
Ebola Entry Inhibitors Discovered from Maesa perlarius





Abstract
:1. Introduction
2. Results
2.1. Stem Extract of M. perlarius Is Identified as a Potential Anti-EBOV Lead
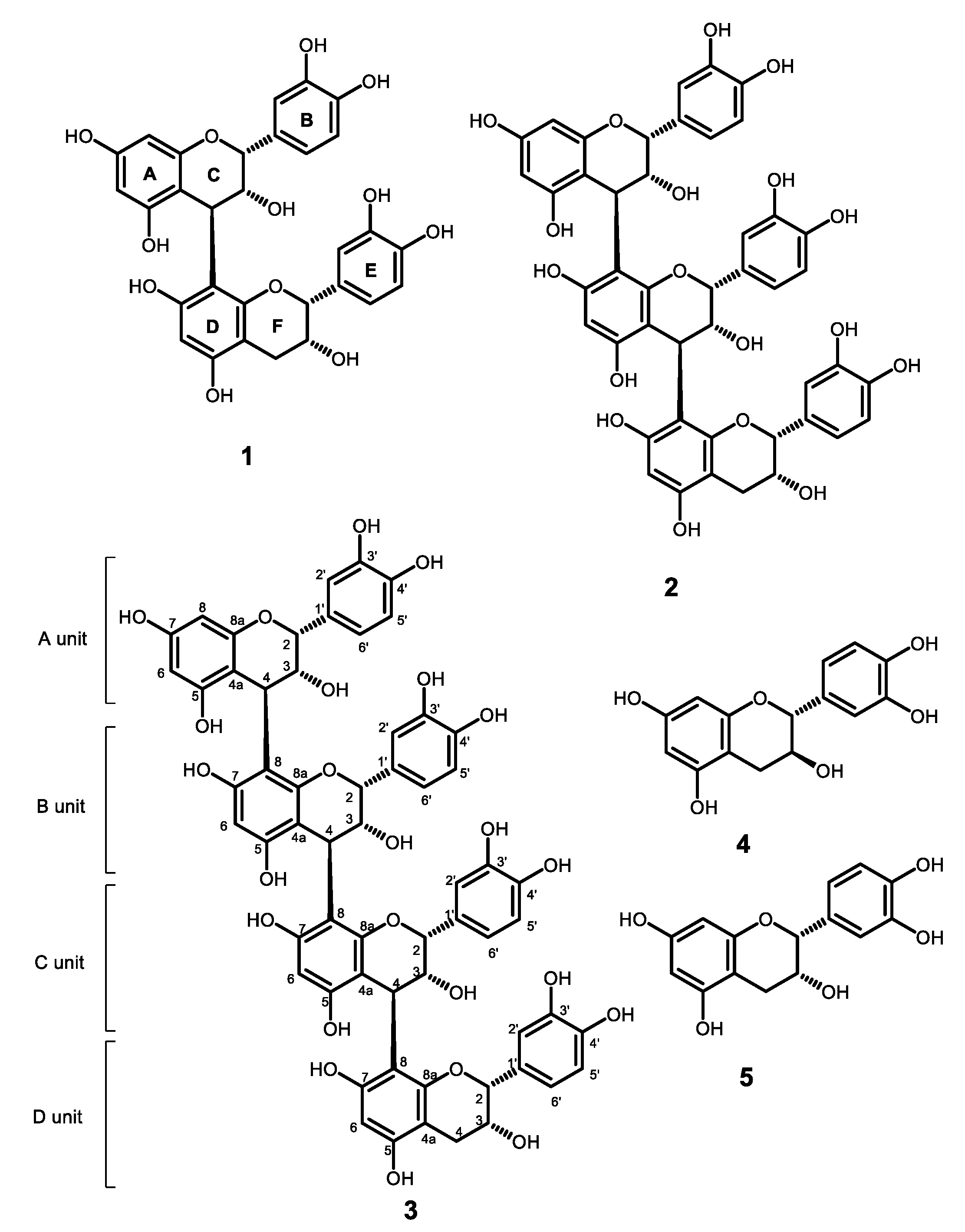
2.2. Flavan-3-Ols and Procyanidins Identified from the Stems of M. perlarius
2.3. Anti-EBOV Activity Evaluation of Proanthocyanidins and Flavan-3-Ols
2.4. Synthesis of ECG Analogues from (−)-Epicatechin to Study the Functional Group Requirement for Anti-EBOV Activity
2.5. Molecular Docking Analysis and Microscale Thermophoresis (MST) Measurement Reveal EBOV-GP as the Potential Molecular Target of B-Type Procyanidins and Flavan-3-Ol Analogues
3. Discussion
4. Materials and Methods
4.1. General
4.2. Plant Materials
4.3. Extraction and Isolation
4.4. Synthesis of ECG Derivatives
4.5. Bioactivity Evaluation Using EBOV Pseudotyped Virus
4.5.1. Cell Cultures
4.5.2. Cytotoxicity Assay
4.5.3. Production of EBOV and VSV Pseudovirions
4.5.4. Pseudovirion Inhibition Screening Assay
4.6. Antiviral Assay against Infectious Ebola Virus
4.7. Molecular Docking Analysis
4.7.1. Preparation of Ligands for Docking
4.7.2. Preparation of Proteins for Docking
4.7.3. Molecular Docking Using AutoDock Vina
4.8. Microscale Thermophoresis (MST) Experiment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Centers for Disease Control and Prevention (CDC). Signs and Symptoms (Ebola Virus). Available online: https://www.cdc.gov/vhf/ebola/symptoms/index.html (accessed on 1 February 2022).
- World Health Organization (WHO). Ebola Virus Disease. World Health Organization. Available online: https://www.afro.who.int/health-topics/ebola-virus-disease (accessed on 1 February 2022).
- Centers for Disease Control and Prevention (CDC). History of Ebola Virus Disease (EVD) Outbreaks. Available online: https://www.cdc.gov/vhf/ebola/history/chronology.html?CDC_AA_refVal=https%3A%2F%2Fwww.cdc.gov%2Fvhf%2Febola%2Foutbreaks%2Fhistory%2Fchronology.html (accessed on 1 February 2022).
- Schafer, A.; Xiong, R.; Cooper, L.; Nowar, R.; Lee, H.; Li, Y.; Ramirez, B.E.; Peet, N.P.; Caffrey, M.; Thatcher, G.R.J.; et al. Evidence for distinct mechanisms of small molecule inhibitors of filoviral entry. PLoS Pathog. 2021, 17, e1009312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.J.; Rumschlag-Booms, E.; Guan, Y.F.; Wang, D.Y.; Liu, K.L.; Li, W.F.; Nguyen, V.H.; Cuong, N.M.; Soejarto, D.D.; Fong, H.H.S.; et al. Potent inhibitor of drug-resistant HIV-1 strains identified from the medicinal plant Justicia gendarussa. J. Nat. Prod. 2017, 80, 1798–1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumschlag-Booms, E.; Zhang, H.; Soejarto, D.D.; Fong, H.H.S.; Rong, L. Development of an antiviral screening protocol: One-Stone-Two-Birds. J. Antivir. Antiretrovir. 2011, 3, 8–10. [Google Scholar] [CrossRef] [Green Version]
