
CHERP Regulates the Alternative Splicing of pre-mRNAs in the Nucleus

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
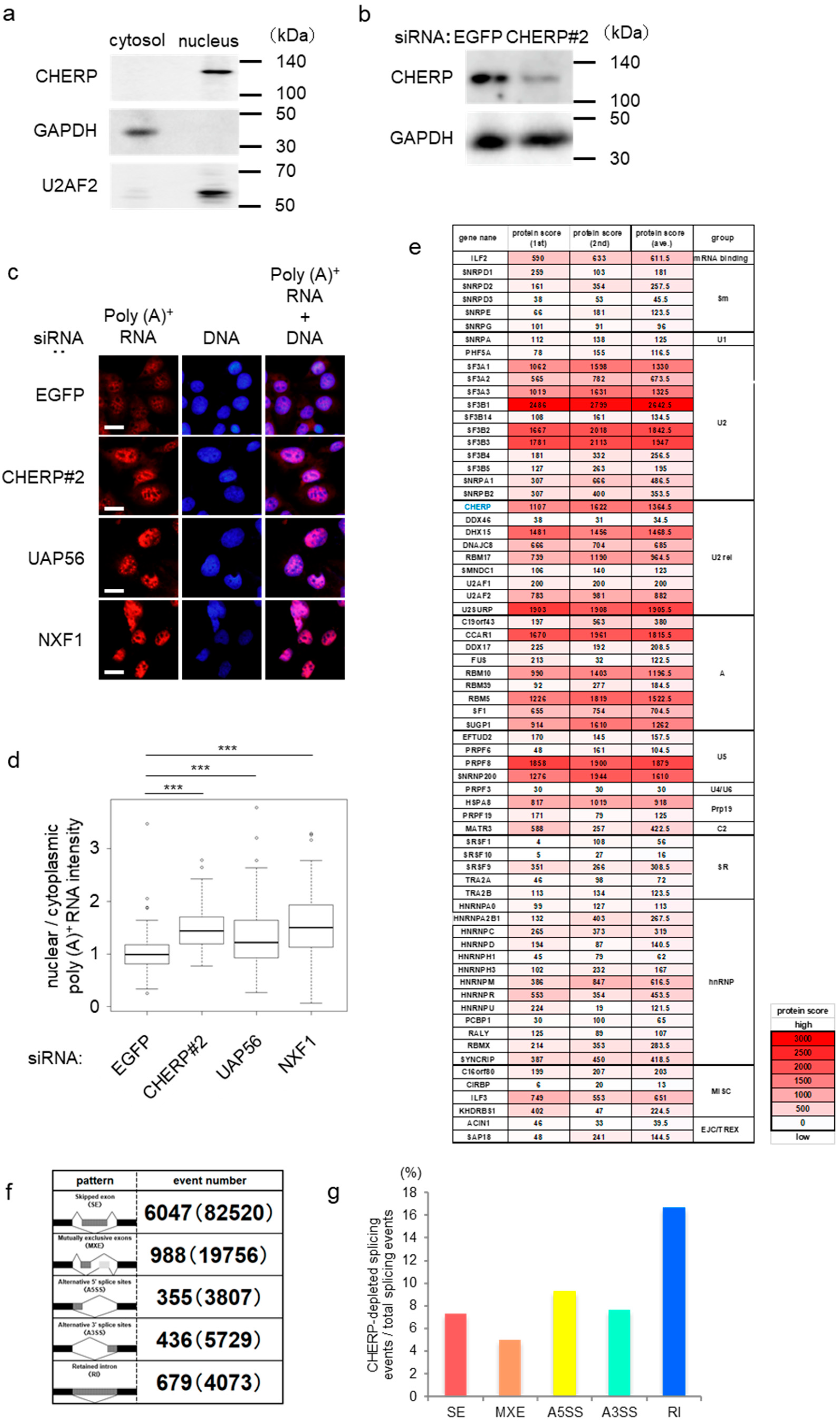
2.1. CHERP Is Involved in Splicing in the Nucleus
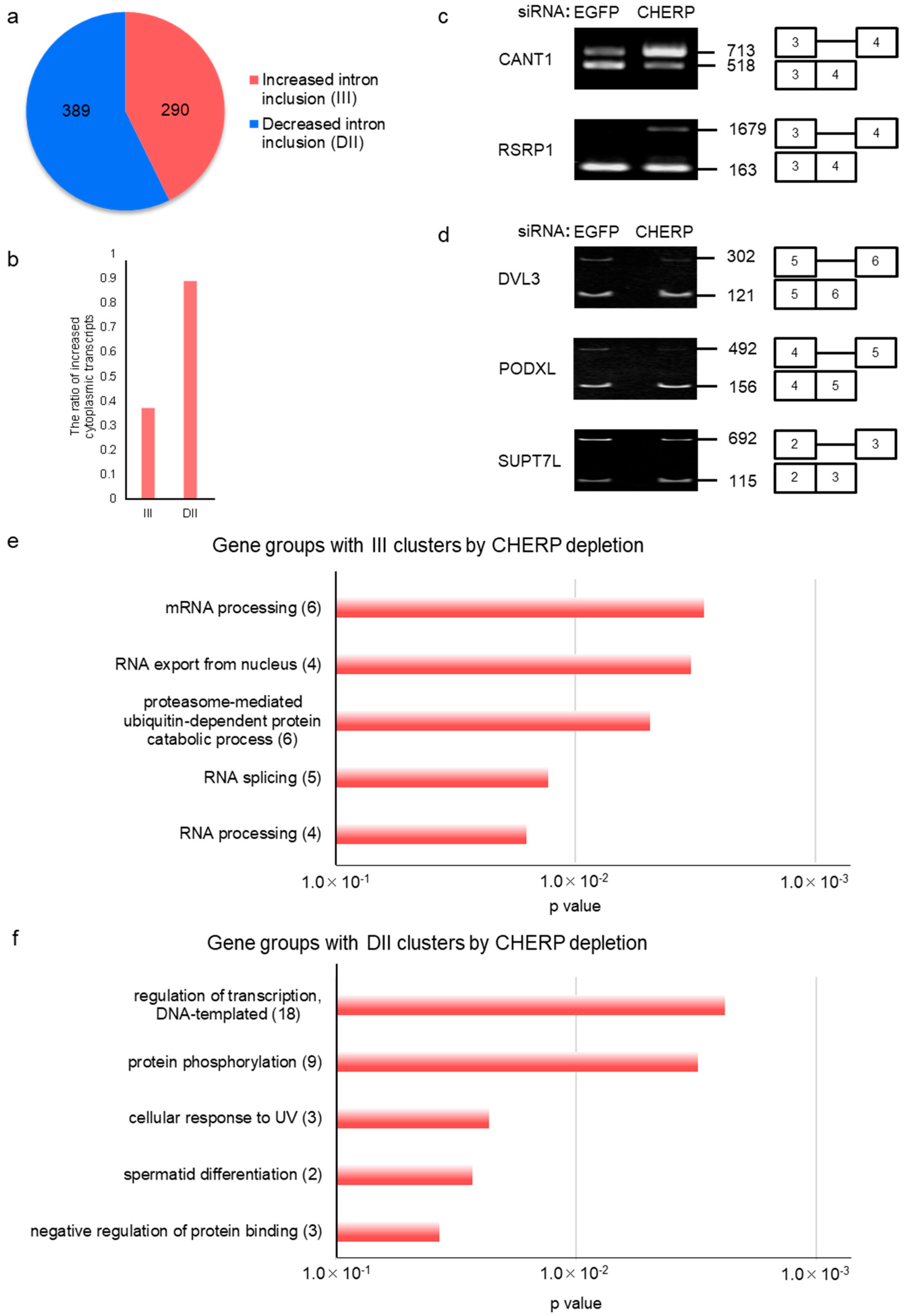
2.2. Identification and Characterization of Intron Retention after CHERP Depletion
2.3. CHERP Regulates Intron Retention in the Target Transcripts
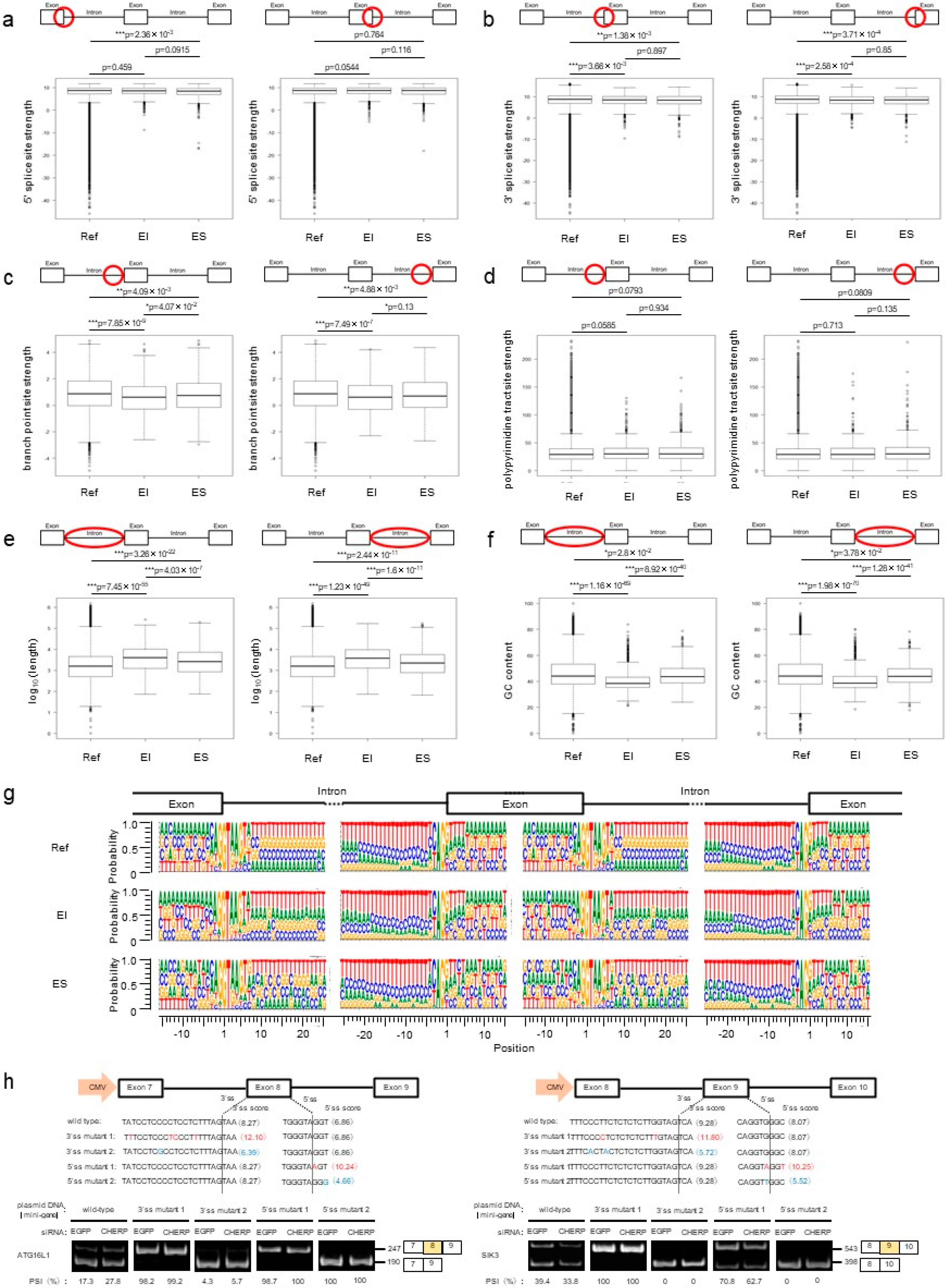
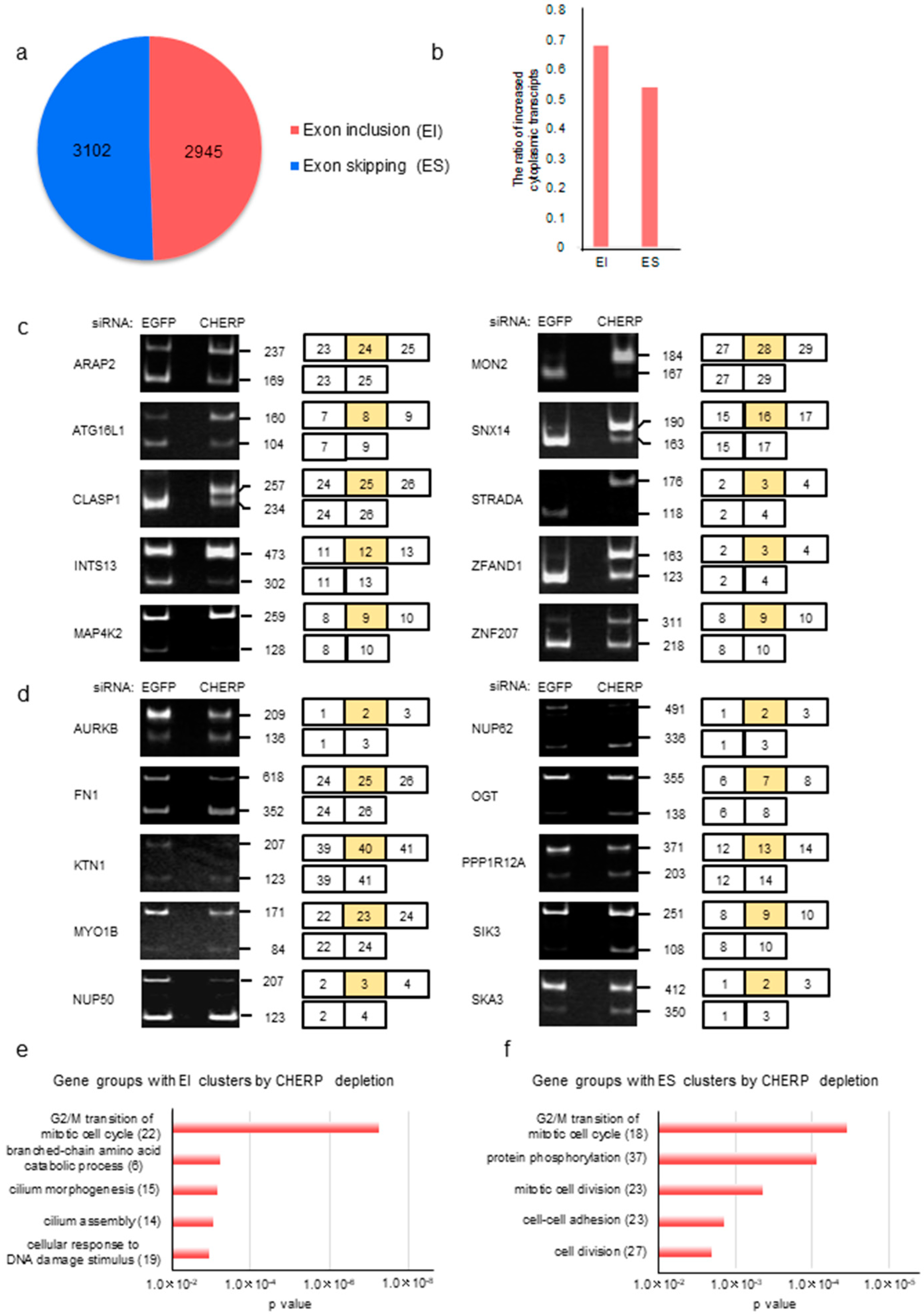
2.4. Identification and Characterization of Cassette Exons Regulated by CHERP
2.5. CHERP Regulates Cassette Exon Inclusion and Skipping in Target Genes
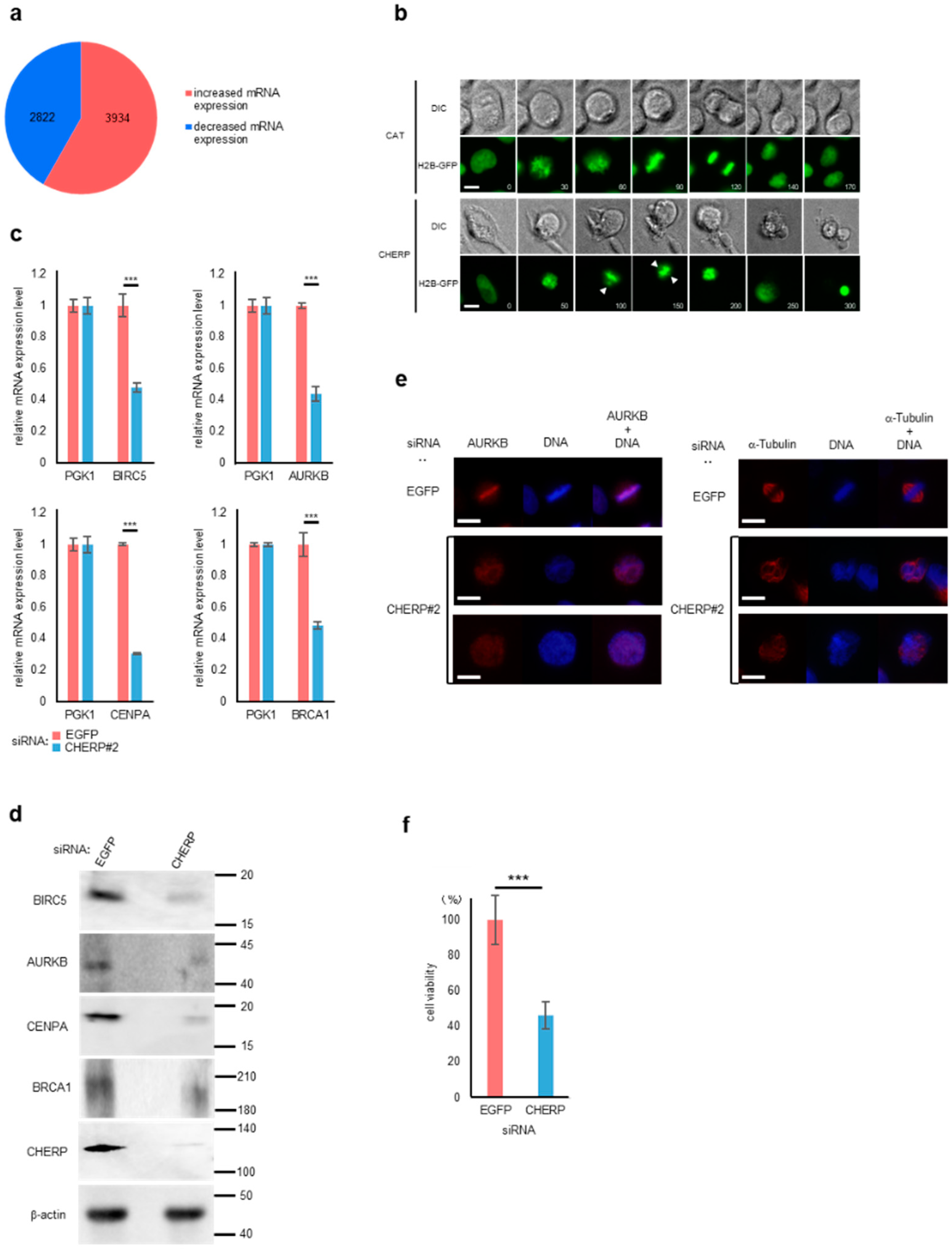
2.6. CHERP Regulates Cell Survival and Cell Death
3. Discussion
4. Materials and Methods
4.1. Reagents and Instruments
4.2. Plasmid DNAs, siRNAs and Oligo DNA Primers
4.3. Antibodies
4.4. Construction of Plasmid DNAs
4.5. Cell Culture
4.6. Establishment of Stable Cell Lines
4.7. Immunostaining
4.8. RNA-FISH
4.9. Preparation of RNA from Cytoplasmic, Nuclear and Whole-Cell Fractions
4.10. Protein Preparation from Cytoplasmic and Nuclear Extracts
4.11. Immunoprecipitations
4.12. Silver Staining
4.13. LC-MS/MS Analyses
4.14. Western Blots
4.15. RNA Isolation, cDNA Synthesis and PCR
4.16. Preparation of RNA-seq Library
4.17. Bioinformatic Analyses
4.18. Quantitative PCR (qPCR)
4.19. Minigene Splicing Reporter Assay
4.20. Live Cell Imaging
4.21. MTT Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Chen, M.; Manley, J.L. Mechanisms of alternative splicing regulation: Insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol. 2009, 10, 741–754. [Google Scholar] [CrossRef]
- Fu, X.D.; Ares, M. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Matera, A.G.; Wang, Z. A day in the life of the spliceosome. Nat. Rev. Mol. Cell Biol. 2014, 15, 108–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The Spliceosome: Design Principles of a Dynamic RNP Machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [Green Version]
