
Combined Transcriptomic and Proteomic Profiling of E. coli under Microaerobic versus Aerobic Conditions: The Multifaceted Roles of Noncoding Small RNAs and Oxygen-Dependent Sensing in Global Gene Expression Control

 ,
,  and
and
Abstract
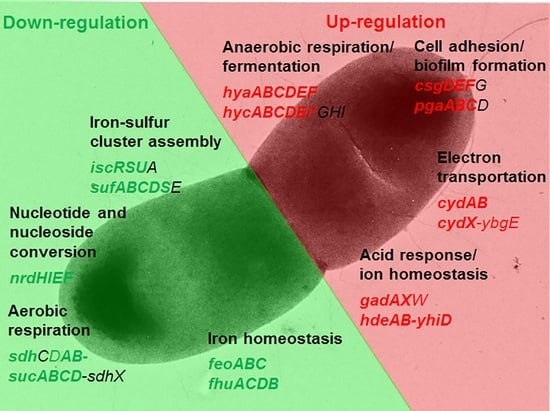
1. Introduction
2. Results
2.1. Generation and Quality Assessment of Aerobic and Microaerobic Transcriptomes of E. coli Cultures Grown on Defined Media
2.2. Defining Major Gene Clusters Involved in Adaptation to Microaerobiosis
2.2.1. Identification of DEGs under Microaerobic Versus Aerobic Conditions
2.2.2. Functional Clusters of DEGs
2.2.3. Prophage- and Phage-Related Genes
2.2.4. Transcription Factor Genes
2.3. Identification of Differentially Expressed sRNAs and Northern-Blot-Based Validation
2.4. Proteome Analysis Corroborates Differential Protein Abundance under Changing Oxygen Availability
2.4.1. Identification of Differentially Abundant Proteins under Microaerobic Versus Aerobic Conditions
2.4.2. Clusters of Proteins with Increased and Decreased Abundance
3. Discussion
4. Materials and Methods
4.1. Bacterial Strain and Growth Conditions
4.2. RNA Isolation
4.3. RNA-seq Analysis
4.4. Northern Blotting
4.5. Western Blot
4.6. Protein Extraction, iTRAQ Labeling, and LC-MS/MS
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sassone-Corsi, M.; Raffatellu, M. No vacancy: How beneficial microbes cooperate with immunity to provide colonization resistance to pathogens. J. Immunol. 2015, 194, 4081–4087. [Google Scholar] [CrossRef] [PubMed]
- Karp, P.D.; Ong, W.K.; Paley, S.; Billington, R.; Caspi, R.; Fulcher, C.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Midford, P.E.; et al. The EcoCyc Database. EcoSal Plus 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Stewart, K.L.; Stewart, A.M.; Bobik, T.A. Prokaryotic Organelles: Bacterial Microcompartments in E. coli and Salmonella. EcoSal Plus 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.R.; Shin, J.H.; Cho, J.S.; Yang, D.; Lee, S.Y. Systems Metabolic Engineering of Escherichia coli. EcoSal Plus 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Alexeeva, S.; Hellingwerf, K.J.; Teixeira de Mattos, M.J. Requirement of ArcA for redox regulation in Escherichia coli under microaerobic but not anaerobic or aerobic conditions. J. Bacteriol. 2003, 185, 204–209. [Google Scholar] [CrossRef]
- Salmon, K.A.; Hung, S.P.; Steffen, N.R.; Krupp, R.; Baldi, P.; Hatfield, G.W.; Gunsalus, R.P. Global gene expression profiling in Escherichia coli K12: Effects of oxygen availability and ArcA. J. Biol. Chem. 2005, 280, 15084–15096. [Google Scholar] [CrossRef]
- Salmon, K.; Hung, S.P.; Mekjian, K.; Baldi, P.; Hatfield, G.W.; Gunsalus, R.P. Global gene expression profiling in Escherichia coli K12. The effects of oxygen availability and FNR. J. Biol. Chem. 2003, 278, 29837–29855. [Google Scholar] [CrossRef]
- Constantinidou, C.; Hobman, J.L.; Griffiths, L.; Patel, M.D.; Penn, C.W.; Cole, J.A.; Overton, T.W. A Reassessment of the FNR Regulon and Transcriptomic Analysis of the Effects of Nitrate, Nitrite, NarXL, and NarQP as Escherichia coli K12 Adapts from Aerobic to Anaerobic Growth. J. Biol. Chem. 2006, 281, 4802–4815. [Google Scholar] [CrossRef]
- Kang, Y.; Weber, K.D.; Qiu, Y.; Kiley, P.J.; Blattner, F.R. Genome-Wide Expression Analysis Indicates that FNR of Escherichia coli K-12 Regulates a Large Number of Genes of Unknown Function. J. Bacteriol. 2005, 187, 1135–1160. [Google Scholar] [CrossRef]
- Beauchene, N.A.; Myers, K.S.; Chung, D.; Park, D.M.; Weisnicht, A.M.; Keles¸, S.; Kiley, P.J. Impact of anaerobiosis on expression of the iron-responsive Fur and RyhB regulons. MBio 2015, 6, e01947-e15. [Google Scholar] [CrossRef]
- Beauchene, N.A.; Mettert, E.L.; Moore, L.J.; Keles, S.; Emily RWilley, E.R.; Patricia JKiley, P.J. O2 availability impacts iron homeostasis in Escherichia coli. Proc. Natl. Acad. Sci. USA 2017, 114, 12261–12266. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Lo, T.-M.; Sitompul, J.; Chang, M.W. Systems-level analysis of Escherichia coli response to silver nanoparticles: The roles of anaerobic respiration in microbial resistance. Biochem. Biophys. Res. Commun. 2012, 424, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Partridge, J.D.; Scott, C.; Tang, Y.; Poole, R.K.; Green, J. Escherichia coli Transcriptome Dynamics during the Transition from Anaerobic to Aerobic Conditions. J. Biol. Chem. 2006, 281, 27806–27815. [Google Scholar] [CrossRef] [PubMed]
- Bayramoglu, B.; Toubiana, D.; Gillor, O. Genome-wide transcription profiling of aerobic and anaerobic Escherichia coli biofilm and planktonic cultures. FEMS Microbiol. Lett. 2017, 364, fnx006. [Google Scholar] [CrossRef][Green Version]
- Bui, T.T.; Selvarajoo, K. Attractor Concepts to Evaluate the Transcriptome-wide Dynamics Guiding Anaerobic to Aerobic State Transition in Escherichia coli. Sci. Rep. 2020, 10, 5878. [Google Scholar] [CrossRef]
