowski, P.; Metra, M.; Anker, S.D.; Cleland, J.G.; Dickstein, K.; et al. Identifying pathophysiological mechanisms in heart failure with reduced versus preserved ejection fraction. J. Am. Coll. Cardiol. 2018, 72, 1081–1090. [Google Scholar] [CrossRef]
- Hulsmans, M.; Sager, H.B.; Roh, J.D.; Valero-Munoz, M.; Houstis, N.E.; Iwamoto, Y.; Sun, Y.; Wilson, R.M.; Wojtkiewicz, G.; Tricot, B.; et al. Cardiac macrophages promote diastolic dysfunction. J. Exp. Med. 2018, 215, 423–440. [Google Scholar] [CrossRef]
- Shen, J.L.; Xie, X.J. Insight into the Pro-inflammatory and Profibrotic Role of Macrophage in Heart Failure with Preserved Ejection Fraction. J. Cardiovasc. Pharmacol. 2020, 76, 276–285. [Google Scholar] [CrossRef]
- Chiong, M.; Wang, Z.V.; Pedrozo, Z.; Cao, D.J.; Troncoso, R.; Ibacache, M.; Criollo, A.; Nemchenko, A.; Hill, J.A.; Lavandero, S. Cardiomyocyte death: Mechanisms and translational implications. Cell Death Dis. 2011, 2, e244. [Google Scholar] [CrossRef]
- Dunlay, S.M.; Roger, V.L.; Redfield, M.M. Epidemiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2017, 14, 591–602. [Google Scholar] [CrossRef]
- Yap, J.; Cabrera-Fuentes, H.A.; Irei, J.; Hausenloy, D.J.; Boisvert, W.A. Role of Macrophages in Cardioprotection. Int. J. Mol. Sci. 2019, 20, 2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.J.; Koplev, S.; Fisher, E.A.; Tabas, I.; Björkegren, J.L.M.; Doran, A.C.; Kovacic, J.C. Macrophage Trafficking, Inflammatory Resolution, and Genomics in Atherosclerosis: JACC Macrophage in CVD Series (Part 2). J. Am. Coll. Cardiol. 2018, 72, 2181–2197. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Ding, A. Nonresolving inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabas, I.; Glass, C.K. Anti-inflammatory therapy in chronic disease: Challenges and opportunities. Science 2013, 339, 166–172. [Google Scholar] [CrossRef] [Green Version]
- Tabas, I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 2010, 10, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Linton, M.F.; Babaev, V.R.; Huang, J.; Linton, E.F.; Tao, H.; Yancey, P.G. Macrophage Apoptosis and Efferocytosis in the Pathogenesis of Atherosclerosis. Circ. J. 2016, 80, 2259–2268. [Google Scholar] [CrossRef] [Green Version]
- Schrijvers, D.M.; De Meyer, G.R.; Kockx, M.M.; Herman, A.G.; Martinet, W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1256–1261. [Google Scholar] [CrossRef] [Green Version]
- Hansson, G.K.; Libby, P.; Tabas, I. Inflammation and plaque vulnerability. J. Intern. Med. 2015, 278, 483–493. [Google Scholar] [CrossRef]
- Libby, P.; Tabas, I.; Fredman, G.; Fisher, E.A. Inflammation and its resolution as determinants of acute coronary syndromes. Circ. Res. 2014, 114, 1867–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmonds, S.J.; Cuijpers, I.; Heymans, S.; Jones, E.A.V. Cellular and molecular differences between HFpEF and HFrEF: A step ahead in an improved pathological understanding. Cells 2020, 9, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutka, M.; Bobiński, R.; Ulman-Włodarz, I.; Hajduga, M.; Bujok, J.; Pająk, C.; Ćwiertnia, M. Various aspects of inflammation in heart failure. Heart Fail. Rev. 2020, 25, 537–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrigley, B.J.; Lip, G.Y.H.; Shantsila, E. The role of monocytes and inflammation in the pathophysiology of heart failure. Eur. J. Heart Fail. 2015, 13, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- van Wezenbeek, J.; Canada, J.M.; Ravindra, K.; Carbone, S.; Trankle, C.R.; Kadariya, D.; Buckley, L.F.; Del Buono, M.; Billingsley, H.; Viscusi, M.; et al. C-reactive protein and N-terminal pro-brain natriuretic peptide levels correlate with impaired cardiorespiratory fitness in patients with heart failure across a wide range of ejection fraction. Front. Cardiovasc. Med. 2018, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.P.; Jyoti Patel, J. Therapeutic approaches targeting inflammation in cardiovascular disorders. Biology 2018, 7, 49. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Bauersachs, J.; Langer, H.F. Immune mechanisms in heart failure. Eur. J. Heart Fail. 2017, 19, 1379–1389. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.M.; Akkerhuis, K.M.; Battes, L.C.; van Vark, L.C.; Hillege, H.L.; Paulus, W.J.; Boersma, E.; Kardys, I. Biomarkers of heart failure with normal ejection fraction: A systematic review. Eur. J. Heart Fail. 2013, 15, 1350–1362. [Google Scholar] [CrossRef]