- Black, D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, M.E.; Charenton, C.; Nagai, K. RNA Splicing by the Spliceosome. Annu. Rev. Biochem. 2020, 89, 359–388. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Query, C.C.; Sharp, P.A. Splicing of Precursors to mRNA by the Spliceosome, 2nd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1999; pp. 525–560. [Google Scholar]
- Kim, E.; Goren, A.; Ast, G. Alternative splicing: Current perspectives. BioEssays 2008, 30, 38–47. [Google Scholar] [CrossRef]
- Keren, H.; Lev-Maor, G.; Ast, G. Alternative splicing and evolution: Diversification, exon definition and function. Nat. Rev. Genet. 2010, 11, 345–355. [Google Scholar] [CrossRef]
- Zhou, Z.; Fu, X.D. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma 2013, 122, 191–207. [Google Scholar] [CrossRef]
- Geuens, T.; Bouhy, D.; Timmerman, V. The hnRNP family: Insights into their role in health and disease. Hum. Genet. 2016, 135, 851–867. [Google Scholar] [CrossRef] [Green Version]
- Maris, C.; Dominguez, C.; Allain, F.H.T. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 2005, 272, 2118–2131. [Google Scholar] [CrossRef] [PubMed]
- Laplante, J.M.; O’Rourke, F.; Lu, X.; Fein, A.; Olsen, A.; Feinstein, M.B. Cloning of human Ca2+ homoeostasis endoplasmic reticulum protein (CHERP): Regulated expression of antisense cDNA depletes CHERP, inhibits intracellular Ca2+ mobilization and decreases cell proliferation. Biochem. J. 2000, 348, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.; Sharma, P.; Ignatchenko, A.; MacLennan, D.H.; Kislinger, T.; Gramolini, A.O. Identification of novel ryanodine receptor 1 (RyR1) protein interaction with calcium homeostasis endoplasmic reticulum protein (CHERP). J. Biol. Chem. 2011, 286, 17060–17068. [Google Scholar] [CrossRef] [Green Version]