- Von Wulffen, J.; Ulmer, A.; Jäger, G.; Sawodny, O.; Feuer, R. Rapid Sampling of Escherichia coli After Changing Oxygen Conditions Reveals Transcriptional Dynamics. Genes 2017, 8, 90. [Google Scholar] [CrossRef]
- Lin-Chao, S.; Cohen, S.N. The rate of processing and degradation of antisense RNAI regulates the replication of ColE1-type plasmids in vivo. Cell 1991, 65, 1233–1242. [Google Scholar] [CrossRef]
- Blattner, F.R.; Plunkett, G., III; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The Complete Genome Sequence of Escherichia coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef]
- Snel, B.; Lehmann, G.; Bork, P.; Huynen, M.A. STRING: A web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 2000, 28, 3442–3444. [Google Scholar] [CrossRef]
- von Mering, C.; Huynen, M.; Jaeggi, D.; Schmidt, S.; Bork, P.; Snel, B. STRING: A database of predicted functional associations between proteins. Nucleic Acids Res. 2003, 31, 258–261. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612, Erratum in Nucleic Acids Res. 2021, 49, 10800. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [PubMed]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef] [PubMed]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S. AmiGO Hub, Web Presence Working Group. AmiGO: Online access to ontology and annotation data. Bioinformatics 2009, 25, 288–289. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Junker, V.L.; Apweiler, R.; Bairoch, A. Representation of functional information in the Swiss-Prot data bank. Bioinformatics 1999, 15, 1066–1067. [Google Scholar] [CrossRef]
- Gattiker, A.; Michoud, K.; Rivoire, C.; Auchincloss, A.H.; Coudert, E.; Lima, T.; Kersey, P.; Pagni, M.; Sigrist, C.J.; Lachaize, C.; et al. Automated annotation of microbial proteomes in Swiss-Prot. Comput. Biol. Chem. 2003, 27, 49–58. [Google Scholar] [CrossRef]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Santos-Zavaleta, A.; Salgado, H.; Gama-Castro, S.; Sánchez-Pérez, M.; Gómez-Romero, L.; Ledezma-Tejeida, D.; García-Sotelo, J.S.; Alquicira-Hernández, K.; José Muñiz-Rascado, L.; Peña-Loredo, P.; et al. RegulonDB v 10.5: Tackling challenges to unify classic and high throughput knowledge of gene regulation in E. coli K-12. Nucleic Acids Res. 2019, 47, D212–D220. [Google Scholar] [CrossRef] [PubMed]
- Keseler, I.M.; Mackie, A.; Santos-Zavaleta, A.; Billington, R.; Bonavides-Martinez, C.; Caspi, R.; Fulcher, C.; Gama-Castro, S.; Kothari, A.; Krummenacker, M.; et al. The EcoCyc database: Reflecting new knowledge about Escherichia coli K-12. Nucleic Acids Res. 2017, 45, D543–D550. [Google Scholar] [CrossRef] [PubMed]
- Touchon, M.; Hoede, C.; Tenaillon, O.; Barbe, V.; Baeriswyl, S.; Bidet, P.; Bingen, E.; Bonacorsi, S.; Bouchier, C.; Bouvet, O.; et al. Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet. 2009, 5, e1000344. [Google Scholar] [CrossRef]
- Bobay, L.-M.; Rocha, E.P.; Touchon, M. The adaptation of temperate bacteriophages to their host genomes. Mol. Biol. Evol. 2012, 30, 737–751. [Google Scholar] [CrossRef]
- Edlin, G.; Lin, L.; Bitner, R. Reproductive fitness of P1, P2, and Mu lysogens of Escherichia coli. J. Virol. 1977, 21, 560–564. [Google Scholar] [CrossRef]
- Barondess, J.J.; Beckwfth, J. A bacterial virulence determinant encoded by lysogenic coliphage lambda. Nature 1990, 346, 871–874. [Google Scholar] [CrossRef]
- Wang, X.; Kim, Y.; Ma, Q.; Hong, S.H.; Pokusaeva, K.; Sturino, J.M.; Wood, T.K. Cryptic prophages help bacteria cope with adverse environments. Nat. Commun. 2010, 1, 147. [Google Scholar] [CrossRef]
- Ragunathan, P.T.; Vanderpool, C.K. Cryptic-prophage-encoded small protein DicB protects Escherichia coli from phage infection by inhibiting inner membrane receptor proteins. J. Bacteriol. 2019, 201, e00475-19. [Google Scholar] [CrossRef]
- Madikonda, A.K.; Shaikh, A.; Khanra, S.; Yakkala, H.; Yellaboina, S.; Lin-Chao, S.; Siddavattam, D. Metabolic remodeling in Escherichia coli MG1655. A prophage e14-encoded small RNA, co293, post-transcriptionally regulates transcription factors HcaR and FadR. FEBS J. 2020, 287, 4767–4782. [Google Scholar] [CrossRef]
- Faubladier, M.; Bouché, J.P. Division inhibition gene dicF of Escherichia coli reveals a widespread group of prophage sequences in bacterial genomes. J. Bacteriol. 1994, 176, 1150–1156. [Google Scholar] [CrossRef] [PubMed]
- Murashko, O.N.; Lin-Chao, S. Escherichia coli responds to environmental changes using enolasic degradosomes and stabilized DicF sRNA to alter cellular morphology. Proc. Natl. Acad. Sci. USA 2017, 114, E8025–E8034. [Google Scholar] [CrossRef] [PubMed]
- Braun, M.; Killmann, H.; Maier, E.; Benz, R.; Braun, V. Diffusion through channel derivatives of the Escherichia coli FhuA transport protein. Eur. J. Biochem. 2002, 269, 4948–4959. [Google Scholar] [CrossRef] [PubMed]
- Ishihama, A.; Shimada, T.; Yamazaki, Y. Transcription profile of Escherichia coli: Genomic SELEX search for regulation targets of transcription factors. Nucleic Acids Res. 2016, 44, 2058–2074. [Google Scholar] [CrossRef] [PubMed]