- Li, W.F.; Wang, J.; Zhang, J.J.; Song, X.; Ku, C.F.; Zou, J.; Li, J.X.; Rong, L.J.; Pan, L.T.; Zhang, H.J. Henrin A: A new anti-HIV ent-kaurane diterpene from Pteris henryi. Int. J. Mol. Sci. 2015, 16, 27978–27987. [Google Scholar] [CrossRef]
- Zhang, H.J.; Rumschlag-Booms, E.; Guan, Y.F.; Liu, K.L.; Wang, D.Y.; Li, W.F.; Nguyen, V.H.; Cuong, N.M.; Soejarto, D.D.; Fong, H.H.S.; et al. Anti-HIV diphyllin glycosides from Justicia gendarussa. Phytochemistry 2017, 136, 94–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.Y.; Wang, D.Y.; Ku, K.F.; Zhao, Y.; Cheng, H.; Liu, K.L.; Rong, L.J.; Zhang, H.J. Anti-HIV lignans from Justicia procumbens. Chin. J. Nat. Med. 2019, 17, 945–952. [Google Scholar] [CrossRef]
- Kos, J.; Ku, C.F.; Kapustikova, I.; Oravec, M.; Zhang, H.J.; Jampilek, J. 8-Hydroxyquinoline-2-carboxanilides as antiviral agents against avian influenza virus. ChemistrySelect 2019, 4, 4582–4587. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Sang, W.; Ma, K.; Axmacher, J.C. Securing a future for China’s wild plant resources. Bioscience 2011, 61, 720–725. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.W. Ancient History of Lingnan, 1st ed.; Guangdong People’s Publishing House: Guangzhou, China, 2014. [Google Scholar]
- Maesa perlarius (Loureiro) Merrill. Available online: http://www.efloras.org/florataxon.aspx?flora_id=2&taxon_id=200016864 (accessed on 1 February 2022).
- Wiart, C. Maesa perlarius (Lour.) Merr. In Medicinal Plants of Asia and the Pacific; Wiart, C., Ed.; CRC Press: Boca Raton, FL, USA, 2006; pp. 67–68. [Google Scholar]
- Sindambiwe, J.B.; Baldé, A.M.; De Bruyne, T.; Pieters, L.; Van denHeuvel, H.; Claeys, M.; Van den Berghe, D.A.; Vlietinck, A.J. Triterpenoid saponins from Maesa lanceolata. Phytochemistry 1996, 41, 269–277. [Google Scholar] [CrossRef]
- Untiwachwuttikul, P.; Pancharoen, O.; Mahubusarakam, W.; Wiriyachitra, P.; Taylor, W.C.; Bubb, W.A.; Towers, G.H.N. A triterpenoid saponin from Maesa ramentacea. Phytochemistry 1997, 44, 491–495. [Google Scholar] [CrossRef]
- Apers, S.; De Bruyne, T.E.; Claeys, M.; Vlietinck, A.J.; Pieters, L.A.C. New acylated triterpenoid saponins from Maesa lanceolata. Phytochemistry 1999, 52, 1121–1131. [Google Scholar] [CrossRef]