- Lin-Moshier, Y.; Sebastian, P.J.; Higgins, L.A.; Sampson, N.D.; Hewitt, J.E.; Marchant, J.S. Re-evaluation of the role of Calcium Homeostasis Endoplasmic Reticulum Protein (CHERP) in cellular calcium signaling. J. Biol. Chem. 2013, 288, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Sasaki-Osugi, K.; Imoto, C.; Takahara, T.; Shibata, H.; Maki, M. Nuclear ALG-2 protein interacts with Ca2+ homeostasis endoplasmic reticulum protein (CHERP) Ca2+-dependently and participates in regulation of alternative splicing of inositol trisphosphate receptor type 1 (IP3R1) Pre-mRNA. J. Biol. Chem. 2013, 288, 33361–33375. [Google Scholar] [CrossRef] [Green Version]
- De Maio, A.; Yalamanchili, H.K.; Adamski, C.J.; Gennarino, V.A.; Liu, Z.; Qin, J.; Jung, S.Y.; Richman, R.; Orr, H.; Zoghbi, H.Y. RBM17 Interacts with U2SURP and CHERP to Regulate Expression and Splicing of RNA-Processing Proteins. Cell Rep. 2018, 25, 726–736.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimoto, R.; Kaida, D.; Furuno, M.; Burroughs, A.M.; Noma, S.; Suzuki, H.; Kawamura, Y.; Hayashizaki, Y.; Mayeda, A.; Yoshida, M. Global analysis of pre-mRNA subcellular localization following splicing inhibition by spliceostatin A. RNA 2017, 23, 47–57. [Google Scholar] [CrossRef]
- Legrain, P.; Rosbash, M. Some cis- and trans-acting mutants for splicing target pre-mRNA to the cytoplasm. Cell 1989, 57, 573–583. [Google Scholar] [CrossRef]
- Okamura, M.; Yamanaka, Y.; Shigemoto, M.; Kitadani, Y.; Kobayashi, Y.; Kambe, T.; Nagao, M.; Kobayashi, I.; Okumura, K.; Masuda, S. Depletion of mRNA export regulator DBP5/ DDX19, GLE1 or IPPK that is a key enzyme for the production of IP6, resulting in differentially altered cytoplasmic mRNA expression and specific cell defect. PLoS ONE 2018, 13, e0197165. [Google Scholar] [CrossRef]
- Zamore, P.D.; Green, M.R. Identification, purification, and biochemical characterization of U2 small nuclear ribonucleoprotein auxiliary factor. Proc. Natl. Acad. Sci. USA 1989, 86, 9243–9247. [Google Scholar] [CrossRef] [Green Version]
