1. Introduction
Currently available antiviral drugs are mainly based on direct-acting antivirals (DAAs), with only a few exceptions, such as interferon-based treatments and some host-directed antiviral (HDA) approaches in AIDS therapy. These DAAs, however, share certain disadvantages that often substantially limit their therapeutic benefit: (i) a common tendency to induce drug-resistant virus mutants; (ii) inhibitory activity in a virus-specific, but not in a broader manner spanning a spectrum of viruses and (iii) a limited clinical efficacy in several cases. Generally, antiviral DAA-type drugs are continuously subject to a developmental process of refinement and a stepwise replacement by next-generation candidates. A major improvement may be achieved by expanding options of antiviral treatment by integrating HDAs and drug combination treatment schemes. Very recently, the use of pharmaceutical kinase inhibitors (PKIs) in antiviral drug research, originally developed for cancer and inflammatory disease therapy, showed very promising success [
1,
2,
3,
4,
5]. As a current example, the PKI maribavir (MBV) has been approved for the therapy of human cytomegalovirus (HCMV) post-transplant disease (FDA, Nov. 2021) after the successful termination of clinical phase III studies ([
6,
7]; NCT02931539, NCT02927067, NCT00497796, NCT00411645). Thus, MBV constitutes the first PKI in antiviral treatment. Concerning the further application of PKIs, the area of antiherpesviral prophylaxis and therapy appears especially promising. This is due to an increasing amount of published data indicating that for some human pathogenic herpesviruses, potential target kinases of antiviral PKIs are not restricted to virus-encoded protein kinases, but also include virus-supportive regulatory host kinases, thus representing potential new antiviral targets.
Human cytomegalovirus (HCMV) represents the prototype species of
Betaherpesvirinae and an opportunistic human pathogen with a predominant, worldwide distribution. Seroprevalence ranges between 40% and 95% in the adult human population, dependent on socio-geographic factors [
8,
9]. In the immunocompetent host, infections with HCMV typically cause only mild symptoms or remain asymptomatic [
10]. In immunocompromised or immunonaïve individuals, however, HCMV infection can lead to significant morbidity and mortality, specifically in patients under antitumoral chemotherapy, stem cell/organ transplantation or coinfection with human immunodeficiency virus type 1 (HIV-1). Moreover, congenital HCMV infection (cCMV), which has been underestimated for a long time, is the main infection-based risk during pregnancy [
11,
12,
13]. Specifically, cCMV is responsible for a variety of symptoms from mild to severe or even life-threatening in the unborn or infants, mainly manifesting as acute or late-onset embryonal developmental defects [
14]. Notably, among the current repertoire of approved anti-HCMV drugs, only a small selection can be used for cCMV therapy, prevention and control. For the most part, the approved drugs are nucleoside/nucleotide or pyrophosphate analogs which intervene with the activity of viral genome replication, i.e., ganciclovir (GCV), its oral prodrug valganciclovir (VGCV), foscarnet (FOS) and cidofovir (CDV) [
15,
16]. Recently, letermovir (LMV, Prevymis
®), an inhibitor of the viral terminase, has been clinically approved, albeit exclusively for the prophylaxis of HCMV infection in recipients of hematopoietic stem cell transplantation [
17,
18]. Now, after decades of development, maribavir (MBV/Livtencity
®) could also be added to the panel of available anti-HCMV drugs. Nevertheless, this panel still faces substantial limitations, such as the induction of viral drug resistance and, in the case of VGCV standard therapy, severe side effects including nephrotoxicity, myelotoxicity and anemia, often limiting their therapeutic compatibility and use in long-term treatments [
19,
20].