- Hartupee, J.; Mann, D.L. Positioning of inflammatory biomarkers in the heart failure landscape. J. Cardiovasc. Transl. Res. 2013, 6, 485–492. [Google Scholar] [CrossRef] [Green Version]
- Rauchhaus, M.; Doehner, W.; Francis, D.P.; Davos, C.; Kemp, M.; Liebenthal, C.; Niebauer, J.; Hooper, J.; Volk, H.D.; Coats, A.J.; et al. Plasma cytokine parameters and mortality in patients with chronic heart failure. Circulation 2000, 102, 3060–3067. [Google Scholar] [CrossRef] [Green Version]
- Collier, P.; Watson, C.J.; Voon, V.; Phelan, D.; Jan, A.; Mak, G.; Martos, R.; Baugh, J.A.; Ledwidge, M.T.; McDonald, K.M. Can emerging biomarkers of myocardial remodelling identify asymptomatic hypertensive patients at risk for diastolic dysfunction and diastolic heart failure? Eur. J. Heart Fail. 2011, 13, 1087–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalogeropoulos, A.; Georgiopoulou, V.; Psaty, B.M.; Rodondi, N.; Smith, A.L.; Harrison, D.G.; Liu, Y.; Hoffmann, U.; Bauer, D.C.; Newman, A.B.; et al. Inflammatory markers and incident heart failure risk in older adults: The Health ABC (Health, Aging, and Body Composition) study. J. Am. Coll. Cardiol. 2010, 55, 2129–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Boer, R.A.; Lok, D.J.; Jaarsma, T.; van der Meer, P.; Voors, A.A.; Hillege, H.L.; van Veldhuisen, D.J. Predictive value of plasma galectin-3 levels in heart failure with reduced and preserved ejection fraction. Ann. Med. 2011, 43, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Henter, J.I.; Elinder, G.; Söder, O.; Hansson, M.; Andersson, B.; Andersson, U. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood 1991, 78, 2918–2922. [Google Scholar] [CrossRef] [PubMed]
- Henter, J.I.; Andersson, B.; Elinder, G.; Jakobson, A.; Lübeck, P.O.; Söder, O. Elevated circulating levels of interleukin-1 receptor antagonist but not IL-1 agonists in hemophagocytic lymphohistiocytosis. Med. Pediatr. Oncol. 1996, 27, 21–25. [Google Scholar] [CrossRef]
- Sumegi, J.; Barnes, M.G.; Nestheide, S.V.; Molleran-Lee, S.; Villanueva, J.; Zhang, K.; Risma, K.A.; Grom, A.A.; Filipovich, A.H. Gene expression profiling of peripheral blood mononuclear cells from children with active hemophagocytic lymphohistiocytosis. Blood 2011, 117, e151–e160. [Google Scholar] [CrossRef] [Green Version]
- Orange, J.S. The lytic NK cell immunological synapse and sequential steps in its formation. Adv. Exp. Med. Biol. 2007, 601, 225–233. [Google Scholar] [CrossRef]
- Jenkins, M.R.; Rudd-Schmidt, J.A.; Lopez, J.A.; Ramsbottom, K.M.; Mannering, S.I.; Andrews, D.M.; Voskoboinik, I.; Trapani, J.A. Failed CTL/NK cell killing and cytokine hypersecretion are directly linked through prolonged synapse time. J. Exp. Med. 2015, 212, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Behrens, E.M.; Cron, R.Q. Kill or be killed. J. Immunol. 2015, 194, 5041–5043. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Behrens, E.M.; Atkinson, T.P.; Shakoory, B.; Grom, A.A.; Cron, R.Q. Genetic defects in cytolysis in macrophage activation syndrome. Curr. Rheumatol. Rep. 2014, 16, 439. [Google Scholar] [CrossRef]
