
Molecular Interactions of Tannic Acid with Proteins Associated with SARS-CoV-2 Infectivity

 , ,
, ,
Abstract
:1. Introduction
2. Results
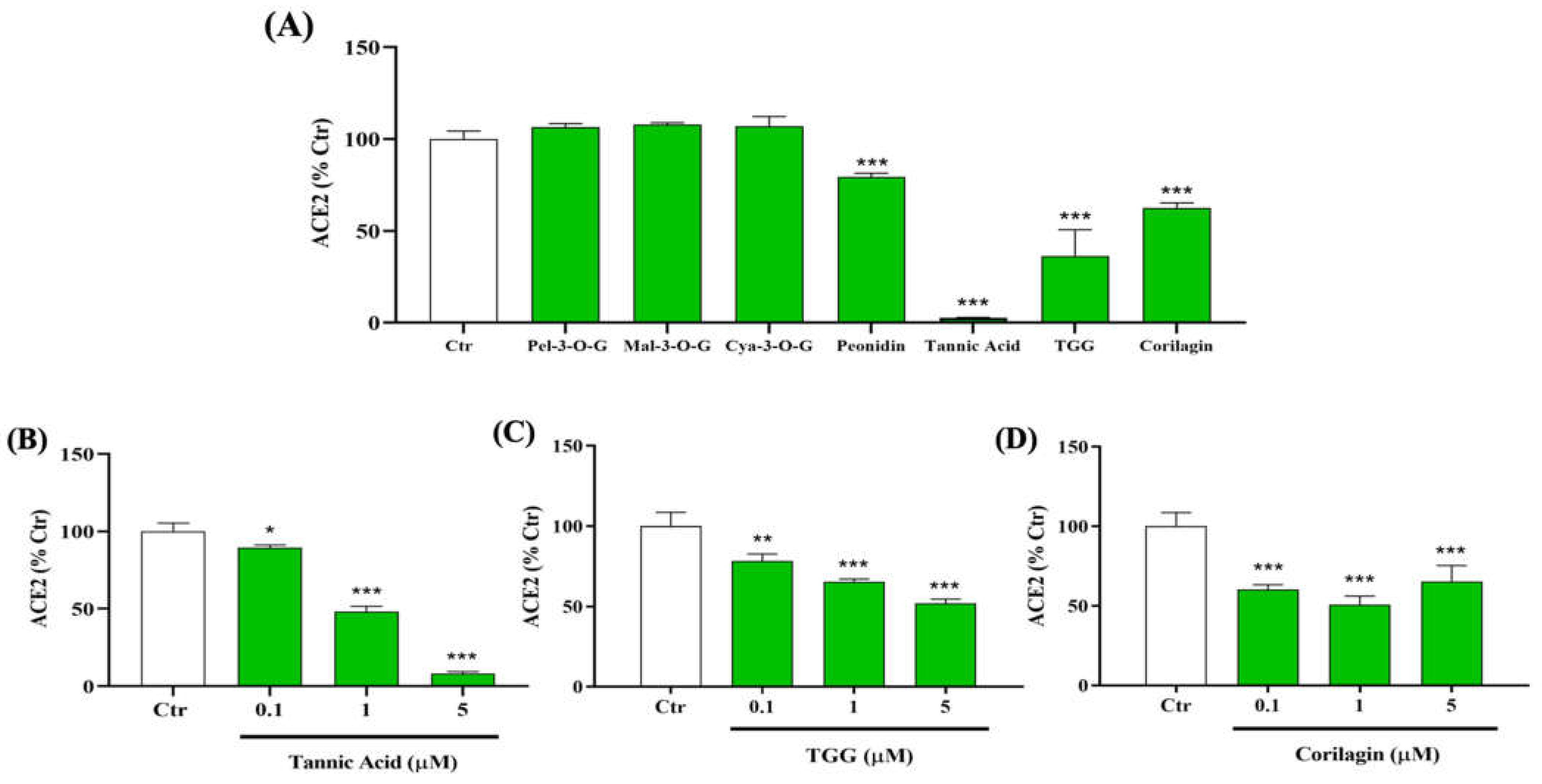