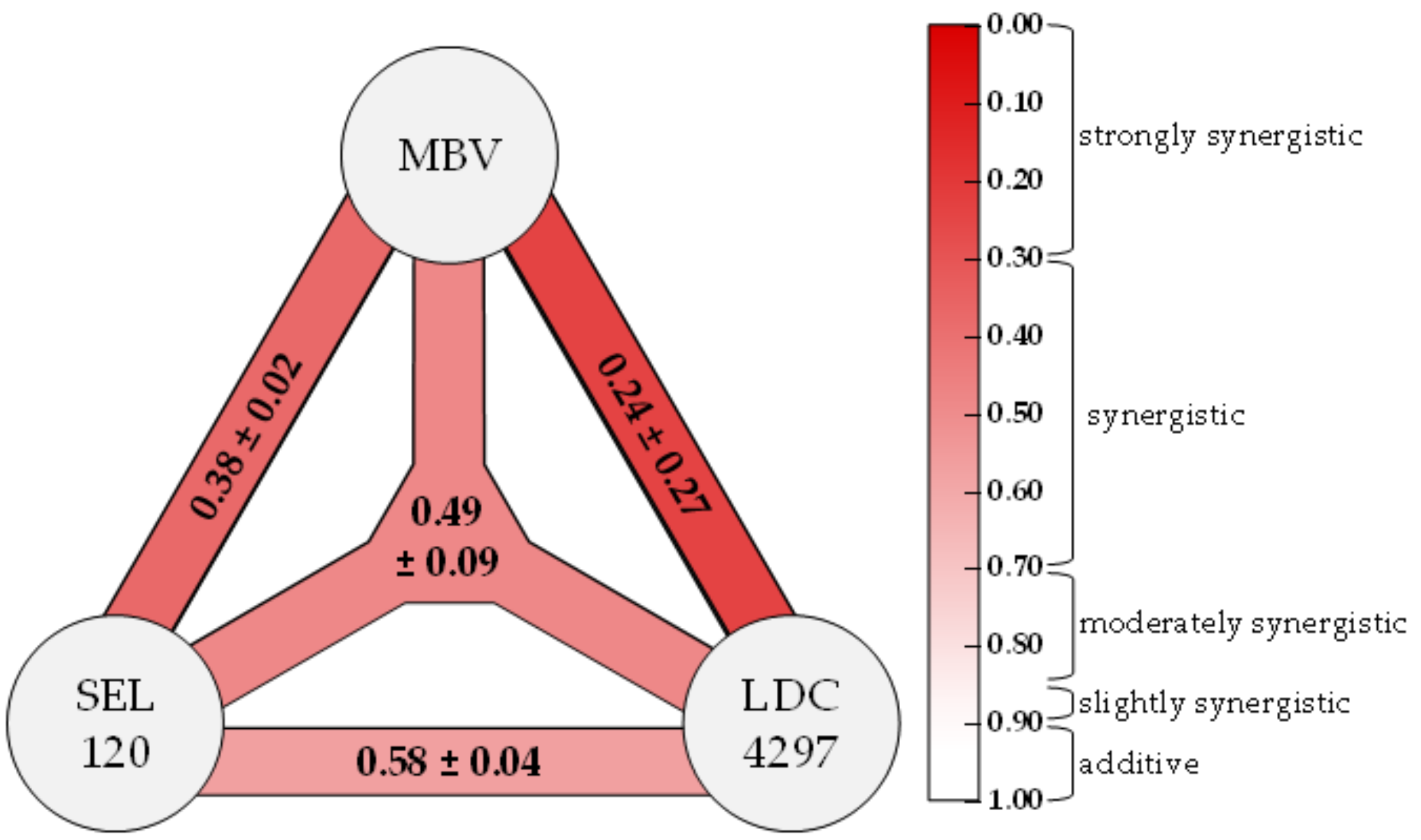
Our approach focuses on novel drug application schemes and targeting strategies, i.e., the development of mechanistically new antiviral drug candidates and so far unexploited targeting approaches. The HCMV-specific antiviral potential of the CDK7 inhibitor LDC4297 has already been demonstrated in our previous studies. Specifically, its mode of action (MoA) has been characterized as a complex inhibitory effect resulting from interference with CDK7 activity, i.e., an inhibition of the immediate early phase of viral replication and cell cycle modulation through altered Rb phosphorylation [
21]. For the HDA LDC4297, a strong synergistic drug interaction with DAA MBV was described in our previous work [
1]. This synergism between two PKIs was further characterized in the current study, including investigations into its molecular mechanistic parameters. Furthermore, we extended our investigations into the promising antiviral activity of additional CDK inhibitors, vCDK/pUL97 inhibitors and, in particular, new synergistic drug combinations.
3. Discussion
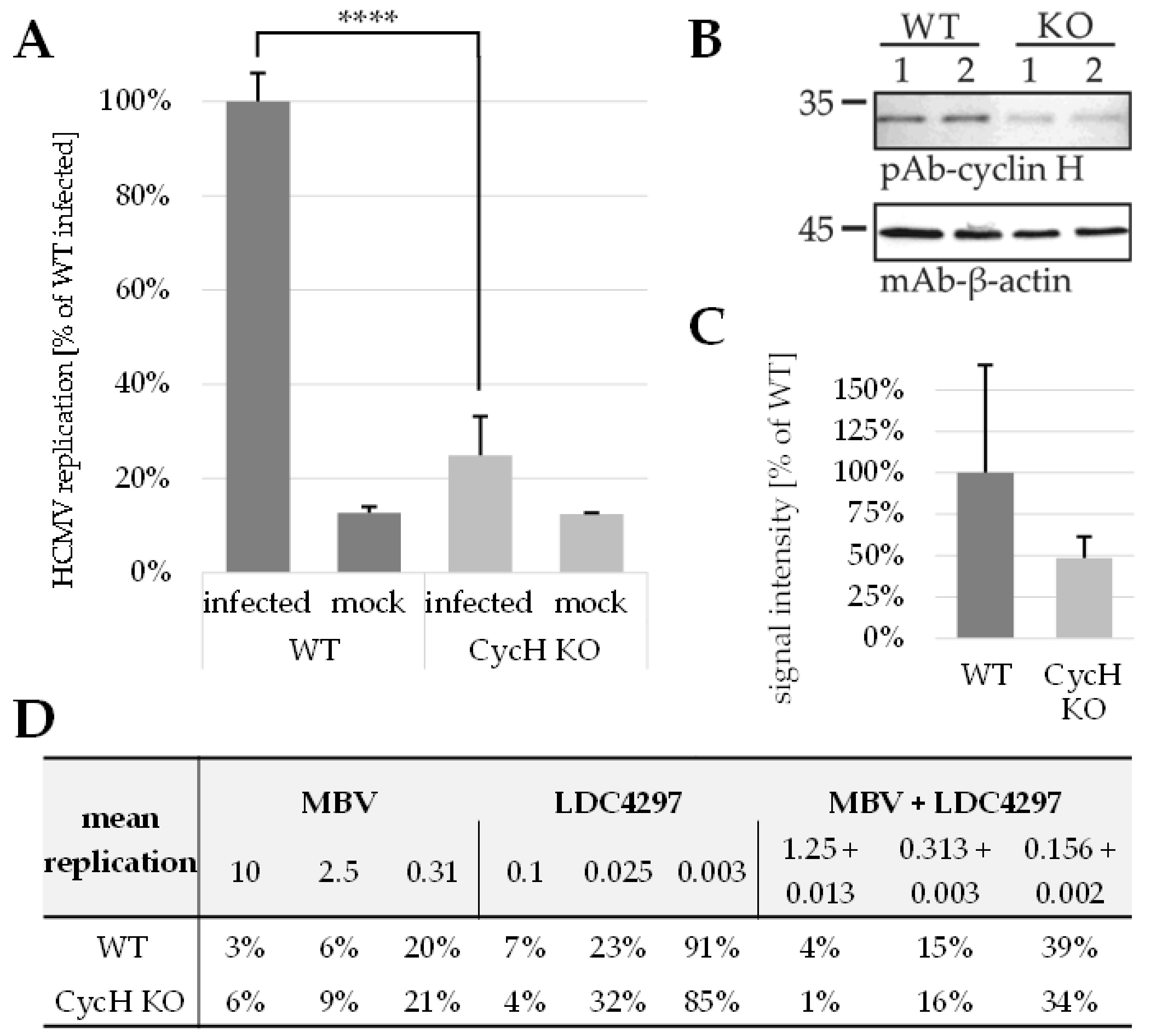
The previous reports of our group and other researchers underlined the multifaceted functional roles of CDK–cyclin complexes in herpesviral replication. Specifically, distinct CDKs and the cytomegaloviral CDK ortholog pUL97 have been validated as promising targets of novel antiviral strategies. Recent findings demonstrated that combinatorial drug treatments with clinically relevant PKIs can exert promising anti-HCMV activity. This aspect was initially underlined by the identification of the true synergistic effect of drug combinations directed against human CDK7 and viral pUL97. Here, we strongly substantiated these findings with a broader panel of compounds showing highly attractive antiviral efficacies and MoA. In essence, we demonstrated that drug synergy was exerted by pairs of PKIs from various chemical classes directed to pUL97 and CDK7 in the absence of putative amplification of cytotoxicity. These data revealed a substantial reduction in drug dosages, thereby further illustrating the benefit of drug combination over single-drug treatment. Moreover, drug synergism was also achieved through specific combinations of two host-directed PKIs (directed against CDK2, CDK7, CDK8 and/or CDK9). Interestingly, the new data also demonstrated that the HCMV-induced upregulation of CDK7 expression levels