- Hegele, A.; Kamburov, A.; Grossmann, A.; Sourlis, C.; Wowro, S.; Weimann, M.; Will, C.L.; Pena, V.; Lührmann, R.; Stelzl, U. Dynamic Protein-Protein Interaction Wiring of the Human Spliceosome. Mol. Cell 2012, 45, 567–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, E.Y.; DeKelver, R.C.; Lo, M.C.; Nguyen, T.A.; Matsuura, S.; Boyapati, A.; Pandit, S.; Fu, X.D.; Zhang, D.E. SON Controls Cell-Cycle Progression by Coordinated Regulation of RNA Splicing. Mol. Cell 2011, 42, 185–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín, E.; Vivori, C.; Rogalska, M.; Herrero-Vicente, J.; Valcárcel, J. Alternative splicing regulation of cell-cycle genes by SPF45/SR140/CHERP complex controls cell proliferation. RNA 2021, 27, 1557–1576. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Göke, J.; Sachs, F.; Jacques, P.É.; Liang, H.; Feng, B.; Bourque, G.; Bubulya, P.A.; Ng, H.H. SON connects the splicing-regulatory network with pluripotency in human embryonic stem cells. Nat. Cell Biol. 2013, 15, 1141–1152. [Google Scholar] [CrossRef] [Green Version]
- Damianov, A.; Ying, Y.; Lin, C.H.; Lee, J.A.; Tran, D.; Vashisht, A.A.; Bahrami-Samani, E.; Xing, Y.; Martin, K.C.; Wohlschlegel, J.A.; et al. Rbfox Proteins Regulate Splicing as Part of a Large Multiprotein Complex LASR. Cell 2016, 165, 606–619. [Google Scholar] [CrossRef] [Green Version]
- Ying, Y.; Wang, X.J.; Vuong, C.K.; Lin, C.H.; Damianov, A.; Black, D.L. Splicing Activation by Rbfox Requires Self-Aggregation through Its Tyrosine-Rich Domain. Cell 2017, 170, 312–323.e10. [Google Scholar] [CrossRef] [Green Version]