- Waters, L.S.; Storz, G. Regulatory RNAs in bacteria. Cell 2009, 136, 615–628. [Google Scholar] [CrossRef]
- Vogel, J.; Luisi, B.F. Hfq and its constellation of RNA. Nat. Rev. Microbiol. 2011, 9, 578–589. [Google Scholar] [CrossRef]
- Unwin, R.D.; Griffiths, J.R.; Whetton, A.D. Simultaneous analysis of relative protein expression levels across multiple samples using iTRAQ isobaric tags with 2D nano LC–MS/MS. Nat. Protoc. 2010, 5, 1574–1582. [Google Scholar] [CrossRef]
- Christoforou, A.L.; Lilley, K.S. Isobaric tagging approaches in quantitative proteomics: The ups and downs. Anal. Bioanal. Chem. 2012, 404, 1029–1037. [Google Scholar] [CrossRef]
- Matsushita, K.; Ohnishi, T.; Kaback, H.R. NADH-ubiquinone oxidoreductases of the Escherichia coli aerobic respiratory chain. Biochemistry 1987, 26, 7732–7737. [Google Scholar] [CrossRef]
- Outten, F.W.; Huffman, D.L.; Hale, J.A.; O’Halloran, T.V. The Independent cue and cus Systems Confer Copper Tolerance during Aerobic and Anaerobic Growth in Escherichia coli. J. Biol. Chem. 2001, 276, 30670–30677. [Google Scholar] [CrossRef]
- Santiago, A.G.; Chen, T.-Y.; Genova, L.A.; Jung, W.; Thompson, A.M.G.; McEvoy, M.M.; Chen, P. Adaptor protein mediates dynamic pump assembly for bacterial metal efflux. Proc. Natl. Acad. Sci. USA 2017, 114, 6694–6699. [Google Scholar] [CrossRef]
- Zengel, J.M.; Lindahl, L. Diverse mechanisms for regulating ribosomal protein synthesis in Escherichia coli. Prog. Nucleic Acid Res. Mol. Biol. 1994, 47, 331–370. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Chang, S.J.; Lin, P.H.; Averina, O.V.; Kaberdin, V.R.; Lin-Chao, S. Regulation of ribonuclease E activity by the L4 ribosomal protein of Escherichia coli. Proc. Natl. Acad. Sci. USA 2009, 106, 864–869. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Murashko, O.N.; Lin-Chao, S. Posttranscriptional Regulation of tnaA by Protein-RNA Interaction Mediated by Ribosomal Protein L4 in Escherichia coli. J. Bacteriol. 2020, 202, e00799-19. [Google Scholar] [CrossRef] [PubMed]
- Warner, J.R.; McIntosh, K.B. How common are extraribosomal functions of ribosomal proteins? Mol. Cell 2009, 34, 3–11. [Google Scholar] [CrossRef]
- Durand, S.; Guillier, M. Transcriptional and Post-transcriptional Control of the Nitrate Respiration in Bacteria. Front. Mol. Biosci. 2021, 8, 667758. [Google Scholar] [CrossRef]
- Kumar, R.; Shimizu, K. Transcriptional regulation of main metabolic pathways of cyoA, cydB, fnr, and fur gene knockout Escherichia coli in C-limited and N-limited aerobic continuous cultures. Microb. Cell Factories 2011, 10, 3. [Google Scholar] [CrossRef]
- Lynch, A.S.; Lin, E.C. Transcriptional control mediated by the ArcA two-component response regulator protein of Escherichia coli: Characterization of DNA binding at target promoters. J. Bacteriol. 1996, 178, 6238–6249. [Google Scholar] [CrossRef]
- Park, H.; McGibbon, L.C.; Potts, A.H.; Yakhnin, H.; Romeo, T.; Babitzke, P. Translational Repression of the RpoS Antiadapter IraD by CsrA Is Mediated via Translational Coupling to a Short Upstream Open Reading Frame. MBio 2017, 8, e01355-17. [Google Scholar] [CrossRef]
- Yang, H.; Liu, M.Y.; Romeo, T. Coordinate genetic regulation of glycogen catabolism and biosynthesis in Escherichia coli via the CsrA gene product. J. Bacteriol. 1996, 178, 1012–1017. [Google Scholar] [CrossRef]
- Rahimpour, M.; Montero, M.; Almagro, G.; Viale, A.M.; Sevilla, Á.; Cánovas, M.; Muñoz, F.J.; Baroja-Fernández, E.; Bahaji, A.; Eydallin, G.; et al. GlgS, described previously as a glycogen synthesis control protein, negatively regulates motility and biofilm formation in Escherichia coli. Biochem. J. 2013, 452, 559–573. [Google Scholar] [CrossRef] [PubMed]
- Potts, A.H.; Guo, Y.; Ahmer, B.M.M.; Romeo, T. Role of CsrA in stress responses and metabolism important for Salmonella virulence revealed by integrated transcriptomics. PLoS ONE 2019, 14, e0211430. [Google Scholar] [CrossRef] [PubMed]
- Potts, A.H.; Vakulskas, C.A.; Pannuri, A.; Akhnin, H.; Babitzke, P.; Romeo, T. Global role of the bacterial post-transcriptional regulator CsrA revealed by integrated transcriptomics. Nat. Commun. 2017, 8, 1596. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Babitzke, P.; Kushner, S.R.; Romeo, T. Identification of a novel regulatory protein (CsrD) that targets the global regulatory RNAs CsrB and CsrC for degradation by RNase, E. Genes Dev. 2006, 20, 2605–2617. [Google Scholar] [CrossRef] [PubMed]
- Pulvermacher, S.C.; Stauffer, L.T.; Stauffer, G.V. The role of the small regulatory RNA GcvB in GcvB/mRNA posttranscriptional regulation of oppA and dppA in Escherichia coli. FEMS Microbiol. Lett. 2008, 281, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Lalaouna, D.; Eyraud, A.; Devinck, A.; Prévost, K.; Massé, E. GcvB small RNA uses two distinct seed regions to regulate an extensive targetome. Mol. Microbiol. 2019, 111, 473–486. [Google Scholar] [CrossRef]