was completely abrogated by synergistic drug treatment. Another mechanistic aspect of these synergistic drug combinations was illustrated by the use of a HCMV ΔUL97 deletion mutant and cyclin H knock-out cells, confirming the quality of this targeting strategy. Finally, we provided the first evidence for a triple PKI combination possessing synergistic potency in terms of anti-HCMV activity.
During the last two decades, a number of specific pharmacological CDK inhibitors have been developed and approved for cancer treatment. The further development of these CDK-directed PKIs for the treatment of viral infections may represent a novel effective therapeutic strategy to combat old and emerging viruses [
4]. In general, viruses rely on the host cell for resources to create a favorable environment of viral replication. To this end, they use a variety of mechanisms to reprogram and control cellular activities, with the manipulation of the host cell cycle being one of the most frequent points of action. This complex and interactive way of controlling kinase-driven cell cycle machinery and signaling pathways introduces many opportunities for viral manipulation [
4,
56]. As far as the function of CDK7 in cell cycle regulation is concerned, the trimeric complex CDK7–cyclin H-MAT1, commonly called the CDK-activating kinase (CAK) complex [
57], fulfils a CDK-activating step. Moreover, CDK7 represents a component of the general transcription factor TFIIH and is involved in the phosphorylation of serine residues of the RNA polymerase II C-terminal domain (RNAP II-CTD). Interestingly, we and others showed that phosphorylation levels within the RNAP II-CTD are unaffected when CDK7 activity is inhibited in fibroblasts [
21]. This finding suggests that CDK7 might not be essential for global RNAP II-driven transcription but that its functional deficiency in RNAP II regulation may be compensated by other kinase activities. In contrast, the loss of CDK7 activity is critical for cell cycle control since it leads to a lack of normal CAK function and induces cell cycle arrest. Interestingly, the mode of antiviral activity of the CDK7-specific inhibitor LDC4297 already manifests at the immediate early (IE) phase of HCMV replication. Since the progression of lytic HCMV replication is strictly dependent on IE gene expression, this specific drug activity is translated into a drastic limitation of all downstream viral replication events [
21].
