
Production of an Active, Human Membrane Protein in Saccharomyces cerevisiae: Full-Length FICD



Abstract
:1. Introduction
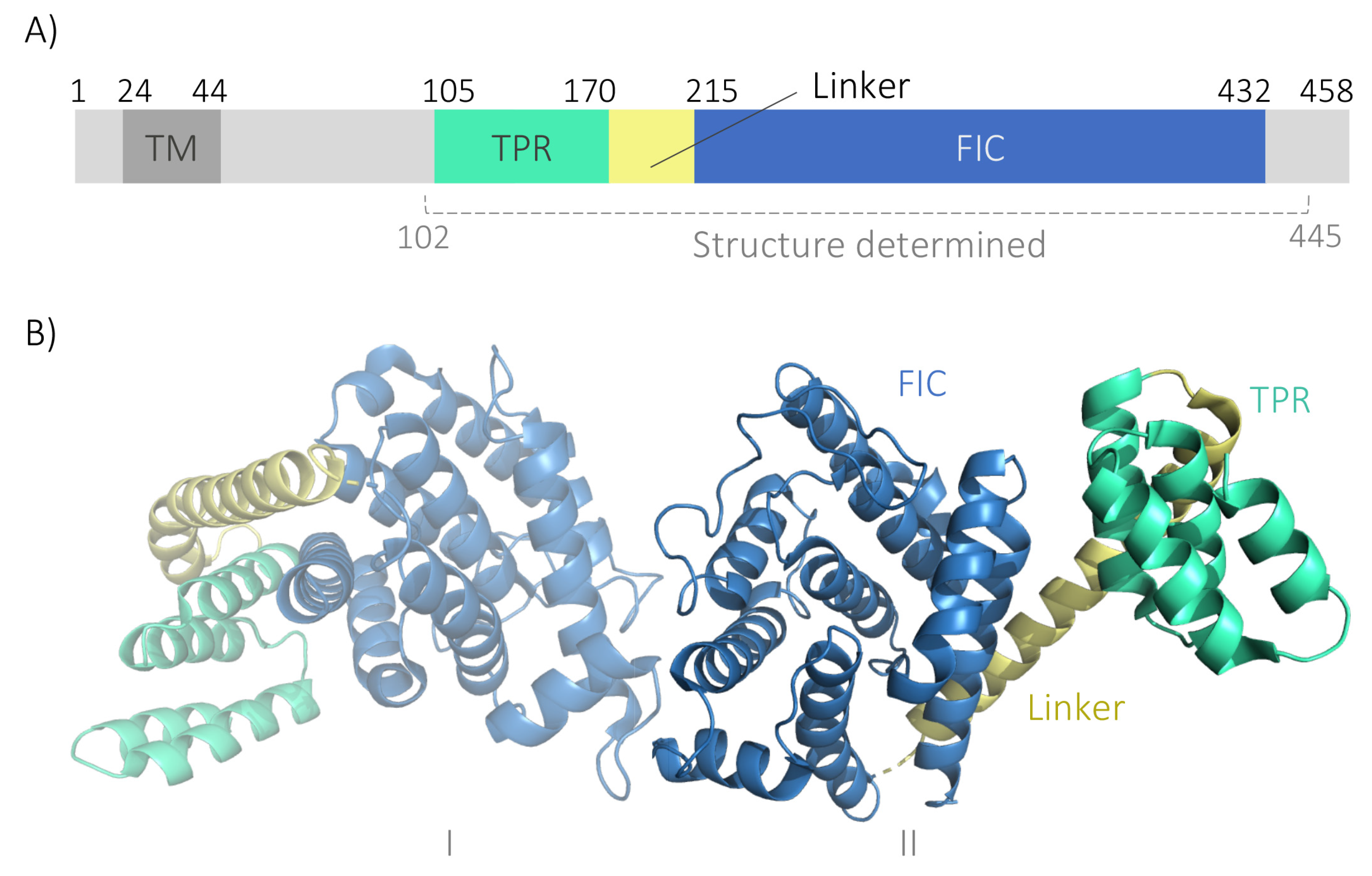
2. Results
2.1. Recombinant GFP-FICD Is Expressed to a High Density in S. cerevisiae
2.2. GFP-FICD Likely Localises to the Golgi
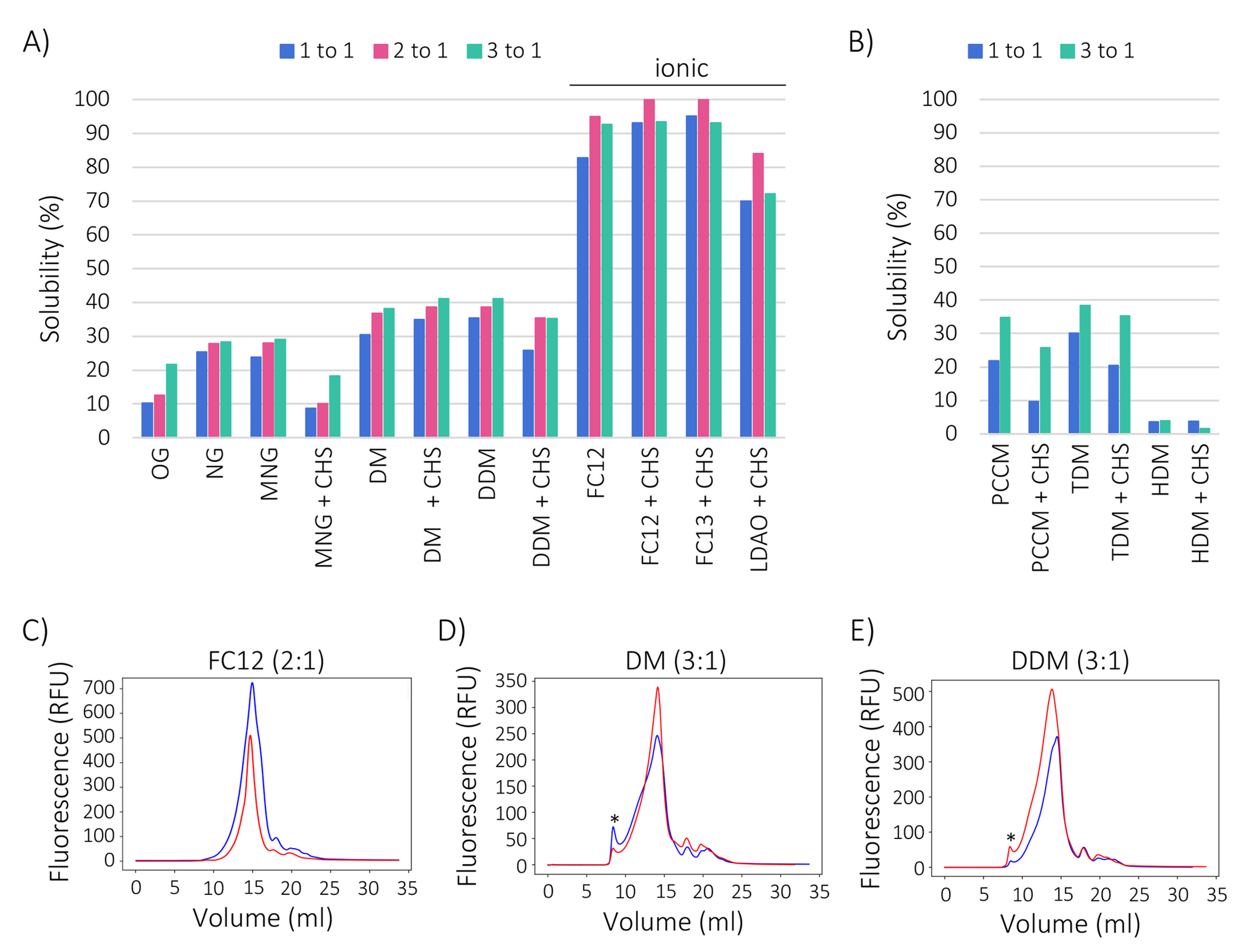
2.3. Zwitterionic Detergents Recover More GFP-FICD Than Non-Ionic Detergents
2.4. Cholesteryl Hemisuccinate Improves GFP-FICD Homogeneity
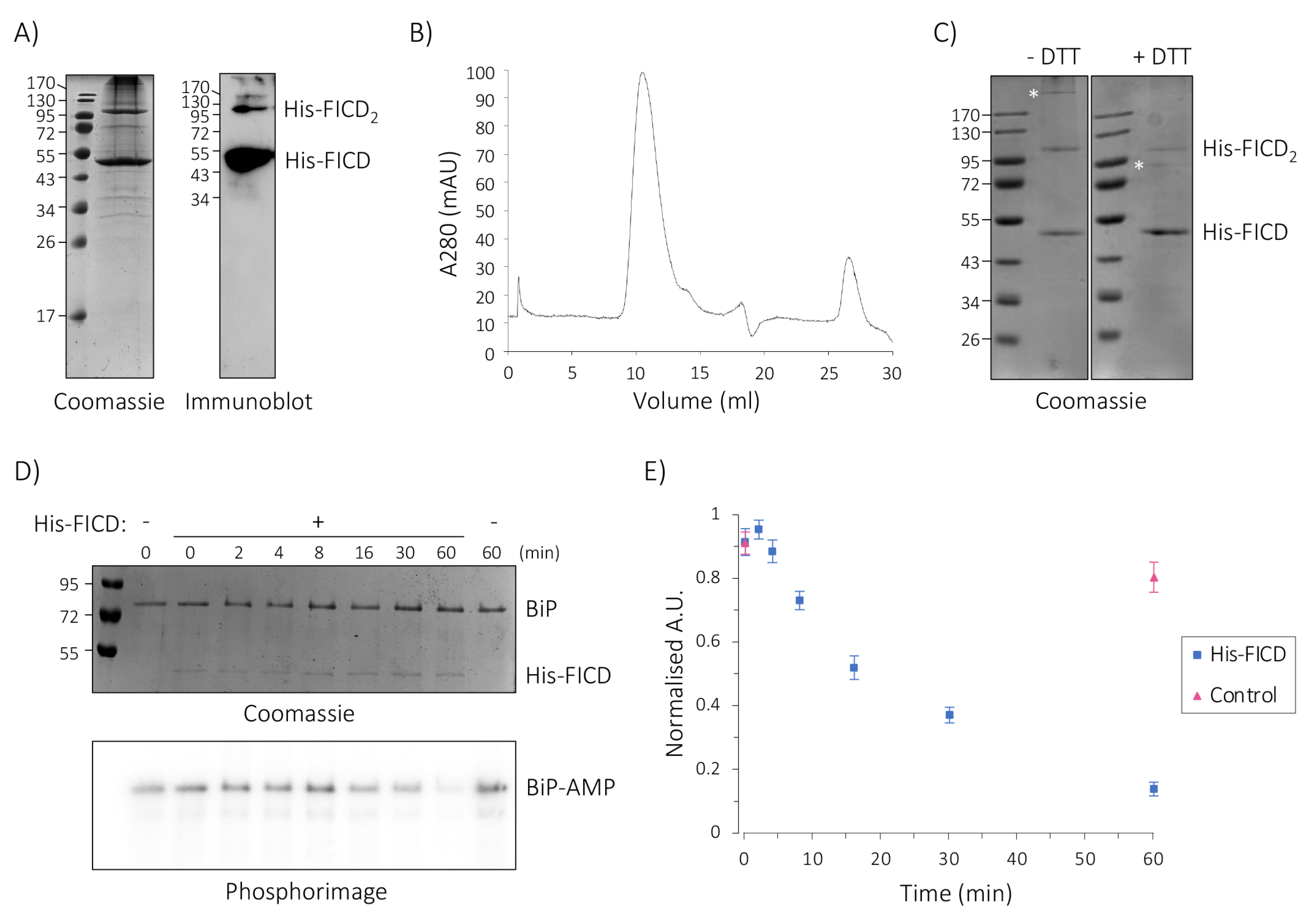
2.5. His-FICD Can Be Purified in a Folded Conformation
2.6. Purified Full-Length His-FICD Is an Active deAMPylase
3. Discussion
4. Materials and Methods
4.1. Yeast Strains
4.2. Construction of S. cerevisiae Expression Plasmids
4.3. Protein Expression in S. cerevisiae
4.4. Isolation of Yeast Membranes
4.5. Detergent Screen
4.6. Large-Scale Purification of His8-FICD
4.7. Whole-Cell Fluorescence and Live Cell Bioimaging
4.8. Quantification of His8-GFP-TEV-FICD Expression
4.9. SDS-PAGE, in-Gel Fluorescence, and Immunoblotting
4.10. Construction of E. coli Expression Plasmids
4.11. Expression and Lysis of Soluble Proteins in E. coli
4.12. Purification of Soluble Proteins
4.13. Preparation of AMPylated BiP
4.14. DeAMPylation Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Garcia-Pino, A.; Zenkin, N.; Loris, R. The many faces of Fic: Structural and functional aspects of Fic enzymes. Trends Biochem. Sci. 2014, 39, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Faber, P.W.; Barnes, G.T.; Srinidhi, J.; Chen, J.; Gusella, J.F.; MacDonald, M.E. Huntingtin interacts with a family of WW domain proteins. Hum. Mol. Genet. 