- Keiler, K.C.; Waller, P.R.; Sauer, R.T. Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA. Science 1996, 271, 990–993. [Google Scholar] [CrossRef]
- Lin-Chao, S.; Wei, C.L.; Lin, Y.T. RNase E is required for the maturation of ssrA RNA and normal ssrA RNA peptide-tagging activity. Proc. Natl. Acad. Sci. USA 1999, 96, 12406–12411. [Google Scholar] [CrossRef]
- Wu, Y.-H.; Hu, C.-W.; Chien, C.-W.; Chen, Y.-J.; Huang, H.-C.; Juan, H.-F. Quantitative Proteomic Analysis of Human Lung Tumor Xenografts Treated with the Ectopic ATP Synthase Inhibitor Citreoviridin. PLoS ONE 2013, 8, e70642. [Google Scholar] [CrossRef][Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Upregulated Genes | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cluster | Operon | Functional Subcategory | Transcriptional Regulator * | Translational Regulator * | ||||
| Activator | Inhibitor | Dual ** | Activator | Inhibitor | ||||
| Anaerobic respiration/fermentation | Gene name | hyaABCDEF | Hydrogenase | AppY, ArcA, YdeO | Fis, IscR, NarL, NarP | - | - | - |
| RNA log2 fold change | 3.89_4.15_3.18_2.33_2.66_1.99 | |||||||
| Protein ratio | X_1.39_X_X_X_X | |||||||
| Gene name | hycABCDEFGHI | Energy production/transport | FhlA, IHF, ModE | NsrR | - | - | - | |
| RNA log2 fold change | 4.82_3.71_2.95_3.17_2.53_1.94_X_X_X | |||||||
| Protein ratio | X_X_X_X_X_X_X_X | |||||||
| Biofilm formation, gastrointestinal tract adaptation | Gene name | csgDEFG | Curli assembly | BasR, BolA, Cra, Crp, CsgD, IHF, MlrA, OmpR, ppGpp, RcdA | - | - | - | GcvB, McaS, OmrA, OmrB, RprA, RybB, RydC, Hfq, Rne |
| RNA log2 fold change | 3.19_4.58_4.10_X | |||||||
| Protein ratio | X_X_X_X | |||||||
| Gene name | pgaABCD | Synthesis of polysacharides | Nac, NhaR | OmpR | - | - | CsrA | |
| RNA log2 fold change | 2.42_2.36_2.68_X | |||||||
| Protein ratio | X_X_X_X | |||||||
| Gene name | oppABCDF | Oligopeptide transport | Nac | Fur, Lrp, ModE | ArcA | spermidine | GcvB, Hfq | |
| RNA log2 fold change | 3.09_2.98_2.86_2.75_2.71 | |||||||
| Protein ratio | 1.60_1.10_1.67_1.93_X | |||||||
| Acid stress resistance | Gene name | gadAX; gadXW | Acid stress regulators | AdiY, ArcA, GadE-RcsB, GadX, PhoB, ppGpp | Nac, CRP, Fis, FNR, H-NS, RcsB, RutR, TorR | GadW | GadY | - |
| RNA log2 fold change | 6.25_2.48; 2.48_1.64 | |||||||
| Protein ratio | 1.68_X; X_X | |||||||
| Gene name | gadBC | Resistance to low pH | AdiY, GadE, GadX, RcsB, ppGpp | Lrp, CRP, Fis, FliZ | GadW | - | - | |
| RNA log2 fold change | 5.35_4.69 | |||||||
| Protein ratio | 2.11_1.53 | |||||||
| Gene name | hdeAB-yhiD | Periplasmic acid stress chaperones | GadE, RcsB, PhoP, ppGpp, TorR | - | GadW, GadX | - | - | |
| RNA log2 fold change | 5.42_5.84_5.21 | |||||||
| Protein ratio | 1.79_2.92_X | |||||||
| Gene name | hdeD | Acid resistance protein | GadE, GadX, RcsB, PhoP, ppGpp | H-NS | - | - | CyaR, RprA, Hfq | |
| RNA log2 fold change | 5.87 | |||||||
| Protein ratio | X | |||||||
| Microaerobic respiration | Gene name | cydAB | Cytochrome biosynthesis | Nac, ArcA, Cra, HypT | H-NS | FNR | - | - |
| RNA log2 fold change | 2.74_2.74 | |||||||
| Protein ratio | 2.19_1.45 | |||||||
| Gene name | cydX-ybgE | Energy production/transport | Nac | - | - | - | - | |
| RNA log2 fold change | 2.83_1.98 | |||||||
| Protein ratio | X_X | |||||||
| Downregulated genes | ||||||||
| Cluster | Operon | Functional subcategory | Transcriptional regulator | Translational regulator | ||||
| activator | inhibitor | dual* | activator | inhibitor | ||||
| Cation efflux | Gene name | cusCFBA | Copper/silver efflux system | CusR, HprR, PhoB | - | - | - | - |
| RNA log2 fold change | −4.77_−4.54_−4.32_−3.40 | |||||||
| Protein ratio | 1.18_1.72_1.36_1.53 | |||||||
| Iron homeostasis | Gene name | fhuACDB | Fe3+ transport | - | Fur | - | - | - |
| RNA log2 fold change | −3.84_−2.87_−2.45_−2.01 | |||||||
| Protein ratio | X_−1.30_X_X | |||||||
| Gene name | fepA-entD | Enterobactin transporter | CRP | Fur | - | - | OmrA, OmrB | |
| RNA log2 fold change | −3.37_X | |||||||
| Protein ratio | X_X | |||||||
| Gene name | fes-ybdZ-entF-fepE | Fe acquisition/incorporation of metal ions | FNR, H-NS | Fur | - | - | - | |
| RNA log2 fold change | −5.04_−4.96_−3.10_−1.69 | |||||||
| Protein ratio | X_X_X_X | |||||||
| Gene name | tonB | Fe acquisition/energy production/siderophore, colicin, bacteriocin transport | - | - | Fur | - | - | |
| RNA log2 fold change | −3.16 | |||||||
| Protein ratio | X | |||||||
| Gene name | feoABC | Fe2+ transport | FNR, OmpR | ArcA, Fur | NagC | - | - | |
| RNA log2 fold change | −3.05_−3.13_−2.68 | |||||||
| Protein ratio | X_X_X | |||||||
| Sulfur-involving pathways | Gene name | sufABCDSE | Fe-S transport protein in Fe-S cluster assembly | IHF, IscR, OxyR, ppGpp | Fur, NsrR | - | - | - |
| RNA log2 fold change | −2.35_−2.17_−2.15_−1.81_−1.64_X | |||||||
| Protein ratio | −1.78_−1.09_−1.27_1.08_−1.20_−1.05 | |||||||
| Gene name | iscRSUA | Fe-S cluster biogenesis | IscR | - | - | - | FnrS, RyhB, Hfq | |