When speculating about the regulatory basis of synergistic antiviral drug interactions, one might think about potentially special features of drug–target interaction. It should be stressed, however, that the drugs applied in this study represent classical ATP-competitive kinase inhibitors, and thus it appears unlikely that the mode of inhibition and the drug binding characteristics may reveal unexpected details. It is much more probable that the emergence of drug synergy stems from the interactive relation between the specific target kinases. Thus, antiviral drug synergy is most probably not determined at the level of drug–target interaction, but at the level of target–target interaction, such as a functional cross-talk between the target kinases. Basically, we suggest three theoretical MoA concepts of kinase interactive relationships associated with drug synergy: (i) two drug-targeted activities are both crucially important, or even absolutely essential, for efficient virus replication, (ii) two drug-targeted activities are functionally related, for instance through an overlapping substrate spectrum, or (iii) two drug-targeted activities are dependent on each other, for instance through mutually activating cross-talk or similar feedback effects. In all these cases, the co-inhibition of two kinase activities, through the respective drug combination treatment, may result in the described synergistic effects leading to strongly increased antiviral efficacy.
Concerning the specific case of antiviral activity exerted by CDK7 inhibitors, we state that at least three mechanistic aspects may have a combined impact on their activities, as based on our previous analyses [
1,
5,
21,
24,
25]. Firstly, the transcription-directed regulatory role of CDK7 activity is considered to be required for efficient HCMV replication. Secondly, its cell cycle-specific CAK master activity is seen to exert an even stronger impact. Thirdly, and possibly more important than previously expected, the direct phosphorylation of viral proteins through CDK-cyclin complexes (M.M. et al., unpublished data) could have an importance for substrate protein activity and the efficiency of lytic viral replication. It should be mentioned that we previously demonstrated the formation of ternary complexes between CDK7, cyclin H and viral pUL97 [
53].
Given this background knowledge, the regulatory role of CDK7 activity for HCMV replication appears to be crucial and should thus be accessible to modern antiviral strategies. In particular, the combined drug targeting of CDK and vCDK/pUL97 activities may enable untapped therapeutic possibilities. It should be emphasized again that, very recently, MBV has been approved as the first kinase inhibitor in the entire field of antiviral therapy, and that further kinase inhibitors like LDC4297 and related analogs are presently being investigated at the preclinical/clinical level. In order to validate the specific benefit of the PKI combination strategy in antiviral treatments, a proof-of-concept has to be achieved in an animal model. So far, the examples of strict algorithm-based calculations of drug synergy through the use of data derived from animal experiments have been rare [
58], and thus we aim to demonstrate our described synergistic PKI combinations in vivo. At present, we are further developing the previously established MCMV/mouse infection model [
1,
24,
59,
60], so that an in vivo proof-of-concept might lift our synergy strategy to the next level of antiviral research. With our combined findings, this study highlights the potential of therapeutic drug combinations of approved, developmental and preclinical PKIs for broadening the scope of future anti-HCMV therapy.
4. Materials and Methods
4.1. Cells and Viruses
Primary human foreskin fibroblasts (HFFs, derived from clinical samples, Children’s Hospital, Erlangen, Germany) were grown in Eagle’s minimal essential medium (MEM) supplemented with 1 × GlutaMAX
TM (both Thermo Fisher Scientific, Waltham, MA, USA), 10 μg/mL gentamicin and 10% fetal bovine serum (FBS, Capricorn, Ebsdorfergrund, Germany). Human embryonic kidney epithelial cells (293Ts, ATCC, Manassas, VA, USA) were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 1 × GlutaMAX
TM (both Thermo Fisher Scientific, Waltham, MA, USA), 10 μg/mL gentamicin and 10% FBS (Capricorn, Ebsdorfergrund, Germany). Cultured cells were maintained at 37 °C, 5% CO
2 and 80% humidity. All cell culture was regularly monitored for absence of mycoplasma contamination (Lonza™ Mycoalert™, Thermo Fisher Scientific, Waltham, MA, USA). Recombinant HCMV AD169 expressing green fluorescent protein (AD169-GFP, [
61]) and recombinant ORF-UL97-deleted HCMV AD169 expressing green fluorescent protein (ΔUL97-GFP BAC213 [
48]) were used for in vitro replication assays.
4.2. Antiviral Compounds
Antiviral drugs were obtained from the following sources: Calbiochem, Darmstadt, Germany (CDK2 Inh II, Gö6976); GPC Biotech AG, Martinsried, Germany (R25); Lead Discovery Center GmbH, Dortmund, Germany (LDC4297); MedChemExpress, Monmouth Junction, NJ, USA (abemaciclib, AZD4573, CVT-313, CYC065, dinaciclib, maribavir, riviciclib, samuraciclib SEL120, SY1365); Tocris, Wiesbaden-Nordenstadt, Germany (SNS032, THAL-SNS032); Vichem Kft, Budapest, Hungary (Ax7396, Vi7392). Stock aliquots were prepared in sterile DMSO (Sigma Aldrich, St. Louis, MO, USA) and stored at −20 °C.