1998, 7, 1463–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khater, S.; Mohanty, D. In silico identification of AMPylating enzymes and study of their divergent evolution. Sci. Rep. 2015, 5, 10804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanyal, A.; Chen, A.J.; Nakayasu, E.S.; Lazar, C.S.; Zbornik, E.A.; Worby, C.A.; Koller, A.; Mattoo, S. A novel link between Fic (filamentation induced by cAMP)-mediated adenylylation/AMPylation and the unfolded protein response. J. Biol. Chem. 2015, 290, 8482–8499. [Google Scholar] [CrossRef] [Green Version]
- Ham, H.; Woolery, A.R.; Tracy, C.; Stenesen, D.; Kramer, H.; Orth, K. Unfolded protein response-regulated Drosophila Fic (dFic) protein reversibly AMPylates BiP chaperone during endoplasmic reticulum homeostasis. J. Biol. Chem. 2014, 289, 36059–36069. [Google Scholar] [CrossRef] [Green Version]
- Kielkowski, P.; Buchsbaum, I.Y.; Kirsch, V.C.; Bach, N.C.; Drukker, M.; Cappello, S.; Sieber, S.A. FICD activity and AMPylation remodelling modulate human neurogenesis. Nat. Commun. 2020, 11, 517. [Google Scholar] [CrossRef] [Green Version]
- Truttmann, M.C.; Wu, Q.; Stiegeler, S.; Duarte, J.N.; Ingram, J.; Ploegh, H.L. HypE-specific nanobodies as tools to modulate HypE-mediated target AMPylation. J. Biol. Chem. 2015, 290, 9087–9100. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, R.; Poderycki, M.J.; Mattoo, S. CryoAPEX—An electron tomography tool for subcellular localization of membrane proteins. J. Cell Sci. 2019, 132, jcs222315. [Google Scholar] [CrossRef] [Green Version]