| RNA log2 fold change | −2.13_−1.67_−1.58_X | |||||||
| Protein ratio | −1.26_−1.76_−1.58_−1.58 | |||||||
| Aerobic respiration | Gene name | sdhCDAB-sucABCD-sdhX | TCA cycle I | Nac, CRP, Fur | FNR | ArcA | - | RybB, RyhB, Spf |
| RNA log2 fold change | X_−1.65_−2.00_−2.01_−2.02_−2.01_−2.08_−2.15_X | |||||||
| Protein ratio | −1.30_−1.12_−1.11_−1.01_−1.80_−1.33_−1.15_−1.41_X | |||||||
| De novo synthesis of nucleosides | Gene name | nrdHIEF | Nucleotide and nucleoside conversions | IscR | Fur, NrdR | - | - | - |
| RNA log2 fold change | −5.89_−4.29_−3.43_−4.20 | |||||||
| Protein ratio | X_X_X_−1.41 | |||||||
| Prophage | Operon/Gene name | RNA (Up/Down) | Protein (Increased/Decreased) | Transcriptional Regulator * | Translational Regulator * | ||||
|---|---|---|---|---|---|---|---|---|---|
| Activator | Inhibitor | Dual | Activator | Inhibitor | Attenuator | ||||
| DPL12 | appY | 1.84 | X | ArcA | DpiA, H-NS | -- | -- | -- | -- |
| ompT | 0.37 | 2.07 | Lrp, PhoP | Nac | -- | -- | OmrA, OmrB, Hfq | -- | |
| rzoD | 3.02 | X | -- | -- | -- | -- | -- | -- | |
| ybcLM | 1.52_1.36 | X_X | -- | -- | -- | -- | -- | -- | |
| ybcV | 1.67 | X | -- | Nac | -- | -- | -- | -- | |
| ybcW | 1.79 | X | -- | -- | -- | -- | -- | -- | |
| ylcH | 2.03 | X | -- | -- | -- | -- | -- | -- | |
| e14 | ymfTLMNR_beeE_jayE_ymfQ_ycfK-tfaP | 0.71_0.91_0.78_X_2.34_1.27_X_0.62_1.86_1.36 | X_X_X_X_X_X_X_X_X_X | -- | -- | -- | -- | -- | -- |
| Qin | cspI | 2.63 | X | BasR | Fis | -- | -- | -- | -- |
| pinQ | 2.36 | X | -- | Nac | -- | -- | -- | -- | |
| ydfV | 4.79 | X | -- | -- | -- | -- | -- | -- | |
| Rac | kilR_ydaE | 2.32_2.04 | X_X | -- | -- | -- | -- | -- | -- |
| sieB | 2.24 | X | -- | -- | -- | -- | -- | -- | |
| ydaGF | 1.53_X | X_X | -- | -- | -- | -- | -- | -- | |
| ynaE | 2.23 | X | GlaR | -- | -- | -- | -- | -- | |
| phage related | fhuA(CDB) | −3.84_(−2.87_−2.45_−2.01) | X_(−1.30_X_X) | -- | Fur | -- | -- | -- | -- |
| TF | RNA (Up/Down) |
|---|---|
| CusR | −1.51 |
| Dps | 1.13 |
| EvgA | 1.3 |
| FecI | −3.15 |
| Fis | −1.11 |
| GadW | 1.64 |
| GadX | 2.48 |
| IscR | −2.13 |
| LeuO | 2.21 |
| PdhR | 1.16 |
| PutA | 1.14 |
| SoxS | −1.12 |
| IscR Regulated Operon/Genes | RNA (Up/Down) | Protein (Increased/Decreased) |
|---|---|---|
| hyaABCDEF | 3.89_4.15_3.18_2.33_ 2.65_1.99 | 100 *_1.40_X_X_X_X |
| sufABCDSE | −2.35_−2.17_−2.15_−1.81_−1.64_X | −1.78_−1.09_−1.27_1.08_−1.20_−1.05 |
| ydiU | −2.01 | X |
| nrdHIEF | −5.89_−4.29_−3.43_−4.20 | X_X_X_−1.41 |
| torT | 1.67 | X |
| iscRSUA | −2.13_−1.67_−1.58_X | −1.26_−1.76_−1.58_−1.52 |
| Fur- Fe2+ Regulated Operon | RNA (Up/Down) | Protein (Increased/Decreased) |
|---|---|---|
| ftnA | 4.27 | 2.4 |
| fumB | 1.7 | X |
| ompF | 0.64 | −1.62 |
| sdhCDAB-sucABCD | −1.51_−1.65_−2.00_−2.01_−2.02_−2.01_−2.08_−2.15 | −1.30_−1.12_−1.11_−1.01_−1.80_−1.33_−1.15_−1.41 |
| tonB | −3.16 | X |
| entS | −4.65 | X |
| gdhA | 1.1 | 1.92 |
| glnK-amtB | 3.70_3.42 | 4.42_3.94 |
| aspC | 0.01 | 1.73 |
| cirA | −2.5 | 1.27 |
| entCEBAH | −7.21_−6.18_−5.70_−4.07_−3.90 | −1.49_X_X_X_−1.17 |
| exbBD | −4.20_−4.18 | −1.60_−1.11 |
| exbD | −4.18 | −1.11 |
| fecIR | −3.15_−2.58 | X_X |
| feoABC | −3.05_−3.13_−2.68 | X_X_X |
| fepA-entD | −3.37_−0.98 | X_X |
| fepB | −3.91 | X |
| fepDGC | −3.96_−3.37_−4.44 | X_X_X |
| fes-ybdZ-entF-fepE | −5.04_−4.96_−3.10_−1.69 | X_X_X_X |
| fhuACDB | −3.84_−2.87_−2.45_−2.01 | X_−1.30_X−X |
| fhuE | −3.12 | X |
| fhuF | −6.12 | X |
| fumC | −1.6 | −1.59 |
| hmp | 0.6 | 1.65 |
| nac | 3.7 | X |
| ndh | 2.21 | 1.32 |
| nrdHIEF | −5.89_−4.29_−3.43_−4.20 | X_X_X_−1.41 |
| oppABCDF | 3.09_2.98_2.86_2.75_2.71 | 1.60_1.10_1.67_1.93_X |
| ryhB | −3.04 | X |
| sodA | −5.8 | −2.2 |
| sufABCDSE | −2.35_−2.17_−2.15_−1.81_−1.64_−1.57 | −1.78_−1.09_−1.27_1.09_−1.20_−1.05 |
| yjjZ | −2.03 | X |
| yqjH | −3.4 | −1.29 |
| Increased Proteins | |||||
|---|---|---|---|---|---|
| Biological Function | Operon | Translational Regulator * | |||
| Activator | Inhibitor | Attenuator | |||
| ATP metabolic process | Gene name | cydAB | -- | -- | -- |
| Protein ratio | 2.19_1.45 | ||||
| RNA log2 fold change | 2.74_2.74 | ||||
| Gene name | ldhA | -- | -- | -- | |
| Protein ratio | 1.93 | ||||
| RNA log2 fold change | 1.97 | ||||
| Gene name | atpBEFHAGDC | -- | -- | -- | |
| Protein ratio | 1.28_1.57_1.45_1.67_1.50_1.43_1.67_1.63 | ||||
| RNA log2 fold change | 0.29_0.14_0.20_0.18_0.20_0.17_0.17_0.45 | ||||
| Gene name | epd-pgk-fbaA | -- | -- | -- | |
| Protein ratio | −1.13_1.54_1.50 | ||||
| RNA log2 fold change | −0.10_0.82_0.70 | ||||
| Gene name | gpmI-envC-yibQ | -- | -- | -- | |
| Protein ratio | 1.68_X_X | ||||
| RNA log2 fold change | 0.62_−0.22_−0.33 | ||||
| Gene name | eno | -- | -- | -- | |
| Protein ratio | 1.80 | ||||
| RNA log2 fold change | 1.35 | ||||
| Gene name | tpiA | -- | -- | -- | |
| Protein ratio | 1.73 | ||||
| RNA log2 fold change | 0.51 | ||||
| Gene name | pfkA | -- | -- | -- | |
| Protein ratio | 1.53 | ||||
| RNA log2 fold change | 0.35 | ||||