4.3. Antibodies
The following antibodies were used in this study: monoclonal mouse anti-CDK7 (Sc-56284; Santa Cruz Biotechnology, Dallas, TX, USA), polyclonal rabbit anti-UL97 (courtesy of Dr. Donald M. Coen, Boston, MA, USA), Alexa 555 goat anti-rabbit (A21429), Alexa 488 goat anti-mouse (A11029, both Thermo Fisher Scientific, Waltham, MA, USA).
4.4. Determination of Cell Viability by Neutral Red Uptake Assay
Drug induced cytotoxicity in HFFs was measured as a reduction of cell viability determined by neutral red uptake assay (NRA), as described previously [
62,
63]. Briefly, HFFs treated with compounds for 7 d were incubated with a final concentration of 40 μg/mL neutral red (Sigma Aldrich, St. Louis, MO, USA) for 3 h. Incorporated neutral red was released from the cells by incubation with destaining solution (50% ethanol, 49% H
2O, 1% acetic acid) and subsequently quantitated in a microplate reader (PerkinElmer, Waltham, MA, USA) by fluorescence measurement using 560/630 nm for excitation/emission, respectively.
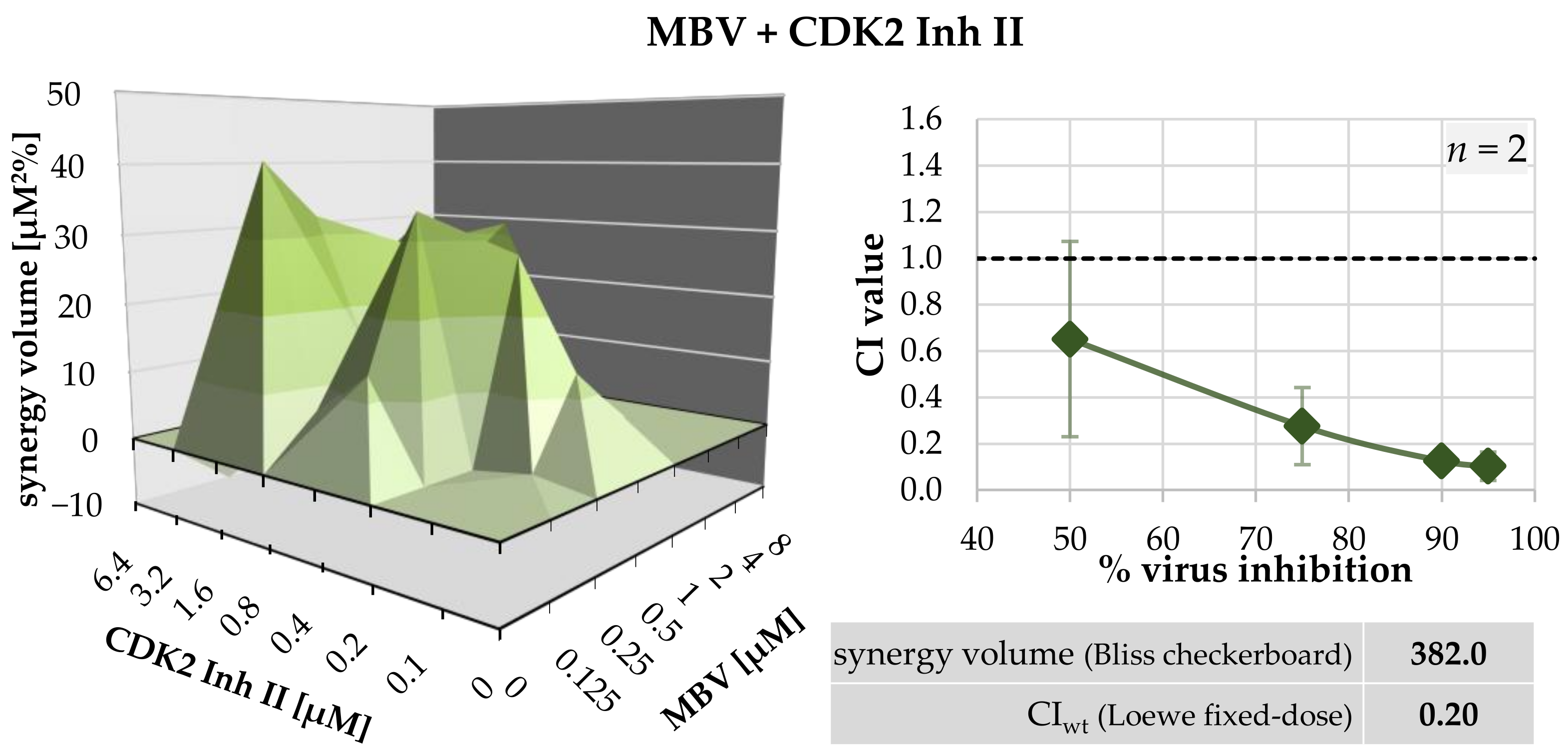
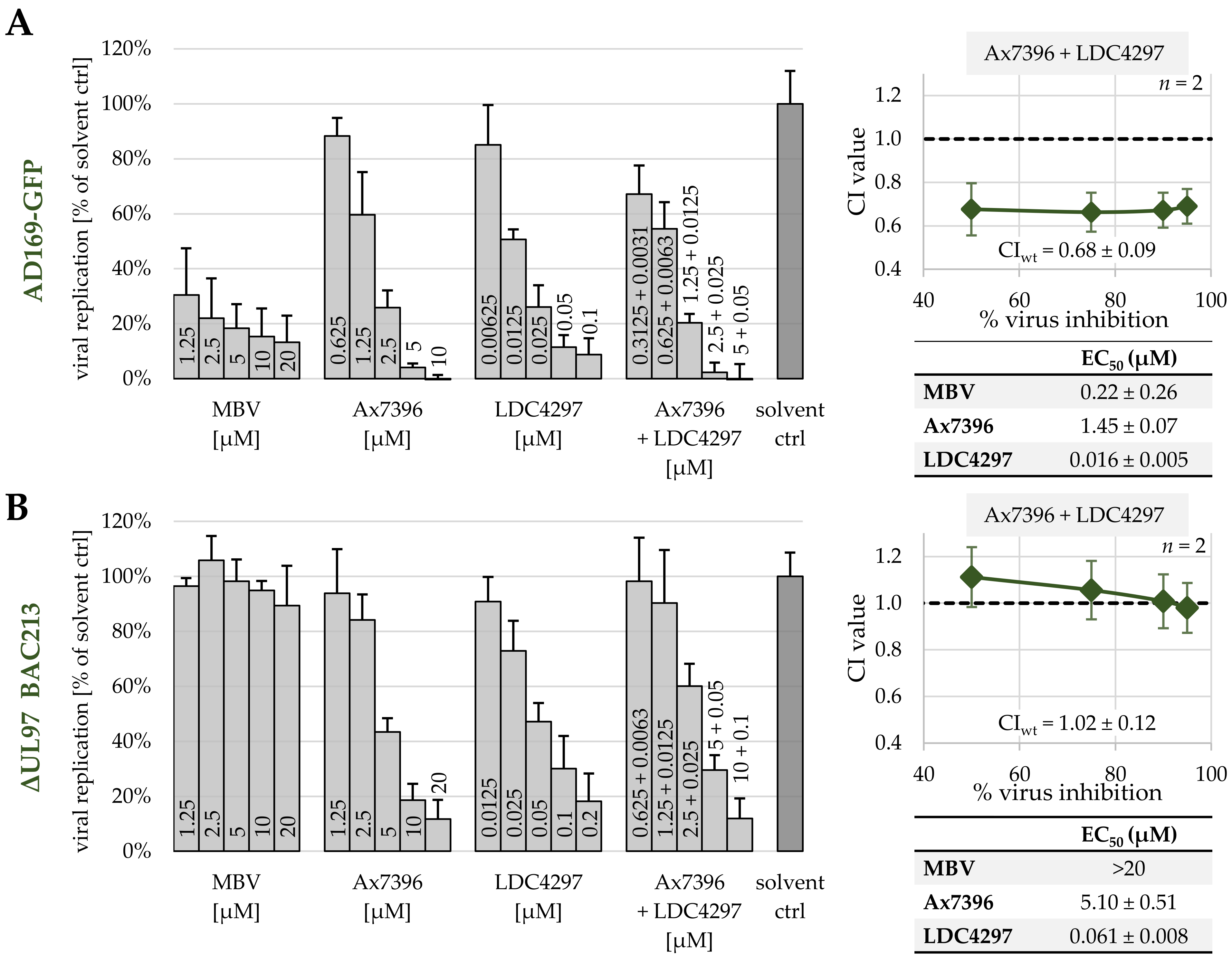
4.5. Drug Interaction Assessment via Bliss Independence Checkerboard Assay Adapted to HCMV-GFP In Vitro Infection
Bliss-based drug interaction was assessed using an adapted protocol of the HCMV GFP-based replication assay described previously [
1,
21,
61]. HFFs were seeded at app. 1.2 × 10
4 cells/well in 96-well culture plates (three plates per assay) and infected on the following day with HCMV AD169-GFP [
61] in a dilution resulting in 25% GFP-positive cells at 7 d p.i. (i.e., 1 × TCID
257d) or remained mock-infected. After a virus adsorption phase of 90 min, the inoculum was replaced by medium supplemented with a matrix of drug combinations in different concentration ratios, solvent control or medium for mock-infected wells. Standard protocol tested a matrix of 8 × 8 concentrations, beginning with ~8 × EC
50 and 1:2 dilutions. All infections were performed in biological triplicates. Cells were lysed by the addition of 100 µL lysis buffer/well 7 d p.i., and cell suspensions were mixed and transferred to another 96-well plate. Centrifugation was performed at 3000 rpm for 15 min and clear lysates were subjected to automated GFP quantitation in a Victor X4 microplate reader (PerkinElmer, Waltham, MA, USA). Measured values were entered into the MacSynergy II software [
64] and results were presented as surface graphs illustrating synergy volume with a 95% confidence interval over the three biological replicates.