- Preissler, S.; Rato, C.; Perera, L.; Saudek, V.; Ron, D. FICD acts bifunctionally to AMPylate and de-AMPylate the endoplasmic reticulum chaperone BiP. Nat. Struct. Mol. Biol. 2017, 24, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Perera, L.A.; Rato, C.; Yan, Y.; Neidhardt, L.; McLaughlin, S.H.; Read, R.J.; Preissler, S.; Ron, D. An oligomeric state-dependent switch in the ER enzyme FICD regulates AMPylation and deAMPylation of BiP. EMBO J. 2019, 38, e102177. [Google Scholar] [CrossRef]
- Perera, L.A.; Preissler, S.; Zaccai, N.R.; Prévost, S.; Devos, J.M.; Haertlein, M.; Ron, D. Structures of a deAMPylation complex rationalise the switch between antagonistic catalytic activities of FICD. Nat. Commun. 2021, 12, 5004. [Google Scholar] [CrossRef]
- Bunney, T.D.; Cole, A.R.; Broncel, M.; Esposito, D.; Tate, E.W.; Katan, M. Crystal structure of the human, FIC-domain containing protein HYPE and implications for its functions. Structure 2014, 22, 1831–1843. [Google Scholar] [CrossRef] [Green Version]
- Preissler, S.; Rato, C.; Chen, R.; Antrobus, R.; Ding, S.; Fearnley, I.M.; Ron, D. AMPylation matches BiP activity to client protein load in the endoplasmic reticulum. Elife 2015, 4, e12621. [Google Scholar] [CrossRef] [PubMed]
- Preissler, S.; Ron, D. Early Events in the Endoplasmic Reticulum Unfolded Protein Response. Cold Spring Harb. Perspect. Biol. 2019, 11, a033894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieber, S.A.; Cappello, S.; Kielkowski, P. From Young to Old: AMPylation Hits the Brain. Cell Chem. Biol. 2020, 27, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Truttmann, M.C.; Pincus, D.; Ploegh, H.L. Chaperone AMPylation modulates aggregation and toxicity of neurodegenerative disease-associated polypeptides. Proc. Natl. Acad. Sci. USA 2018, 115, E5008–E5017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.; Ham, H.; Liu, X.; Sugiura, Y.; Orth, K.; Kramer, H. Visual neurotransmission in Drosophila requires expression of Fic in glial capitate projections. Nat. Neurosci. 