- Manguro, L.O.A.; Midiwo, J.O.; Tietze, L.F.; Hao, P. Triterpene saponins of Maesa lanceolata leaves. Arkivoc 2011, 2011, 172–198. [Google Scholar] [CrossRef] [Green Version]
- Germonprez, N.; Van Puyvelde, L.; Maes, L.; Van Tri, M.; De Kimpe, N. New pentacyclic triterpene saponins with strong anti-leishmanial activity from the leaves of Maesa balansae. Tetrahedron 2004, 60, 219–228. [Google Scholar] [CrossRef]
- Wall, M.E.; Wani, M.C.; Gaetano, K.; Manikumar, G.; Taylor, H.; McGivney, R. Plant antimutagenic agents, 4. Isolation and structure elucidation of maesol, an inactive constituent of Maesa spp. J. Nat. Prod. 1988, 51, 1226–1231. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.P.; Shurtleff, A.C.; Costantino, J.A.; Tritsch, S.R.; Retterer, C.; Spurgers, K.B.; Bavari, S. HSPA5 is an essential host factor for Ebola virus infection. Antiviral Res. 2014, 109, 171–174. [Google Scholar] [CrossRef]
- Pleško, S. In Silico Study of Plant polyphenols’ interactions with VP24–Ebola virus matrix protein. Acta Chim. Slov. 2015, 62, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.E.; Saphire, E.O. Ebolavirus glycoprotein structure and mechanism of entry. Future Virol. 2009, 4, 621–635. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Ren, J.; Harlos, K.; Jones, D.M.; Zeltina, A.; Bowden, T.A.; Padilla-Parra, S.; Fry, E.E.; Stuart, D.I. Toremifene interacts with and destabilizes the Ebola virus glycoprotein. Nature 2016, 535, 169–172. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Zhao, Y.; Fry, E.E.; Stuart, D.I. Target identification and mode of action of four chemically divergent drugs against Ebola virus infection. J. Med. Chem. 2018, 61, 724–733. [Google Scholar] [CrossRef]
- Zhao, Y.; Ren, J.; Fry, E.E.; Xiao, J.; Townsend, A.R.; Stuart, D.I. Structures of Ebola virus glycoprotein complexes with tricyclic antidepressant and antipsychotic drugs. J. Med. Chem. 2018, 61, 4938–4945. [Google Scholar] [CrossRef]
- Shaikh, F.; Zhao, Y.; Alvarez, L.; Iliopoulou, M.; Lohans, C.; Schofield, C.J.; Padilla-Parra, S.; Siu, S.W.I.; Fry, E.E.; Ren, J.; et al. Structure-based in silico screening identifies a potent ebolavirus inhibitor from a traditional Chinese medicine library. J. Med. Chem. 2019, 62, 2928–2937. [Google Scholar] [CrossRef] [Green Version]
- Crystal Structure of Ebola Glycoprotein in Complex with Toremifene. Available online: https://www.wwpdb.org/pdb?id=pdb_00005jq7 (accessed on 1 February 2022).