- Cheah, P.Y.; Wong, Y.H.; Koh, P.K.; Loi, C.; Chew, M.H.; Tang, C.L. A novel indel in exon 9 of APC upregulates a “skip exon 9” isoform and causes very severe familial adenomatous polyposis. Eur. J. Hum. Genet. 2014, 22, 833–836. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.; Moon, H.; Loh, T.J.; Jang, H.N.; Liu, Y.; Zhou, J.; Ohn, T.; Zheng, X.; Shen, H. Splicing inhibition of U2AF65 leads to alternative exon skipping. Proc. Natl. Acad. Sci. USA 2015, 112, 9926–9931. [Google Scholar] [CrossRef] [Green Version]
- Echeverria, G.V.; Cooper, T.A. Muscleblind-like 1 activates insulin receptor exon 11 inclusion by enhancing U2AF65 binding and splicing of the upstream intron. Nucleic Acids Res. 2014, 42, 1893–1903. [Google Scholar] [CrossRef] [Green Version]
- Anczuków, O.; Akerman, M.; Cléry, A.; Wu, J.; Shen, C.; Shirole, N.H.; Raimer, A.; Sun, S.; Jensen, M.A.; Hua, Y.; et al. SRSF1-Regulated Alternative Splicing in Breast Cancer. Mol. Cell 2015, 60, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Anczuków, O.; Rosenberg, A.Z.; Akerman, M.; Das, S.; Zhan, L.; Karni, R.; Muthuswamy, S.K.; Krainer, A.R. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat. Struct. Mol. Biol. 2012, 19, 220–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calarco, J.A.; Superina, S.; O’Hanlon, D.; Gabut, M.; Raj, B.; Pan, Q.; Skalska, U.; Clarke, L.; Gelinas, D.; van der Kooy, D.; et al. Regulation of Vertebrate Nervous System Alternative Splicing and Development by an SR-Related Protein. Cell 2009, 138, 898–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dauksaite, V.; Gotthardt, M. Molecular basis of titin exon exclusion by RBM20 and the novel titin splice regulator PTB4. Nucleic Acids Res. 2018, 46, 5227–5238. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.; Kazan, H.; Chan, E.T.; Castillo, L.P.; Chaudhry, S.; Talukder, S.; Blencowe, B.J.; Morris, Q.; Hughes, T.R. Rapid and systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nat. Biotechnol. 2009, 27, 667–670. [Google Scholar] [CrossRef] [PubMed]