2012, 15, 871–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanyal, A.; Dutta, S.; Camara, A.; Chandran, A.; Koller, A.; Watson, B.G.; Sengupta, R.; Ysselstein, D.; Montenegro, P.; Cannon, J.; et al. Alpha-Synuclein Is a Target of Fic-Mediated Adenylylation/AMPylation: Possible Implications for Parkinson’s Disease. J. Mol. Biol. 2019, 431, 2266–2282. [Google Scholar] [CrossRef]
- McCaul, N.; Porter, C.M.; Becker, A.; Tang, C.-H.A.; Wijne, C.; Chatterjee, B.; Bousbaine, D.; Bilate, A.; Hu, C.-C.A.; Ploegh, H.; et al. Deletion of mFICD AMPylase alters cytokine secretion and affects visual short-term learning in vivo. J. Biol. Chem. 2021, 297, 100991. [Google Scholar] [CrossRef]
- Hendershot, L.M. The ER function BiP is a master regulator of ER function. Mt. Sinai J. Med. 2004, 71, 289–297. [Google Scholar]
- Preissler, S.; Rohland, L.; Yan, Y.; Chen, R.; Read, R.J.; Ron, D. AMPylation targets the rate-limiting step of BiP’s ATPase cycle for its functional inactivation. Elife 2017, 6, e29428. [Google Scholar] [CrossRef]
- Fauser, J.; Gulen, B.; Pogenberg, V.; Pett, C.; Pourjafar-Dehkordi, D.; Krisp, C.; Hopfner, D.; Konig, G.; Schluter, H.; Feige, M.J.; et al. Specificity of AMPylation of the human chaperone BiP is mediated by TPR motifs of FICD. Nat. Commun. 2021, 12, 2426. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System; Version 2.1.1; Schrödinger, LLC.: New York, NY, USA, 2018.