4.6. Drug Interaction Assessment via Loewe Additivity Fixed-Dose Assay Adapted to HCMV-GFP In Vitro Infection
Loewe additivity was assessed using an adapted protocol of the HCMV GFP-based replication assay [
21,
61] described previously [
1]. HFFs were seeded at app. 1.6 × 10
5 cells/well in 12-well culture plates (four plates per assay) and infected on the following day with HCMV AD169-GFP [
61] in a dilution resulting in 25% GFP-positive cells at 7 d p.i. (i.e., 1× TCID
257d). After virus adsorption, the inoculum was replaced by medium supplemented with single compound, compound combination or solvent control. Standard protocol comprised a serial dilution of six concentrations/single compound, starting with app. 4–8 × EC
50, and a 1:2 serial dilution of eight concentrations of the combination, starting with a combination of half of the highest concentrations of both single dilution series. All infections were performed in biological duplicates. Cells were lysed by the addition of 200 µL lysis buffer/well 7 d p.i., and cell suspensions were mixed and transferred to a 96-well plate. Centrifugation was performed at 3000 rpm for 15 min and clear lysates were subjected to automated GFP quantitation in a Victor X4 microplate reader (PerkinElmer, Waltham, MA, USA). Antiviral efficacy (mean of duplicate measurement of biological duplicates) was expressed as the percentage of solvent control and entered into the CompuSyn software (Version 1.0 [
65]; ComboSyn, Inc., Paramus, NJ, USA). Only experiments with an r value > 0.90 and EC
50 values close to previously determined concentrations were accepted.
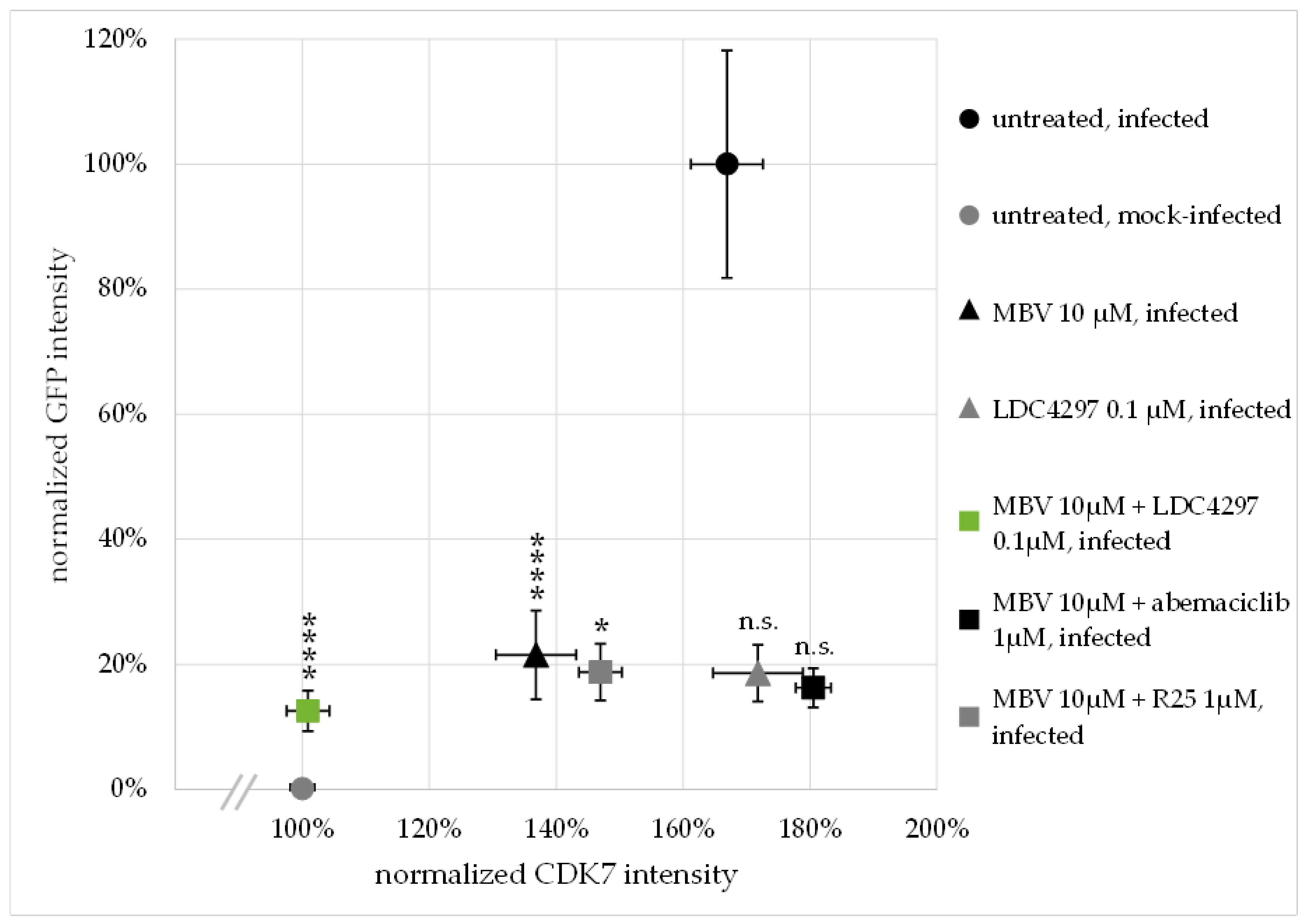
4.7. Indirect Immunofluorescence Assay Utilizing Confocal Laser-Scanning Microscopy
For immunofluorescence detection, app. 2 × 10
5 HFFs/well were grown on coverslips in six-well culture plates and used for infection with HCMV AD169-GFP [
61] at an MOI of 1 or remained mock-infected. After virus adsorption, the inoculum was replaced by medium supplemented with single compound, compound combination or solvent control. At 7 d p.i., cells were fixed with 10% formalin solution (Sigma Aldrich, St. Louis, MO, USA; 10 min, room temperature) and permeabilized by incubation with 0.2% Triton X-100 solution (Roth, Karlsruhe, Germany; 20 min, 4 °C). Nonspecific staining was blocked by incubation with 2 mg/mL human γ-globulin Cohn fraction II, (Merck, Darmstadt, Germany; 30 min, 37 °C). Indirect immunofluorescence staining was performed by incubation with primary antibody diluted in PBS (60 min, 37 °C), washing and subsequent incubation with diluted dye-conjugated secondary antibody (30 min, 37 °C). After further washing steps, cells were mounted with VECTASHIELD