| Gene name | pfkB | -- | -- | -- | |
| Protein ratio | 1.50 | ||||
| RNA log2 fold change | 0.27 | ||||
| Gene name | glk | -- | -- | -- | |
| Protein ratio | 1.91 | ||||
| RNA log2 fold change | 0.75 | ||||
| Pyruvate metabolic process | Gene name | ispDF | -- | -- | -- |
| Protein ratio | 1.96_1.09 | ||||
| RNA log2 fold change | −0.39_−0.27 | ||||
| Gene name | epd-pgk-fbaA | -- | -- | -- | |
| Protein ratio | −1.13_1.54_1.50 | ||||
| RNA log2 fold change | −0.10_0.82_0.70 | ||||
| Gene name | gpmI-envC-yibQ | -- | -- | -- | |
| Protein ratio | 1.68_X_X | ||||
| RNA log2 fold change | 0.62_−0.22_−0.33 | ||||
| Gene name | eno | -- | -- | -- | |
| Protein ratio | 1.80 | ||||
| RNA log2 fold change | 1.35 | ||||
| Gene name | tpiA | -- | -- | -- | |
| Protein ratio | 1.73 | ||||
| RNA log2 fold change | 0.51 | ||||
| Gene name | pfkA | -- | -- | -- | |
| Protein ratio | 1.53 | ||||
| RNA log2 fold change | 0.35 | ||||
| Gene name | pfkB | -- | -- | -- | |
| Protein ratio | 1.50 | ||||
| RNA log2 fold change | 0.27 | ||||
| Gene name | glk | -- | -- | -- | |
| Protein ratio | 1.91 | ||||
| RNA log2 fold change | 0.75 | ||||
| Glycolytic process | Gene name | epd-pgk-fbaA | -- | -- | -- |
| Protein ratio | −1.13_1.54_1.50 | ||||
| RNA log2 fold change | −0.10_0.82_0.70 | ||||
| Gene name | gpmI-envC-yibQ | -- | -- | -- | |
| Protein ratio | 1.68_X_X | ||||
| RNA log2 fold change | 0.62_−0.22_−0.33 | ||||
| Gene name | eno | -- | -- | -- | |
| Protein ratio | 1.80 | ||||
| RNA log2 fold change | 1.35 | ||||
| Gene name | tpiA | -- | -- | -- | |
| Protein ratio | 1.73 | ||||
| RNA log2 fold change | 0.51 | ||||
| Gene name | pfkA | -- | -- | -- | |
| Protein ratio | 1.53 | ||||
| RNA log2 fold change | 0.35 | ||||
| Gene name | pfkB | -- | -- | -- | |
| Protein ratio | 1.50 | ||||
| RNA log2 fold change | 0.27 | ||||
| Gene name | glk | -- | -- | -- | |
| Protein ratio | 1.91 | ||||
| RNA log2 fold change | 0.75 | ||||
| Glucose metabolic process | Gene name | sdaA | -- | -- | -- |
| Protein ratio | 2.07 | ||||
| RNA log2 fold change | 0.78 | ||||
| Gene name | focA-pflB | -- | -- | -- | |
| Protein ratio | X_2.03 | ||||
| RNA log2 fold change | 1.01_1.47 | ||||
| Gene name | pckA | -- | -- | -- | |
| Protein ratio | 1.60 | ||||
| RNA log2 fold change | 0.69 | ||||
| Gene name | epd-pgk-fbaA | -- | -- | -- | |
| Protein ratio | −1.13_1.54_1.50 | ||||
| RNA log2 fold change | −0.10_0.82_0.70 | ||||
| Gene name | gpmI-envC-yibQ | -- | -- | -- | |
| Protein ratio | 1.68_X_X | ||||
| RNA log2 fold change | 0.62_−0.22_−0.33 | ||||
| Gene name | tpiA | -- | -- | -- | |
| Protein ratio | 1.73 | ||||
| RNA log2 fold change | 0.51 | ||||
| Gene name | pfkA | -- | -- | -- | |
| Protein ratio | 1.53 | ||||
| RNA log2 fold change | 0.35 | ||||
| Purine-containing compound metabolic process | Gene name | allDC-ylbA | -- | -- | -- |
| Protein ratio | X-X−1.51 | ||||
| RNA log2 fold change | 0.25_0.47_0.56 | ||||
| Gene name | atpBEFHAGDC | -- | -- | -- | |
| Protein ratio | 1.28_1.57_1.45_1.67_1.50_1.43_1.67_1.63 | ||||
| RNA log2 fold change | 0.29_0.14_0.20_0.18_0.20_0.17_0.17_0.45 | ||||
| Gene name | ackA-pta | -- | SdhX | -- | |
| Protein ratio | 1.99_−1.13 | ||||
| RNA log2 fold change | 1.19–0.97 | ||||
| Gene name | epd-pgk-fbaA | -- | -- | -- | |
| Protein ratio | −1.13_1.54_1.50 | ||||
| RNA log2 fold change | −0.10_0.82_0.70 | ||||
| Gene name | gpmI-envC-yibQ | -- | -- | -- | |
| Protein ratio | 1.68_X_X | ||||
| RNA log2 fold change | 0.62_−0.22_−0.33 | ||||
| Gene name | eno | -- | -- | -- | |
| Protein ratio | 1.80 | ||||
| RNA log2 fold change | 1.35 | ||||
| Gene name | tpiA | -- | -- | -- | |
| Protein ratio | 1.73 | ||||
| RNA log2 fold change | 0.51 | ||||
| Gene name | pfkA | -- | -- | -- | |
| Protein ratio | 1.53 | ||||
| RNA log2 fold change | 0.35 | ||||
| Gene name | pfkB | -- | -- | -- | |
| Protein ratio | 1.50 | ||||
| RNA log2 fold change | 0.27 | ||||
| Gene name | glk | -- | -- | -- | |
| Protein ratio | 1.91 | ||||
| RNA log2 fold change | 0.75 | ||||
| Ion homeostasis | Gene name | gadAX | GadY (onto GadX) | -- | -- |
| Protein ratio | 1.68_X | ||||
| RNA log2 fold change | 6.25_2.48 | ||||
| Gene name | cusCFBA | -- | -- | -- | |
| Protein ratio | 1.18_1.72_1.36_1.53 | ||||
| RNA log2 fold change | −4.77_−4.54_−4.32_−3.40 | ||||
| Gene name | copA | -- | -- | -- | |
| Protein ratio | 1.67 | ||||
| RNA log2 fold change | −1.11 | ||||
| Gene name | chaA | -- | -- | -- | |
| Protein ratio | 1.77 | ||||
| RNA log2 fold change | 0.55 | ||||
| Gene name | gadBC | -- | -- | -- | |
| Protein ratio | 2.12_1.53 | ||||
| RNA log2 fold change | 5.35_4.69 | ||||
| Gene name | bfd-bfr | -- | RyhB | -- | |
| Protein ratio | X_1.59 | ||||
| RNA log2 fold change | −3.99_1.32 | ||||
| Gene name | adiA | -- | -- | -- | |
| Protein ratio | 1.78 | ||||
| RNA log2 fold change | 0.31 | ||||
| Gene name | ftnA | -- | -- | -- | |
| Protein ratio | 2.40 | ||||
| RNA log2 fold change | 4.27 | ||||