- Fagerberg, L.; Jonasson, K.; von Heijne, G.; Uhlen, M.; Berglund, L. Prediction of the human membrane proteome. Proteomics 2010, 10, 1141–1149. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Stevens, T.J.; Arkin, I.T. Do more complex organisms have a greater proportion of membrane proteins in their genomes? Proteins 2000, 39, 417–420. [Google Scholar] [CrossRef]
- White, S.H. Membrane Proteins of Known Structure. Available online: https://blanco.biomol.uci.edu/mpstruc/ (accessed on 6 October 2021).
- Tusnady, G.E.; Dosztanyi, Z.; Simon, I. Transmembrane proteins in the Protein Data Bank: Identification and classification. Bioinformatics 2004, 20, 2964–2972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.T. Do transmembrane protein superfolds exist? FEBS Lett. 1998, 423, 281–285. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Rost, B. Comparing function and structure between entire proteomes. Protein Sci. 2001, 10, 1970–1979. [Google Scholar] [CrossRef] [Green Version]
- Orr-Weaver, T.L.; Szostak, J.W. Fungal recombination. Microbiol. Rev. 1985, 49, 33–58. [Google Scholar] [CrossRef]
- Bomholt, J.; Helix-Nielsen, C.; Scharff-Poulsen, P.; Pedersen, P.A. Recombinant production of human Aquaporin-1 to an exceptional high membrane density in Saccharomyces cerevisiae. PLoS ONE 2013, 8, e56431. [Google Scholar] [CrossRef] [Green Version]
- Molbaek, K.; Scharff-Poulsen, P.; Helix-Nielsen, C.; Klaerke, D.A.; Pedersen, P.A. High yield purification of full-length functional hERG K+ channels produced in Saccharomyces cerevisiae. Microb. Cell Fact. 2015, 14, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, P.A.; Rasmussen, J.H.; Joorgensen, P.L. Expression in high yield of pig alpha 1 beta 1 Na,K-ATPase and inactive mutants D369N and D807N in Saccharomyces cerevisiae. J. Biol. Chem. 1996, 271, 2514–2522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharff-Poulsen, P.; Pedersen, P.A. Saccharomyces cerevisiae-based platform for rapid production and evaluation of eukaryotic nutrient transporters and transceptors for biochemical studies and crystallography. PLoS ONE 2013, 8, e76851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preisler, S.S.; Wiuf, A.D.; Friis, M.; Kjaergaard, L.; Hurd, M.; Becares, E.R.; Nurup, C.N.; Bjoerkskov, F.B.; Szathmáry, Z.; Gourdon, P.E.; et al. Saccharomyces cerevisiae as a superior host for overproduction of prokaryotic integral membrane proteins. Curr. Res. Struct. Biol. 2021, 3, 51–71. [Google Scholar] [CrossRef]
- Bjorkskov, F.B.; Krabbe, S.L.; Nurup, C.N.; Missel, J.W.; Spulber, M.; Bomholt, J.; Molbaek, K.; Helix-Nielsen, C.; Gotfryd, K.; Gourdon, P.; et al. Purification and functional comparison of nine human Aquaporins produced in Saccharomyces cerevisiae for the purpose of biophysical characterization. Sci. Rep. 2017, 7, 16899. [Google Scholar] [CrossRef] [Green Version]
- Hansen, M.K.; Tams, J.W.; Fahrenkrug, J.; Pedersen, P.A. Functional expression of rat VPAC1 receptor in Saccharomyces cerevisiae. Recept. Channels 1999, 6, 271–281. [Google Scholar]
- Lenoir, G.; Menguy, T.; Corre, F.; Montigny, C.; Pedersen, P.A.; Thines, D.; le Maire, M.; Falson, P. Overproduction in yeast and rapid and efficient purification of the rabbit SERCA1a Ca(2+)-ATPase. Biochim. Biophys. Acta 2002, 1560, 67–83. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, K.; Klaerke, D.A.; Calloe, K.; Lowrey, L.; Pedersen, P.A.; Gourdon, P.; Gotfryd, K. Purification of Functional Human TRP Channels Recombinantly Produced in Yeast. Cells 2019, 8, 148. [Google Scholar] [CrossRef] [Green Version]
- Mirzadeh, K.; Martinez, V.; Toddo, S.; Guntur, S.; Herrgard, M.J.; Elofsson, A.; Norholm, M.H.; Daley, D.O. Enhanced Protein Production in Escherichia coli by Optimization of Cloning Scars at the Vector-Coding Sequence Junction. ACS Synth. Biol. 2015, 4, 959–965. [Google Scholar] [CrossRef]
- Szostak, J.W.; Orr-Weaver, T.L.; Rothstein, R.J.; Stahl, F.W. The double-strand-break repair model for recombination. Cell 1983, 33, 25–35. [Google Scholar] [CrossRef]