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sierra, N.; Folio, C.; Robert, X.; Long, M.; Guillon, C.; Álvarez, G. Looking for novel capsid protein multimerization inhibitors of feline immunodeficiency virus. Pharmaceuticals 2018, 11, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MST Starting Guide—Monolith NT. 115. Available online: https://physiology.case.edu/media/eq_manuals/eq_manual_nano-temper_mst_starting_guide.pdf (accessed on 1 February 2022).
- Magnez, R.; Thiroux, B.; Taront, S.; Segaoula, Z.; Quesnel, B.; Thuru, X. PD-1/PD-L1 binding studies using microscale thermophoresis. Sci. Rep. 2017, 7, 17623. [Google Scholar] [CrossRef] [Green Version]
- Lane, T.R.; Ekins, S. Toward the target: Tilorone, quinacrine, and pyronaridine bind to Ebola virus glycoprotein. ACS Med. Chem. Lett. 2020, 11, 1653–1658. [Google Scholar] [CrossRef] [PubMed]
- Aron, P.M.; Kennedy, J.A. Flavan-3-ols: Nature, occurrence and biological activity. Mol. Nutr. Food Res. 2008, 52, 79–104. [Google Scholar] [CrossRef] [PubMed]
- Moini, H.; Rimbach, G.; Packer, L. Molecular aspects of procyanidin biological activity: Disease preventative and therapeutic potentials. Drug Metab. Drug Interact. 2000, 17, 237–259. [Google Scholar] [CrossRef] [PubMed]
- Schoonees, A.; Visser, J.; Musekiwa, A.; Volmink, J. Pycnogenol® (extract of French maritime pine bark) for the treatment of chronic disorders. Cochrane Database Syst. Rev. 2012, 4, CD008294. [Google Scholar] [CrossRef]
- Verstraeten, S.V.; Fraga, C.G.; Oteiza, P.I. Interactions of flavan-3-ols and procyanidins with membranes: Mechanisms and the physiological relevance. Food Funct. 2015, 6, 32–40. [Google Scholar] [CrossRef]
- Hammerstone, J.F.; Lazarus, S.A.; Schmitz, H.H. Procyanidin content and variation in some commonly consumed foods. J. Nutr. 2000, 130, 2086S–2092S. [Google Scholar] [CrossRef]
- Xu, X.; Xie, H.; Wang, Y.; Wei, X. A-type proanthocyanidins from lychee seeds and their antioxidant and antiviral activities. J. Agric. Food Chem. 2010, 58, 11667–11672. [Google Scholar] [CrossRef]
- Cheng, H.Y.; Lin, T.C.; Yang, C.M.; Shieh, D.E.; Lin, C.C. In vitro anti-HSV-2 activity and mechanism of action of proanthocyanidin A-1 from Vaccinium vitis-idaea. J. Sci. Food Agric. 2005, 85, 10–15. [Google Scholar] [CrossRef]
- DeBruyne, T.; Pieters, L.; Witvrouw, M.; DeClercq, E.; Berghe, D.V.; Vlietinck, A.J. Biological evaluation of proanthocyanidin dimers and related polyphenols. J. Nat. Prod. 1999, 62, 954–958. [Google Scholar] [CrossRef]