® mounting medium with DAPI (Vector Laboratories, Burlingame, CA, USA), before glass coverslips were sealed using nail polish. Confocal laser-scanning microscopy was performed with a TCS SP5 microscope using a 63× HCX PL APO CS oil immersion objective lens (Leica Microsystems, Mannheim, Germany). Images were processed using the LAS AF software (version 2.6.0 build 7266; Leica Microsystems, Mannheim, Germany) and edited with Photoshop CS5 (Adobe, San José, CA, USA).
4.8. Quantitation of Intracellular Immunofluorescence Signals
Intensity of GFP and CDK7 signals of cells, fixed and stained as described above, was measured utilizing Fiji/ImageJ (version 1.52p, [
66]). For this purpose, confocal microscopy images taken under identical setting conditions were converted to 16-bit format. Subsequently, the DAPI channel image was converted to binary and outlines of nuclei were used as a template to measure GFP/CDK7 intensity in the respective channels (tool:
Analyze particles; setting:
Size (
inch2) = 0.50 −
Infinity). Outlines of measured objects were visually monitored in order to exclude artefacts or multiple nuclei measured as one. The GFP/CDK7 intensity was further normalized to the DAPI intensity of the respective nucleus in order to exclude z plane variation effects.
4.9. Generation of Lentiviral Transfer Constructs
Cyclin H-specific guide RNAs (gRNAs) were designed using the benchling.com software (Benchling, San Francisco, CA, USA) and the scoring method of Hsu et al., 2013 [
67]. gRNAs were directed to cyclin H exons 1–5 and had on-target scores between 52.8 and 73.4 out of 100 and off-target scores between 43.1 and 82.9. The following gRNAs were used: Cyclin H A: 5′ GCCGCTTCTGACTACTGTTG 3′; Cyclin H B: 5′ GTCCAAGAGGACTCTCCCGG 3′; Cyclin H C: 5′ ATTCTCCAATATGGGATAGC 3′; Cyclin H D: 5′ ACAGTAGTCAGAAGCGGCAC 3′; Cyclin H E: 5′ CCGGGAGAGTCCTCTTGGAC 3‘; Cyclin H F: 5′ CAGTAATGGAATATCACCCC 3′. Cyclin H-specific CRISPR/Cas9 vectors (pLentiCRIPSR-V2) were generated according to the standard Zhang laboratory protocol [
67,
68,
69]. 293T cells were transfected with pLentiCRISPR-V2 together with a packaging plasmid mix coding for HIV-1 gag/pol/rev and the envelope protein G of vesicular stomatitis virus (VSV-G) using Lipofectamine 2000 (Invitrogen, Karlsruhe, Germany). Lentiviral supernatants were harvested 48 h post-transfection, centrifuged for 5 min at 2000 rpm to remove cell debris, filtered and stored in aliquots at −80 °C.
4.10. Transient Cyclin H Knock-Out of HFFs
To achieve a transient knock-out of cyclin H, app. 1.5 × 105 HFFs/well were grown in 12-well culture plates and, starting the following day, incubated for 24 h with 0.5 mL of lentiviral supernatant (a mixture containing all six gRNAs described in 4.9) supplemented with 0.5 mL MEM and 7.5 µg/mL polybrene (Sigma Aldrich, St. Louis, MO, USA) per well. Virus supernatant was replaced by fresh media 24 h later and cells were used for infection experiments (see 4.5 and 4.6) on the following day.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}