- Ajiro, M.; Jia, R.; Yang, Y.; Zhu, J.; Zheng, Z.M. A genome landscape of SRSF3-regulated splicing events and gene expression in human osteosarcoma U2OS cells. Nucleic Acids Res. 2015, 44, 1854–1870. [Google Scholar] [CrossRef]
- Zhou, X.; Wu, W.; Li, H.; Cheng, Y.; Wei, N.; Zong, J.; Feng, X.; Xie, Z.; Chen, D.; Manley, J.L.; et al. Transcriptome analysis of alternative splicing events regulated by SRSF10 reveals position-dependent splicing modulation. Nucleic Acids Res. 2014, 42, 4019–4030. [Google Scholar] [CrossRef]
- Kuroyanagi, H.; Watanabe, Y.; Suzuki, Y. Position-dependent and neuron-specific splicing regulation by the CELF family RNA-binding protein UNC-75 in Caenorhabditis elegans. Nucleic Acids Res. 2013, 41, 4015–4025. [Google Scholar] [CrossRef] [Green Version]
- Bechara, E.G.; Sebestyén, E.; Bernardis, I.; Eyras, E.; Valcárcel, J. RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Mol. Cell 2013, 52, 720–733. [Google Scholar] [CrossRef] [Green Version]
- Imai, T.; Tokunaga, A.; Yoshida, T.; Hashimoto, M.; Mikoshiba, K.; Weinmaster, G.; Nakafuku, M.; Okano, H. The Neural RNA-Binding Protein Musashi1 Translationally Regulates Mammalian numb Gene Expression by Interacting with Its mRNA. Mol. Cell. Biol. 2001, 21, 3888–3900. [Google Scholar] [CrossRef] [Green Version]
- Ruth Zearfoss, N.; Deveau, L.M.; Clingman, C.C.; Schmidt, E.; Johnson, E.S.; Massi, F.; Ryder, S.P. A conserved three-nucleotide core motif defines musashi RNA binding specificity. J. Biol. Chem. 2014, 289, 35530–35541. [Google Scholar] [CrossRef] [Green Version]
- Chawla, G.; Lin, C.-H.; Han, A.; Shiue, L.; Ares, M.; Black, D.L. Sam68 Regulates a Set of Alternatively Spliced Exons during Neurogenesis. Mol. Cell. Biol. 2009, 29, 201–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iijima, T.; Wu, K.; Witte, H.; Hanno-Iijima, Y.; Glatter, T.; Richard, S.; Scheiffele, P. SAM68 regulates neuronal activity-dependent alternative splicing of neurexin-1. Cell 2011, 147, 