- Ma, H.; Kunes, S.; Schatz, P.J.; Botstein, D. Plasmid construction by homologous recombination in yeast. Gene 1987, 58, 201–216. [Google Scholar] [CrossRef]
- Baldari, C.; Cesareni, G. Plasmids pEMBLY: New single-stranded shuttle vectors for the recovery and analysis of yeast DNA sequences. Gene 1985, 35, 27–32. [Google Scholar] [CrossRef]
- Erhart, E.; Hollenberg, C.P. The presence of a defective LEU2 gene on 2 mu DNA recombinant plasmids of Saccharomyces cerevisiae is responsible for curing and high copy number. J. Bacteriol. 1983, 156, 625–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotfryd, K.; Mosca, A.F.; Missel, J.W.; Truelsen, S.F.; Wang, K.; Spulber, M.; Krabbe, S.; Helix-Nielsen, C.; Laforenza, U.; Soveral, G.; et al. Human adipose glycerol flux is regulated by a pH gate in AQP10. Nat. Commun. 2018, 9, 4749. [Google Scholar] [CrossRef] [Green Version]
- Drew, D.; Newstead, S.; Sonoda, Y.; Kim, H.; von Heijne, G.; Iwata, S. GFP-based optimization scheme for the overexpression and purification of eukaryotic membrane proteins in Saccharomyces cerevisiae. Nat. Protoc. 2008, 3, 784–798. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Preisler, S.S.; Zhang, L.; Cui, Y.; Missel, J.W.; Gronberg, C.; Gotfryd, K.; Lindahl, E.; Andersson, M.; Calloe, K.; et al. Structure of the human ClC-1 chloride channel. PLoS Biol. 2019, 17, e3000218. [Google Scholar] [CrossRef] [Green Version]
- Kassem, N.; Araya-Secchi, R.; Bugge, K.; Barclay, A.; Steinocher, H.; Khondker, A.; Wang, Y.; Lenard, A.J.; Burck, J.; Sahin, C.; et al. Order and disorder-An integrative structure of the full-length human growth hormone receptor. Sci. Adv. 2021, 7, eabh3805. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, Z.T.; Tang, S.W.; Zhao, B.S.; Li, H.; Song, J.Z.; Li, D.; Xie, Z. A Validated Set of Fluorescent-Protein-Based Markers for Major Organelles in Yeast (Saccharomyces cerevisiae). mBio 2019, 10, e01691-19. [Google Scholar] [CrossRef]
- Antebi, A.; Fink, G.R. The yeast Ca(2+)-ATPase homologue, PMR1, is required for normal Golgi function and localizes in a novel Golgi-like distribution. Mol. Biol. Cell 1992, 3, 633–654. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, K.; Fink, G. Calcineurin-dependent growth control in Saccharomyces cerevisiae mutants lacking PMC1, a homolog of plasma membrane Ca2+ ATPases. J. Cell Biol. 1994, 124, 351–363. [Google Scholar] [CrossRef]
- Geertsma, E.R.; Groeneveld, M.; Slotboom, D.J.; Poolman, B. Quality control of overexpressed membrane proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 5722–5727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, J.R.; Reithmeier, R.A.F. Detergent interaction with band 3, a model polytopic membrane protein. Biochemistry 1993, 32, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Barth, H.G.; Jackson, C.; Boyes, B.E. Size Exclusion Chromatography. Anal. Chem. 1994, 66, 595–620. [Google Scholar] [CrossRef]
- Wang, D.-N.; Lemieux, M.J.; Boulter, J.M. Purification and Characterization of Transporter Proteins from Human Erythrocyte Membrane. In Membrane Protein Protocols; Humana Press: Totowa, NJ, USA, 2003; Volume 228, pp. 239–256. [Google Scholar]
- Casey, J.R.; Reithmeier, R.A. Analysis of the oligomeric state of Band 3, the anion transport protein of the human erythrocyte membrane, by size exclusion high performance liquid chromatography. Oligomeric stability and origin of heterogeneity. J. Biol. Chem. 1991, 266, 15726–15737. [Google Scholar] [CrossRef]
- Stetsenko, A.; Guskov, A. An Overview of the Top Ten Detergents Used for Membrane Protein Crystallization. Crystals 2017, 7, 197. [Google Scholar] [CrossRef] [Green Version]
- Javitch, J.A. The Ants Go Marching Two by Two: Oligomeric Structure of G-Protein-Coupled Receptors. Mol. Pharmacol. 2004, 66, 1077–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soulié, S.; Møller, J.V.; Falson, P.; le Maire, M. Urea Reduces the Aggregation of Membrane Proteins on Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis. Anal. Biochem. 1996, 236, 363–364. [Google Scholar] [CrossRef]