- Kaufman, K.M.; Linghu, B.; Szustakowski, J.D.; Husami, A.; Yang, F.; Zhang, K.; Filipovich, A.H.; Fall, N.; Harley, J.B.; Nirmala, N.R.; et al. Whole-exome sequencing reveals overlap between macrophage activation syndrome in systemic juvenile idiopathic arthritis and familial hemophagocytic lymphohistiocytosis. Arthritis Rheumatol. 2014, 66, 3486–3495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracaglia, C.; Prencipe, G.; De Benedetti, F. Macrophage Activation Syndrome: Different mechanisms leading to a one clinical syndrome. Pediatr. Rheumatol. Online J. 2017, 15, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrestha, B.; Omran, A.; Rong, P.; Wang, W. Report of a Fatal Pediatric Case of Hemophagocytic Lymphohistiocytosis Associated with Pandemic Influenza A (H1N1) Infection in 2009. Pediatr. Neonatol. 2015, 56, 189–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strippoli, R.; Caiello, I.; De Benedetti, F. Reaching the threshold: A multilayer pathogenesis of macrophage activation syndrome. J. Rheumatol. 2013, 40, 761–767. [Google Scholar] [CrossRef]
- Pascual, V.; Allantaz, F.; Arce, E.; Punaro, M.; Banchereau, J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J. Exp. Med. 2005, 201, 1479–1486. [Google Scholar] [CrossRef]
- Fall, N.; Barnes, M.; Thornton, S.; Luyrink, L.; Olson, J.; Ilowite, N.T.; Gottlieb, B.S.; Griffin, T.; Sherry, D.D.; Thompson, S.; et al. Gene expression profiling of peripheral blood from patients with untreated new-onset systemic juvenile idiopathic arthritis reveals molecular heterogeneity that may predict macrophage activation syndrome. Arthritis Rheum. 2007, 56, 3793–3804. [Google Scholar] [CrossRef] [PubMed]
- Behrens, E.M.; Canna, S.W.; Slade, K.; Rao, S.; Kreiger, P.A.; Paessler, M.; Kambayashi, T.; Koretzky, G.A. Repeated TLR9 stimulation results in macrophage activation syndrome-like disease in mice. J. Clin. Investig. 2011, 121, 2264–2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef]
- Mosser, D.M. The many faces of macrophage activation. J. Leukoc. Biol. 2003, 73, 209–212. [Google Scholar] [CrossRef]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Kratochvill, F.; Neale, G.; Haverkamp, J.M.; Van de Velde, L.A.; Smith, A.M.; Kawauchi, D.; McEvoy, J.; Roussel, M.F.; Dyer, M.A.; Qualls, J.E.; et al. TNF Counterbalances the Emergence of M2 Tumor Macrophages. Cell Rep. 2015, 12, 1902–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gattorno, M.; Piccini, A.; Lasigliè, D.; Tassi, S.; Brisca, G.; Carta, S.; Delfino, L.; Ferlito, F.; Pelagatti, M.A.; Caroli, F.; et al. The pattern of response to anti-interleukin-1 treatment distinguishes two subsets of patients with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2008, 58, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Nigrovic, P.A.; Mannion, M.; Prince, F.H.; Zeft, A.; Rabinovich, C.E.; van Rossum, M.A.; Cortis, E.; Pardeo, M.; Miettunen, P.M.; Janow, G.; et al. Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: Report of forty-six patients from an international multicenter series. Arthritis Rheum. 2011, 63, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Zeft, A.; Hollister, R.; LaFleur, B.; Sampath, P.; Soep, J.; McNally, B.; Kunkel, G.; Schlesinger, M.; Bohnsack, J. Anakinra for systemic juvenile arthritis: The Rocky Mountain experience. J. Clin. Rheumatol. 2009, 15, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, A.A.; Leclercq, S.A.; Yan, A.; Homik, J.E.; Dinarello, C.A. Rapid responses to anakinra in patients with refractory adult-onset Still’s disease. Arthritis Rheum. 2005, 52, 1794–1803. [Google Scholar] [CrossRef]
- Ling, X.B.; Park, J.L.; Carroll, T.; Nguyen, K.D.; Lau, K.; Macaubas, C.; Chen, E.; Lee, T.; Sandborg, C.; Milojevic, D.; et al. Plasma profiles in active systemic juvenile idiopathic arthritis: Biomarkers and biological implications. Proteomics 2010, 10, 4415–4430. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, M.; Yokoyama, T.; Yamada, K.; Kaneda, H.; Wada, H.; Wada, T.; Toma, T.; Ohta, K.; Kasahara, Y.; Yachie, A. Distinct cytokine profiles of systemic-onset juvenile idiopathic arthritis-associated macrophage activation syndrome with particular emphasis on the role of interleukin-18 in its pathogenesis. Rheumatology 2010, 49, 1645–1653. [Google Scholar] [CrossRef] [Green Version]