1601–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Wang, Y.; Liu, Y.; Zhang, C.; Luo, Y.; Guo, R.; Zhan, Z.; Wei, N.; Xie, Z.; Shen, L.; et al. U2-related proteins CHERP and SR140 contribute to colorectal tumorigenesis via alternative splicing regulation. Int. J. Cancer 2019, 145, 2728–2739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, M.J.; Wang, Q.; Kennedy, C.J.; Silver, P.A. An alternative splicing network links cell-cycle control to apoptosis. Cell 2010, 142, 625–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramírez-valle, F.; Badura, M.L.; Braunstein, S.; Narasimhan, M.; Schneider, R.J. Mitotic Raptor Promotes mTORC1 Activity, G 2/M Cell Cycle Progression, and Internal Ribosome Entry Site-Mediated. Mol. Cell. Biol. 2010, 30, 3151–3164. [Google Scholar] [CrossRef] [Green Version]
- Maggioni, D.; Garavello, W.; Rigolio, R.; Pignataro, L.; Gaini, R.; Nicolini, G. Apigenin impairs oral squamous cell carcinoma growth in vitro inducing cell cycle arrest and apoptosis. Int. J. Oncol. 2013, 43, 1675–1682. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, T.; Fujiwara, N.; Yukinaga, H.; Ebisuya, M.; Shiki, T.; Kurihara, T.; Kioka, N.; Kambe, T.; Nagao, M.; Nishida, E.; et al. The Closely Related RNA helicases, UAP56 and URH49, Preferentially Form Distinct mRNA Export Machineries and Coordinately Regulate Mitotic Progression. Mol. Biol. Cell 2010, 21, 1033–1046. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K.; Yamazaki, T.; Harada, K.; Seno, S.; Matsuda, H.; Masuda, S. URH49 exports mRNA by remodeling complex formation and mediating the NXF1-dependent pathway. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194480. [Google Scholar] [CrossRef]