- Casey, A.K.; Moehlman, A.T.; Zhang, J.; Servage, K.A.; Kramer, H.; Orth, K. Fic-mediated deAMPylation is not dependent on homodimerization and rescues toxic AMPylation in flies. J. Biol. Chem. 2017, 292, 21193–21204. [Google Scholar] [CrossRef] [Green Version]
- Granseth, E.; Seppala, S.; Rapp, M.; Daley, D.O.; Von Heijne, G. Membrane protein structural biology—How far can the bugs take us? Mol. Membr. Biol. 2007, 24, 329–332. [Google Scholar] [CrossRef]
- Michelsen, K.; Yuan, H.; Schwappach, B. Hide and run. Arginine-based endoplasmic-reticulum-sorting motifs in the assembly of heteromultimeric membrane proteins. EMBO Rep. 2005, 6, 717–722. [Google Scholar] [CrossRef]
- Roth, D.; Lynes, E.; Riemer, J.; Hansen, H.G.; Althaus, N.; Simmen, T.; Ellgaard, L. A di-arginine motif contributes to the ER localization of the type I transmembrane ER oxidoreductase TMX4. Biochem. J. 2010, 425, 195–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skaar, K.; Korza, H.J.; Tarry, M.; Sekyrova, P.; Högbom, M. Expression and Subcellular Distribution of GFP-Tagged Human Tetraspanin Proteins in Saccharomyces cerevisiae. PLoS ONE 2015, 10, e0134041. [Google Scholar] [CrossRef] [PubMed]
- Drew, D.E.; von Heijne, G.; Nordlund, P.; de Gier, J.-W.L. Green fluorescent protein as an indicator to monitor membrane protein overexpression in Escherichia coli. FEBS Lett. 2001, 507, 220–224. [Google Scholar] [CrossRef] [Green Version]
- Lipfert, J.; Columbus, L.; Chu, V.B.; Lesley, S.A.; Doniach, S. Size and Shape of Detergent Micelles Determined by Small-Angle X-ray Scattering. J. Phys. Chem. B 2007, 111, 12427–12438. [Google Scholar] [CrossRef]
- Dorr, J.M.; Scheidelaar, S.; Koorengevel, M.C.; Dominguez, J.J.; Schafer, M.; van Walree, C.A.; Killian, J.A. The styrene-maleic acid copolymer: A versatile tool in membrane research. Eur. Biophys. J. 2016, 45, 3–21. [Google Scholar] [CrossRef] [Green Version]
- Denisov, I.G.; Sligar, S.G. Nanodiscs for structural and functional studies of membrane proteins. Nat. Struct. Mol. Biol. 2016, 23, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Arkin, I.T. Structural aspects of oligomerization taking place between the transmembrane alpha-helices of bitopic membrane proteins. Biochim. Biophys. Acta 2002, 1565, 347–363. [Google Scholar] [CrossRef] [Green Version]
- Kohlway, A.; Pirakitikulr, N.; Barrera, F.N.; Potapova, O.; Engelman, D.M.; Pyle, A.M.; Lindenbach, B.D. Hepatitis C Virus RNA Replication and Virus Particle Assembly Require Specific Dimerization of the NS4A Protein Transmembrane Domain. J. Virol. 2014, 88, 628–642. [Google Scholar] [CrossRef] [Green Version]
- Bugge, K.; Lindorff-Larsen, K.; Kragelund, B.B. Understanding single-pass transmembrane receptor signaling from a structural viewpoint-what are we missing? FEBS J. 2016, 283, 4424–4451. [Google Scholar] [CrossRef] [Green Version]
- Pahl, M.C.; Askinazi, O.L.; Hamilton, C.; Cheng, I.; Cichewicz, K.; Kuhn, J.; Manohar, S.; Deppmann, C.D. Signalling via Single-Pass Transmembrane Proteins. In eLS; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2013. [Google Scholar]
- Stavropoulos, I.; Khaldi, N.; Davey, N.E.; O’Brien, K.; Martin, F.; Shields, D.C. Protein Disorder and Short Conserved Motifs in Disordered Regions Are Enriched near the Cytoplasmic Side of Single-Pass Transmembrane Proteins. PLoS ONE 2012, 7, e44389. [Google Scholar] [CrossRef] [Green Version]
- Cormack, B.P.; Bertram, G.; Egerton, M.; Gow, N.A.R.; Falkow, S.; Brown, A.J.P. Yeast-enhanced green fluorescent protein (yEGFP): A reporter of gene expression in Candida albicans. Microbiology 1997, 143, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gietz, R.D.; Schiestl, R.H. Frozen competent yeast cells that can be transformed with high efficiency using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2007, 2, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Wickerham, L.J. Taxonomy of Yeasts; US Department of Agriculture: Washington, DC, USA, 1951.