- Kaplanski, G. Interleukin-18: Biological properties and role in disease pathogenesis. Immunol. Rev. 2018, 281, 138–153. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, M.; Inoue, N.; Mizuta, M.; Nakagishi, Y.; Yachie, A. Characteristic elevation of soluble TNF receptor II: I ratio in macrophage activation syndrome with systemic juvenile idiopathic arthritis. Clin. Exp. Immunol. 2018, 191, 349–355. [Google Scholar] [CrossRef] [Green Version]
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018, 24, 739–748. [Google Scholar] [CrossRef]
- de Benedetti, F.; Massa, M.; Robbioni, P.; Ravelli, A.; Burgio, G.R.; Martini, A. Correlation of serum interleukin-6 levels with joint involvement and thrombocytosis in systemic juvenile rheumatoid arthritis. Arthritis Rheum. 1991, 34, 1158–1163. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.J.; Tang, Y.M.; Song, H.; Yang, S.L.; Xu, W.Q.; Zhao, N.; Shi, S.W.; Shen, H.P.; Mao, J.Q.; Zhang, L.Y.; et al. Diagnostic accuracy of a specific cytokine pattern in hemophagocytic lymphohistiocytosis in children. J. Pediatr. 2012, 160, 984–990.e1. [Google Scholar] [CrossRef] [PubMed]
- Strippoli, R.; Carvello, F.; Scianaro, R.; De Pasquale, L.; Vivarelli, M.; Petrini, S.; Bracci-Laudiero, L.; De Benedetti, F. Amplification of the response to Toll-like receptor ligands by prolonged exposure to interleukin-6 in mice: Implication for the pathogenesis of macrophage activation syndrome. Arthritis Rheum. 2012, 64, 1680–1688. [Google Scholar] [CrossRef] [PubMed]
- Musso, T.; Espinoza-Delgado, I.; Pulkki, K.; Gusella, G.L.; Longo, D.L.; Varesio, L. Transforming growth factor beta downregulates interleukin-1 (IL-1)-induced IL-6 production by human monocytes. Blood 1990, 76, 2466–2469. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Kullberg, B.J.; Verschueren, I.; Van Der Meer, J.W. Interleukin-18 induces production of proinflammatory cytokines in mice: No intermediate role for the cytokines of the tumor necrosis factor family and interleukin-1beta. Eur. J. Immunol. 2000, 30, 3057–3060. [Google Scholar] [CrossRef]
- Mazodier, K.; Marin, V.; Novick, D.; Farnarier, C.; Robitail, S.; Schleinitz, N.; Veit, V.; Paul, P.; Rubinstein, M.; Dinarello, C.A.; et al. Severe imbalance of IL-18/IL-18BP in patients with secondary hemophagocytic syndrome. Blood 2005, 106, 3483–3489. [Google Scholar] [CrossRef]
- Maeno, N.; Takei, S.; Imanaka, H.; Yamamoto, K.; Kuriwaki, K.; Kawano, Y.; Oda, H. Increased interleukin-18 expression in bone marrow of a patient with systemic juvenile idiopathic arthritis and unrecognized macrophage-activation syndrome. Arthritis Rheum. 2004, 50, 1935–1938. [Google Scholar] [CrossRef]
- Kawashima, M.; Yamamura, M.; Taniai, M.; Yamauchi, H.; Tanimoto, T.; Kurimoto, M.; Miyawaki, S.; Amano, T.; Takeuchi, T.; Makino, H. Levels of interleukin-18 and its binding inhibitors in the blood circulation of patients with adult-onset Still’s disease. Arthritis Rheum. 2001, 44, 550–560. [Google Scholar] [CrossRef]
- Novick, D.; Schwartsburd, B.; Pinkus, R.; Suissa, D.; Belzer, I.; Sthoeger, Z.; Keane, W.F.; Chvatchko, Y.; Kim, S.H.; Fantuzzi, G.; et al. A novel IL-18BP ELISA shows elevated serum IL-18BP in sepsis and extensive decrease of free IL-18. Cytokine 2001, 14, 334–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novick, D.; Elbirt, D.; Miller, G.; Dinarello, C.A.; Rubinstein, M.; Sthoeger, Z.M. High circulating levels of free interleukin-18 in patients with active SLE in the presence of elevated levels of interleukin-18 binding protein. J. Autoimmun. 2010, 34, 121–126. [Google Scholar] [CrossRef]