- Kurata, M.; Fujiwara, N.; Fujita, K.; Yamanaka, Y.; Seno, S.; Kobayashi, H.; Miyamae, Y.; Takahashi, N.; Shibuya, Y.; Masuda, S. Food-Derived Compounds Apigenin and Luteolin Modulate mRNA Splicing of Introns with Weak Splice Sites. iScience 2019, 22, 336–352. [Google Scholar] [CrossRef]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Bo, L.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.G.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, S.; Park, J.W.; Lu, Z.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef]
- Corvelo, A.; Hallegger, M.; Smith, C.W.J.; Eyras, E. Genome-Wide Association between Branch Point Properties and Alternative Splicing. PLoS Comput. Biol. 2010, 6, e1001016. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Crooks, G.; Hon, G.; Chandonia, J.; Brenner, S. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Huang, H.; Sun, L.; Yang, M.; Pan, C.; Chen, W.; Wu, D.; Lin, Z.; Zeng, C.; Yao, Y.; et al. MiR-21 indicates poor prognosis in tongue squamous cell carcinomas as an apoptosis inhibitor. Clin. Cancer Res. 2009, 15, 3998–4008. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamanaka, Y.; Ishizuka, T.; Fujita, K.-i.; Fujiwara, N.; Kurata, M.; Masuda, S. CHERP Regulates the Alternative Splicing of pre-mRNAs in the Nucleus. Int. J. Mol. Sci. 2022, 23, 2555. https://doi.org/10.3390/ijms23052555
Yamanaka Y, Ishizuka T, Fujita K-i, Fujiwara N, Kurata M, Masuda S. CHERP Regulates the Alternative Splicing of pre-mRNAs in the Nucleus. International Journal of Molecular Sciences. 2022; 23(5):2555. https://doi.org/10.3390/ijms23052555
Chicago/Turabian StyleYamanaka, Yasutaka, Takaki Ishizuka, Ken-ichi Fujita, Naoko Fujiwara, Masashi Kurata, and Seiji Masuda. 2022. "CHERP Regulates the Alternative Splicing of pre-mRNAs in the Nucleus" International Journal of Molecular Sciences 23, no. 5: 2555. https://doi.org/10.3390/ijms23052555
APA StyleYamanaka, Y., Ishizuka, T., Fujita, K.-i., Fujiwara, N., Kurata, M., & Masuda, S. (2022). CHERP Regulates the Alternative Splicing of pre-mRNAs in the Nucleus. International Journal of Molecular Sciences, 23(5), 2555. https://doi.org/10.3390/ijms23052555