- Quosdorf, S.; Schuetz, A.; Kolodziej, H. Different inhibitory potencies of oseltamivir carboxylate, zanamivir, and several tannins on bacterial and viral neuraminidases as assessed in a cell-free fluorescence-based enzyme inhibition assay. Molecules 2017, 22, 1989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Q.; Du, R.; Anantpadma, M.; Schafer, A.; Hou, L.; Tian, J.; Davey, R.A.; Cheng, H.; Rong, L. Identification of ellagic acid from plant Rhodiola rosea L. as an anti-Ebola virus entry inhibitor. Viruses 2018, 10, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikra, C.; Rossos, G.; Hadjikakou, S.K.; Kourkoumelis, N. Molecular docking and structure activity relationship studies of nsaids. What do they reveal about IC50? Lett. Drug Des. Discov. 2017, 14, 949–958. [Google Scholar] [CrossRef]
- Rainard, J.M.; Pandarakalam, G.C.; McElroy, S.P. Using microscale thermophoresis to characterize hits from high-throughput screening: A European lead factory perspective. SLAS Discov. 2018, 23, 225–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makabe, H.; Kamo, T.; Hirota, M.; Mohri, Y.; Sagehashi, M.; Yamada, T.; Hattori, Y.; Morimura, K. An efficient synthesis of procyanidins using equimolar condensation of catechin and/or epicatechin catalyzed by ytterbium triflate. Heterocycles 2009, 79, 549–563. [Google Scholar] [CrossRef] [Green Version]
- Saito, A.; Doi, Y.; Tanaka, A.; Matsuura, N.; Ubukata, M.; Nakajima, N. Systematic synthesis of four epicatechin series procyanidin trimers and their inhibitory activity on the Maillard reaction and antioxidant activity. Bioorg. Med. Chem. 2004, 12, 4783–4790. [Google Scholar] [CrossRef]
- Shoji, T.; Mutsuga, M.; Nakamura, T.; Kanda, T.; Akiyama, H.; Goda, Y. Isolation and structural elucidation of some procyanidins from apple by low-temperature nuclear magnetic resonance. J. Agric. Food Chem. 2003, 51, 3806–3813. [Google Scholar] [CrossRef] [PubMed]
- Tarascou, I.; Barathieu, K.; André, Y.; Pianet, I.; Dufourc, E.J.; Fouquet, E. An improved synthesis of procyanidin dimers: Regio- and stereocontrol of the interflavan bond. Eur. J. Org. Chem. 2006, 5367–5377. [Google Scholar] [CrossRef]
- Hwang, B.Y.; Kim, H.S.; Lee, J.H.; Hong, Y.S.; Ro, J.S.; Lee, K.S.; Lee, J.J. Antioxidant benzoylated flavan-3-ol glycoside from Celastrus orbiculatus. J. Nat. Prod. 2001, 64, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Adam, A.T.; Lee, J.J.; Gasiewicz, T.A.; Henry, E.C.; Rotella, D.P. Towards the discovery of drug-like epigallocatechin gallate analogs as Hsp90 inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 2263–2266. [Google Scholar] [CrossRef]