- Favilli, F.; Anzilotti, C.; Martinelli, L.; Quattroni, P.; De Martino, S.; Pratesi, F.; Neumann, D.; Beermann, S.; Novick, D.; Dinarello, C.A.; et al. IL-18 activity in systemic lupus erythematosus. Ann. N. Y. Acad. Sci. 2009, 1173, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Put, K.; Avau, A.; Brisse, E.; Mitera, T.; Put, S.; Proost, P.; Bader-Meunier, B.; Westhovens, R.; Van den Eynde, B.J.; Orabona, C.; et al. Cytokines in systemic juvenile idiopathic arthritis and haemophagocytic lymphohistiocytosis: Tipping the balance between interleukin-18 and interferon-γ. Rheumatology 2015, 54, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, S.K.; Chu, P.G.; Weiss, L.M. CD163: A specific marker of macrophages in paraffin-embedded tissue samples. Am. J. Clin. Pathol. 2004, 122, 794–801. [Google Scholar] [CrossRef]
- Sakumura, N.; Shimizu, M.; Mizuta, M.; Inoue, N.; Nakagishi, Y.; Yachie, A. Soluble CD163, a unique biomarker to evaluate the disease activity, exhibits macrophage activation in systemic juvenile idiopathic arthritis. Cytokine 2018, 110, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Gullestad, L.; Ueland, T.; Vinge, L.E.; Finsen, A.; Yndestad, A.; Aukrust, P. Inflammatory cytokines in heart failure: Mediators and markers. Cardiology 2012, 122, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Kostopoulou, M.; Nikolopoulos, D.; Parodis, I.; Bertsias, G. Cardiovascular Disease in Systemic Lupus Erythematosus: Recent Data on Epidemiology, Risk Factors and Prevention. Curr. Vasc. Pharmacol. 2020, 18, 549–565. [Google Scholar] [CrossRef]
- Clin spectrum Gavand, P.E.; Serio, I.; Arnaud, L.; Costedoat-Chalumeau, N.; Carvelli, J.; Dossier, A.; Hinschberger, O.; Mouthon, L.; Le Guern, V.; Korganow, A.S.; et al. Clinical spectrum and therapeutic management of systemic lupus erythematosus-associated macrophage activation syndrome: A study of 103 episodes in 89 adult patients. Autoimmun. Rev. 2017, 16, 743–749. [Google Scholar] [CrossRef]
- Dhakal, B.P.; Kim, C.H.; Al-Kindi, S.G.; Oliveira, G.H. Heart failure in systemic lupus erythematosus. Trends Cardiovasc. Med. 2018, 28, 187–197. [Google Scholar] [CrossRef]
- Kim, C.H.; Al-Kindi, S.G.; Jandali, B.; Askari, A.D.; Zacharias, M.; Oliveira, G.H. Incidence and risk of heart failure in systemic lupus erythematosus. Heart 2017, 103, 227–233. [Google Scholar] [CrossRef]
- Bartels, C.M.; Buhr, K.A.; Goldberg, J.W.; Bell, C.L.; Visekruna, M.; Nekkanti, S.; Greenlee, R.T. Mortality and cardiovascular burden of systemic lupus erythematosus in a US population-based cohort. J. Rheumatol. 2014, 41, 680–687. [Google Scholar] [CrossRef]
- Fernández-Nebro, A.; Rúa-Figueroa, Í.; López-Longo, F.J.; Galindo-Izquierdo, M.; Calvo-Alén, J.; Olivé-Marqués, A.; Ordóñez-Cañizares, C.; Martín-Martínez, M.A.; Blanco, R.; Melero-González, R.; et al. Cardiovascular Events in Systemic Lupus Erythematosus: A Nationwide Study in Spain from the RELESSER Registry. Medicine 2015, 94, e1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuna, J.; Żuber, Z.; Chmielewski, G.; Gromadziński, L.; Krajewska-Włodarczyk, M. Role of Distinct Macrophage Populations in the Development of Heart Failure in Macrophage Activation Syndrome. Int. J. Mol. Sci. 2022, 23, 2433. https://doi.org/10.3390/ijms23052433
Kuna J, Żuber Z, Chmielewski G, Gromadziński L, Krajewska-Włodarczyk M. Role of Distinct Macrophage Populations in the Development of Heart Failure in Macrophage Activation Syndrome. International Journal of Molecular Sciences. 2022; 23(5):2433. https://doi.org/10.3390/ijms23052433
Chicago/Turabian StyleKuna, Jakub, Zbigniew Żuber, Grzegorz Chmielewski, Leszek Gromadziński, and Magdalena Krajewska-Włodarczyk. 2022. "Role of Distinct Macrophage Populations in the Development of Heart Failure in Macrophage Activation Syndrome" International Journal of Molecular Sciences 23, no. 5: 2433. https://doi.org/10.3390/ijms23052433