| Decreased proteins | |||||
| Biological function | Operon | Translational regulator * | |||
| activator | inhibitor | attenuator | |||
| Ribosome assembly | Gene name | rpsB-tsf | -- | RpsB | -- |
| Protein ratio | −1.99_−1.03 | ||||
| RNA log2 fold change | −0.28_−0.21 | ||||
| Gene name | rpsMKD-rpoA-rplQ | -- | RpsD | -- | |
| Protein ratio | −1.67_−1.54_−1.55_−1.17_−1.81 | ||||
| RNA log2 fold change | −0.51_−0.63_−0.61_−0.61_−0.70 | ||||
| Gene name | metY-yhbC-nusA-infB-rbfA-truB-rpsO-pnp | -- | -- | -- | |
| Protein ratio | X_−1.70_−1.31_−1.38_−1.22_−1.21_−1.77_−1.12 | ||||
| RNA log2 fold change | 0.29_−0.50_−0.39_−0.39_−0.20_−0.56_−0.50_−0.45_−0.38 | ||||
| Gene name | rpsJ-rplCDWB-rpsS-rplV-rpsC-rplP-rpmC-rpsQ | -- | -- | RplD | |
| Protein ratio | −1.81_−1.88_−1.98_−1.97_−1.97_−1.94_−1.67_−1.84_−1.47_−1.57_−1.76 | ||||
| RNA log2 fold change | −0.67_−0.84_−0.89_−0.80_−0.92_−0.98_−0.86_−0.71_−0.67_−0.66_−0.78 | ||||
| Gene name | rplNXE-rpsNH-rplFR-rpsE-rpmD-rplO-secY-rpmJ | -- | RpsH | -- | |
| Protein ratio | −1.67_−1.68_−1.99_−2.08_−1.57_−1.78_−1.64_−1.63_−1.84_−1.73_−1.13_−1.54 | ||||
| RNA log2 fold change | −0.36_−0.70_−0.62_−0.60_−0.78_−0.70_−0.69_−0.70_−0.62_−0.70_−0.60_−0.34 | ||||
| Gene name | rpsU-dnaG-rpoD | -- | -- | -- | |
| Protein ratio | −1.87_X_1.38 | ||||
| RNA log2 fold change | −0.70_−0.36_−0.16 | ||||
| Gene name | rpsA-ihfB | -- | RpsA | -- | |
| Protein ratio | −1.82_−1.39 | ||||
| RNA log2 fold change | −0.46_−0.00 | ||||
| Gene name | rpsT | -- | -- | -- | |
| Protein ratio | −1.55 | ||||
| RNA log2 fold change | −0.51 | ||||
| Gene name | rpsLG-fusA-tufA | -- | RpsG | -- | |
| Protein ratio | −1.77_−1.68_−1.14_X | ||||
| RNA log2 fold change | −0.42_−0.47_−0.63_−0.35 | ||||
| Gene name | rplKAJL-rpoBC | -- | RplA | -- | |
| Protein ratio | −1.55_−1.49_−1.42_−1.44_−1.13_−1.12 | ||||
| RNA log2 fold change | −0.50_−0.52_−0.65_−0.74_−0.43_−0.46 | ||||
| Gene name | yhbY | -- | -- | -- | |
| Protein ratio | −1.67 | ||||
| RNA log2 fold change | −0.36 | ||||
| Gene name | thrS-infC-rpmI-rplT-pheMST-ihfA | -- | RplT | -- | |
| Protein ratio | −1.03_−1.51_−1.17_−2.19_X_1.44_1.38_−1.36 | ||||
| RNA log2 fold change | −0.26_−0.02_−0.03_−0.14_X_−0.07_−0.11_−0.36 | ||||
| Gene name | rpmBG-mutM | -- | -- | -- | |
| Protein ratio | −1.80_−1.80_X | ||||
| RNA log2 fold change | −0.87_−1.12_−0.93 | ||||
| Gene name | rplY | -- | RplY | -- | |
| Protein ratio | −2.34 | ||||
| RNA log2 fold change | −0.43 | ||||
| Gene name | rlmE-ftsH | -- | -- | -- | |
| Protein ratio | −4.10_−1.09 | ||||
| RNA log2 fold change | −0.17_−0.25 | ||||
| Gene name | yceD-rpmF; rpmF-plsX-fabHDG | -- | -- | -- | |
| Protein ratio | −1.81_−1.62; −1.62_1.03_−1.17_1.18_−1.17 | ||||
| RNA log2 fold change | −0.64_−0.51; −0.51_−0.06_−0.16_0.00_−0.34 | ||||
| Gene name | rplU-rpmA | -- | -- | -- | |
| Protein ratio | −1.89_−1.61 | ||||
| RNA log2 fold change | −0.65_−0.61 | ||||
| Negative regulation of translation | Gene name | hfq-hflXKC | -- | -- | -- |
| Protein ratio | −1.67_X_1.71_1.21 | ||||
| RNA log2 fold change | −0.36_−0.25_−0.28_−0.33 | ||||
| Gene name | rplM-rpsI | -- | RplM | -- | |
| Protein ratio | −1.95_−1.75 | ||||
| RNA log2 fold change | −0.74_−0.64 | ||||
| Gene name | rpsLG-fusA-tufA | -- | RpsG | -- | |
| Protein ratio | −1.77_−1.68_−1.14_X | ||||
| RNA log2 fold change | −0.42_−0.47_−0.63_−0.35 | ||||
| Gene name | rpsMKD-rpoA-rplQ | -- | RpsD | -- | |
| Protein ratio | −1.67_−1.54_−1.55_−1.17_1.67 | ||||
| RNA log2 fold change | −0.51_−0.63_−0.61_−0.61_−0.70 | ||||
| Gene name | rpsJ-rplCDWB-rpsS-rplV-rpsC-rplP-rpmC-rpsQ | -- | -- | RplD | |
| Protein ratio | −1.81_−1.88_−1.98_−1.97_−1.97_−1.94_−1.67_−1.84_−1.47_−1.57_−1.76 | ||||
| RNA log2 fold change | −0.67_−0.84_−0.89_−0.80_−0.92_−0.98_−0.86_−0.71_−0.67_−0.66_−0.78 | ||||
| Gene name | rpsA-ihfB | -- | RpsA | -- | |
| Protein ratio | −1.82_−1.39 | ||||
| RNA log2 fold change | −0.46_−0.00 | ||||
| Gene name | thrS-infC-rpmI-rplT-pheMST-ihfA | -- | RplT | -- | |
| Protein ratio | −1.03_−1.51_−1.17_−2.19_X_1.44_1.38_−1.36 | ||||
| RNA log2 fold change | −0.26_−0.02_−0.03_−0.14_X_−0.07_−0.11_−0.36 | ||||
| Gene name | rplY | -- | RplY | -- | |
| Protein ratio | −2.34 | ||||
| RNA log2 fold change | −0.43 | ||||
| Response to antibiotic | Gene name | pstSCAB-phoU | -- | -- | -- |
| Protein ratio | −1.41_X_X_X_−1.85 | ||||
| RNA log2 fold change | −1.02_−1.01_−0.78_−0.93 | ||||
| Gene name | macAB | -- | -- | -- | |
| Protein ratio | −1.10_−1.82 | ||||
| RNA log2 fold change | 0.10_−0.05 | ||||
| Gene name | amiA-hemF | -- | -- | -- | |
| Protein ratio | −1.68_−1.13 | ||||
| RNA log2 fold change | −1.66_−1.41 | ||||
| Gene name | rpmBG-mutM | -- | -- | -- | |
| Protein ratio | −1.80_−1.80_X | ||||
| RNA log2 fold change | −0.87_−1.12_−0.93 | ||||
| Gene name | efeOB | -- | -- | -- | |
| Protein ratio | −2.97_X | ||||
| RNA log2 fold change | −6.19_−4.74 | ||||
| Gene name | ldtD/ycbB | -- | -- | -- | |
| Protein ratio | −7.34 | ||||