- Huo, C.; Shi, G.; Lam, W.H.; Chen, D.; Cui, Q.C.; Dou, Q.P.; Chan, T.H. Semi-synthesis and proteasome inhibition of D-ring deoxy analogs of (–)-epigallocatechin gallate (EGCG), the active ingredient of green tea extract. Can. J. Chem. 2008, 86, 495–502. [Google Scholar] [CrossRef]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]
- Connor, R.I.; Chen, B.K.; Choe, S.; Landau, N.R. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 1995, 206, 935–944. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Choe, S.; Walker, R.; Di Marzio, P.; Morgan, P.O.; Landau, N.R. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 1995, 69, 6705–6711. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EBOVpp IC50 (µM) | A549 CC50 (µM) | Selectivity Index (SI) b | |
|---|---|---|---|
| Procyanidin B2 (1) | 0.83 | >86.5 | >104 |
| Procyanidin C1 (2) | >11.5 | >11.5 | - |
| Procyanidin tetramer (3) | >11.5 | >11.5 | - |
| (+) Catechin (4) | 36.0 | >34.5 | >0.48 |
| (−) Epicatechin (5) | 22.1 | >172 | >7.79 |
| Procyanidin B1 (6) | 0.95 | >86.5 | >91.1 |
| Procyanidin B3 (7) | 1.52 | >86.5 | >57.1 |
| EGCG (8) | 2.80 | >21.8 | >7.81 |
| ECG (9) | 2.95 | >22.6 | >7.68 |
| Gallocatechin (10) | 12.4 | >163 | >13.1 |
| Epigallocatechin (11) | 5.53 | >163 | >29.5 |
| Toremifene | 0.17 | 2.92 | 17.0 |
| Compound | IC50 (M) a | Docking Score b (kcal/mol) |
|---|---|---|
| Procyanidin B1 (6) | 9.13 × 10−7 | −9.6 |
| Procyanidin B2 (1) | 8.29 × 10−7 | −9.4 |
| EGCG (8) | 2.80 × 10−6 | −9.3 |
| ECG (9) | 2.95 × 10−6 | −9.0 |
| Epigallocatechin (11) | 5.53 × 10−6 | −7.9 |
| Gallocatechin (10) | 1.24 × 10−5 | −7.4 |
| (−) Epicatechin (5) | 2.21 × 10−5 | −8.0 |
| (+) Catechin (4) | 3.60 × 10−5 | −6.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsang, N.Y.; Li, W.-F.; Varhegyi, E.; Rong, L.; Zhang, H.-J. Ebola Entry Inhibitors Discovered from Maesa perlarius. Int. J. Mol. Sci. 2022, 23, 2620. https://doi.org/10.3390/ijms23052620
Tsang NY, Li W-F, Varhegyi E, Rong L, Zhang H-J. Ebola Entry Inhibitors Discovered from Maesa perlarius. International Journal of Molecular Sciences. 2022; 23(5):2620. https://doi.org/10.3390/ijms23052620
Chicago/Turabian StyleTsang, Nga Yi, Wan-Fei Li, Elizabeth Varhegyi, Lijun Rong, and Hong-Jie Zhang. 2022. "Ebola Entry Inhibitors Discovered from Maesa perlarius" International Journal of Molecular Sciences 23, no. 5: 2620. https://doi.org/10.3390/ijms23052620
APA StyleTsang, N. Y., Li, W.-F., Varhegyi, E., Rong, L., & Zhang, H.-J. (2022). Ebola Entry Inhibitors Discovered from Maesa perlarius. International Journal of Molecular Sciences, 23(5), 2620. https://doi.org/10.3390/ijms23052620