- Jorgensen, J.R.; Pedersen, P.A. Role of phylogenetically conserved amino acids in folding of Na,K-ATPase. Biochemistry 2001, 40, 7301–7308. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Rogov, V.V.; Rozenknop, A.; Rogova, N.Y.; Löhr, F.; Tikole, S.; Jaravine, V.; Güntert, P.; Dikic, I.; Dötsch, V. A Universal Expression Tag for Structural and Functional Studies of Proteins. Chembiochem 2012, 13, 959–963. [Google Scholar] [CrossRef]
- Gaut, J.R.; Hendershot, L.M. Mutations within the nucleotide binding site of immunoglobulin-binding protein inhibit ATPase activity and interfere with release of immunoglobulin heavy chain. J. Biol. Chem. 1993, 268, 7248–7255. [Google Scholar] [CrossRef]
- McCarty, J.S.; Walker, G.C. DnaK as a thermometer: Threonine-199 is site of autophosphorylation and is critical for ATPase activity. Proc. Natl. Acad. Sci. USA 1991, 88, 9513–9517. [Google Scholar] [CrossRef] [Green Version]
- Palleros, D.R.; Raid, K.L.; Shi, L.; Welch, W.J.; Fink, A.L. ATP-induced protein Hsp70 complex dissociation requires K+ but not ATP hydrolysis. Nature 1993, 365, 664–666. [Google Scholar] [CrossRef]
- Wei, J.; Gaut, J.R.; Hendershot, L.M. In Vitro Dissociation of BiP-Peptide Complexes Requires a Conformational Change in BiP after ATP Binding but Does Not Require ATP Hydrolysis. J. Biol. Chem. 1995, 270, 26677–26682. [Google Scholar] [CrossRef] [Green Version]
- Laufen, T.; Mayer, M.P.; Beisel, C.; Klostermeier, D.; Mogk, A.; Reinstein, J.; Bukau, B. Mechanism of regulation of Hsp70 chaperones by DnaJ cochaperones. Proc. Natl. Acad. Sci. USA 1999, 96, 5452–5457. [Google Scholar] [CrossRef] [Green Version]
- Petrova, K.; Oyadomari, S.; Hendershot, L.M.; Ron, D. Regulated association of misfolded endoplasmic reticulum lumenal proteins with P58/DNAJc3. EMBO J. 2008, 27, 2862–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preissler, S.; Chambers, J.E.; Crespillo-Casado, A.; Avezov, E.; Miranda, E.; Perez, J.; Hendershot, L.M.; Harding, H.P.; Ron, D. Physiological modulation of BiP activity by trans-protomer engagement of the interdomain linker. Elife 2015, 4, e08961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, L.D.; Foged, M.M.; Albert, A.; Bertelsen, A.B.; Søltoft, C.L.; Robinson, S.D.; Petersen, S.V.; Purcell, A.W.; Olivera, B.M.; Norton, R.S.; et al. The three-dimensional structure of an H-superfamily conotoxin reveals a granulin fold arising from a common ICK cysteine framework. J. Biol. Chem. 2019, 294, 8745–8759. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Oliver, R.C.; Lipfert, J.; Fox, D.A.; Lo, R.H.; Kim, J.J.; Doniach, S.; Columbus, L. Tuning micelle dimensions and properties with binary surfactant mixtures. Langmuir 2014, 30, 13353–13361. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Detergent Name | Properties |
|---|---|---|
| MNG | 2,2-Dioctylpropane-1,3-bis ß-D-maltopyranoside | Non-ionic |
| DM | n-decyl ß-D-maltopyranoside | ″ |
| DDM | n-dodecyl ß-D-maltopyranoside | ″ |
| TDM | n-tridecyl ß-D-maltopyranoside | ″ |
| HDM | n-hexadecyl ß-D-maltopyranoside | ″ |
| OG | n-octyl ß-D-glucopyranoside | ″ |
| NG | n-nonyl-β-D-glucopyranoside | ″ |
| PCCM | 4-trans-(4-trans propylcyclohexyl)-cyclohexyl α-D-maltopyranoside | ″ |
| FC12 | n-dodecylphosphocholine | Ionic |
| FC13 | n-tridecylphosphocholine | ″ |
| LDAO | n-dodecyl-dimethylamine oxide | ″ |
| CHS | cholesteryl hemisuccinate | Ionic |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Virolainen, M.S.; Søltoft, C.L.; Pedersen, P.A.; Ellgaard, L. Production of an Active, Human Membrane Protein in Saccharomyces cerevisiae: Full-Length FICD. Int. J. Mol. Sci. 2022, 23, 2458. https://doi.org/10.3390/ijms23052458
Virolainen MS, Søltoft CL, Pedersen PA, Ellgaard L. Production of an Active, Human Membrane Protein in Saccharomyces cerevisiae: Full-Length FICD. International Journal of Molecular Sciences. 2022; 23(5):2458. https://doi.org/10.3390/ijms23052458
Chicago/Turabian StyleVirolainen, Minttu S., Cecilie L. Søltoft, Per A. Pedersen, and Lars Ellgaard. 2022. "Production of an Active, Human Membrane Protein in Saccharomyces cerevisiae: Full-Length FICD" International Journal of Molecular Sciences 23, no. 5: 2458. https://doi.org/10.3390/ijms23052458