| RNA log2 fold change | −0.11 | ||||
| Gene name | rpsMKD-rpoA-rplQ | -- | RpsD | -- | |
| Protein ratio | −1.67_−1.54_−1.55_−1.17_1.67 | ||||
| RNA log2 fold change | −0.51_−0.63_−0.61_−0.61_−0.70 | ||||
| Gene name | rpsJ-rplCDWB-rpsS-rplV-rpsC-rplP-rpmC-rpsQ | -- | -- | RplD | |
| Protein ratio | −1.81_−1.88_−1.98_−1.97_−1.97_−1.94_−1.67_−1.84_−1.47_−1.57_−1.76 | ||||
| RNA log2 fold change | −0.67_−0.84_−0.89_−0.80_−0.92_−0.98_−0.86_−0.71_−0.67_−0.66_−0.78 | ||||
| Gene name | rplNXE-rpsNH-rplFR-rpsE-rpmD-rplO-secY-rpmJ | -- | RpsH | -- | |
| Protein ratio | −1.67_−1.68_−1.99_−2.08_−1.57_−1.78_−1.64_−1.63_−1.84_−1.73_−1.13_−1.54 | ||||
| RNA log2 fold change | −0.36_−0.70_−0.62_−0.60_−0.78_−0.70_−0.69_−0.70_−0.62_−0.70_−0.60_−0.34 | ||||
| Gene name | rpsLG-fusA-tufA | -- | RpsG | -- | |
| Protein ratio | −1.77_−1.68_−1.14_X | ||||
| RNA log2 fold change | −0.42_−0.47_−0.63_−0.35 | ||||
| Posttranscriptional regulation of gene expression | Gene name | proQ-prc | -- | -- | -- |
| Protein ratio | −1.68_−1.24 | ||||
| RNA log2 fold change | −0.44_−0.34 | ||||
| Gene name | rppH-ptsP | -- | -- | -- | |
| Protein ratio | −1.66_−1.27 | ||||
| RNA log2 fold change | −0.21_−0.11 | ||||
| Gene name | rpsMKD-rpoA-rplQ | -- | RpsD | -- | |
| Protein ratio | −1.67_−1.54_−1.55_−1.17_1.67 | ||||
| RNA log2 fold change | −0.51_−0.63_−0.61_−0.61_−0.70 | ||||
| Gene name | metY-yhbC-nusA-infB-rbfA-truB-rpsO-pnp | -- | -- | -- | |
| Protein ratio | X_−1.70_−1.31_−1.38_−1.22_−1.21_−1.77_−1.12 | ||||
| RNA log2 fold change | 0.29_−0.50_−0.39_−0.39_−0.20_−0.56_−0.50_−0.45_−0.38 | ||||
| Gene name | rpsJ-rplCDWB-rpsS-rplV-rpsC-rplP-rpmC-rpsQ | -- | -- | RplD | |
| Protein ratio | −1.81_−1.88_−1.98_−1.97_−1.97_−1.94_−1.67_−1.84_−1.47_−1.57_−1.76 | ||||
| RNA log2 fold change | −0.67_−0.84_−0.89_−0.80_−0.92_−0.98_−0.86_−0.71_−0.67_−0.66_−0.78 | ||||
| Gene name | rplNXE-rpsNH-rplFR-rpsE-rpmD-rplO-secY-rpmJ | -- | RpsH | -- | |
| Protein ratio | −1.67_−1.68_−1.99_−2.08_−1.57_−1.78_−1.64_−1.63_−1.84_−1.73_−1.13_−1.54 | ||||
| RNA log2 fold change | −0.36_−0.70_−0.62_−0.60_−0.78_−0.70_−0.69_−0.70_−0.62_−0.70_−0.60_−0.34 | ||||
| Gene name | rpsA-ihfB | -- | RpsA | -- | |
| Protein ratio | −1.82_−1.39 | ||||
| RNA log2 fold change | −0.46_−0.00 | ||||
| Gene name | rpsLG-fusA-tufA | -- | RpsG | -- | |
| Protein ratio | −1.77_−1.68_−1.14_X | ||||
| RNA log2 fold change | −0.42_−0.47_−0.63_−0.35 | ||||
| Gene name | thrS-infC-rpmI-rplT-pheMST-ihfA | -- | RplT | -- | |
| Protein ratio | −1.03_−1.51_−1.17_−2.19_X_1.44_1.38_−1.36 | ||||
| RNA log2 fold change | −0.26_−0.02_−0.03_−0.14_X_−0.07_−0.11_−0.36 | ||||
| Gene name | rplY | -- | RplY | -- | |
| Protein ratio | −2.34 | ||||
| RNA log2 fold change | −0.43 | ||||
| Gene name | hfq-hflXKC | -- | -- | -- | |
| Protein ratio | −1.67_X_1.71_1.21 | ||||
| RNA log2 fold change | −0.36_−0.25_−0.28_−0.33 | ||||
| Iron-sulfur cluster assembly | Gene name | iscRSUA | -- | Hfq, RyhB | -- |
| Protein ratio | −1.26_−1.76_−1.58_−1.58 | ||||
| RNA log2 fold change | −2.13_−1.67_−1.58_−1.44 | ||||
| Gene name | erpA | -- | RyhB | -- | |
| Protein ratio | −1.99 | ||||
| RNA log2 fold change | −0.82 | ||||
| Gene name | sufABCDSE | -- | -- | -- | |
| Protein ratio | −1.78_−1.09_−1.27_−0.92_−1.20_−1.05 | ||||
| RNA log2 fold change | −2.35_−2.17_−2.15_−1.81_−1.64_−1.57 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liou, G.-G.; Chao Kaberdina, A.; Wang, W.-S.; Kaberdin, V.R.; Lin-Chao, S. Combined Transcriptomic and Proteomic Profiling of E. coli under Microaerobic versus Aerobic Conditions: The Multifaceted Roles of Noncoding Small RNAs and Oxygen-Dependent Sensing in Global Gene Expression Control. Int. J. Mol. Sci. 2022, 23, 2570. https://doi.org/10.3390/ijms23052570
Liou G-G, Chao Kaberdina A, Wang W-S, Kaberdin VR, Lin-Chao S. Combined Transcriptomic and Proteomic Profiling of E. coli under Microaerobic versus Aerobic Conditions: The Multifaceted Roles of Noncoding Small RNAs and Oxygen-Dependent Sensing in Global Gene Expression Control. International Journal of Molecular Sciences. 2022; 23(5):2570. https://doi.org/10.3390/ijms23052570
Chicago/Turabian StyleLiou, Gunn-Guang, Anna Chao Kaberdina, Wei-Syuan Wang, Vladimir R. Kaberdin, and Sue Lin-Chao. 2022. "Combined Transcriptomic and Proteomic Profiling of E. coli under Microaerobic versus Aerobic Conditions: The Multifaceted Roles of Noncoding Small RNAs and Oxygen-Dependent Sensing in Global Gene Expression Control" International Journal of Molecular Sciences 23, no. 5: 2570. https://doi.org/10.3390/ijms23052570
APA StyleLiou, G.-G., Chao Kaberdina, A., Wang, W.-S., Kaberdin, V. R., & Lin-Chao, S. (2022). Combined Transcriptomic and Proteomic Profiling of E. coli under Microaerobic versus Aerobic Conditions: The Multifaceted Roles of Noncoding Small RNAs and Oxygen-Dependent Sensing in Global Gene Expression Control. International Journal of Molecular Sciences, 23(5), 2570. https://doi.org/10.3390/ijms23052570