2.1. Inhibitory Effect of Polyphenols on the Binding between RBD (N501Y) Spike Protein and Human ACE2
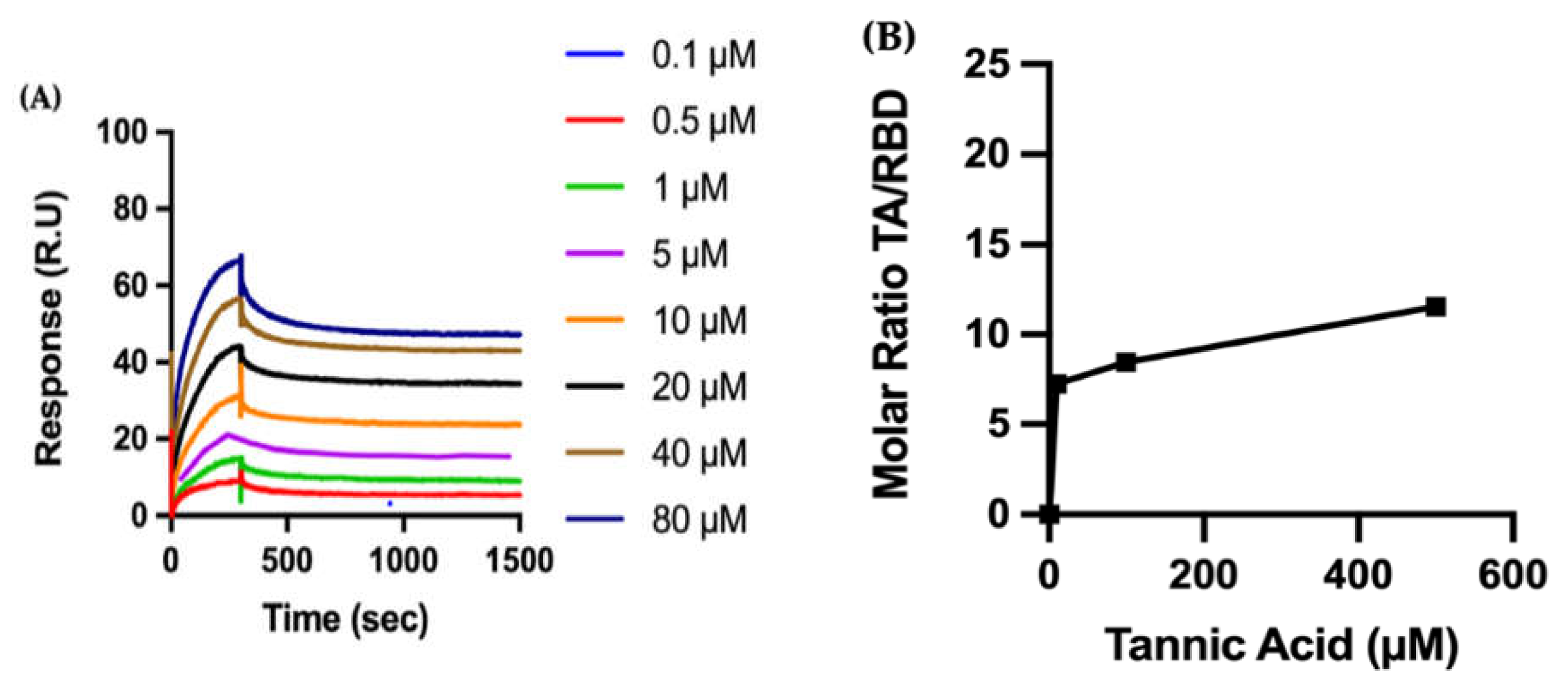
Biophysical Characterization of TA/RBD Interactions
2.2. Polyphenols and TMPRSS2 Enzyme
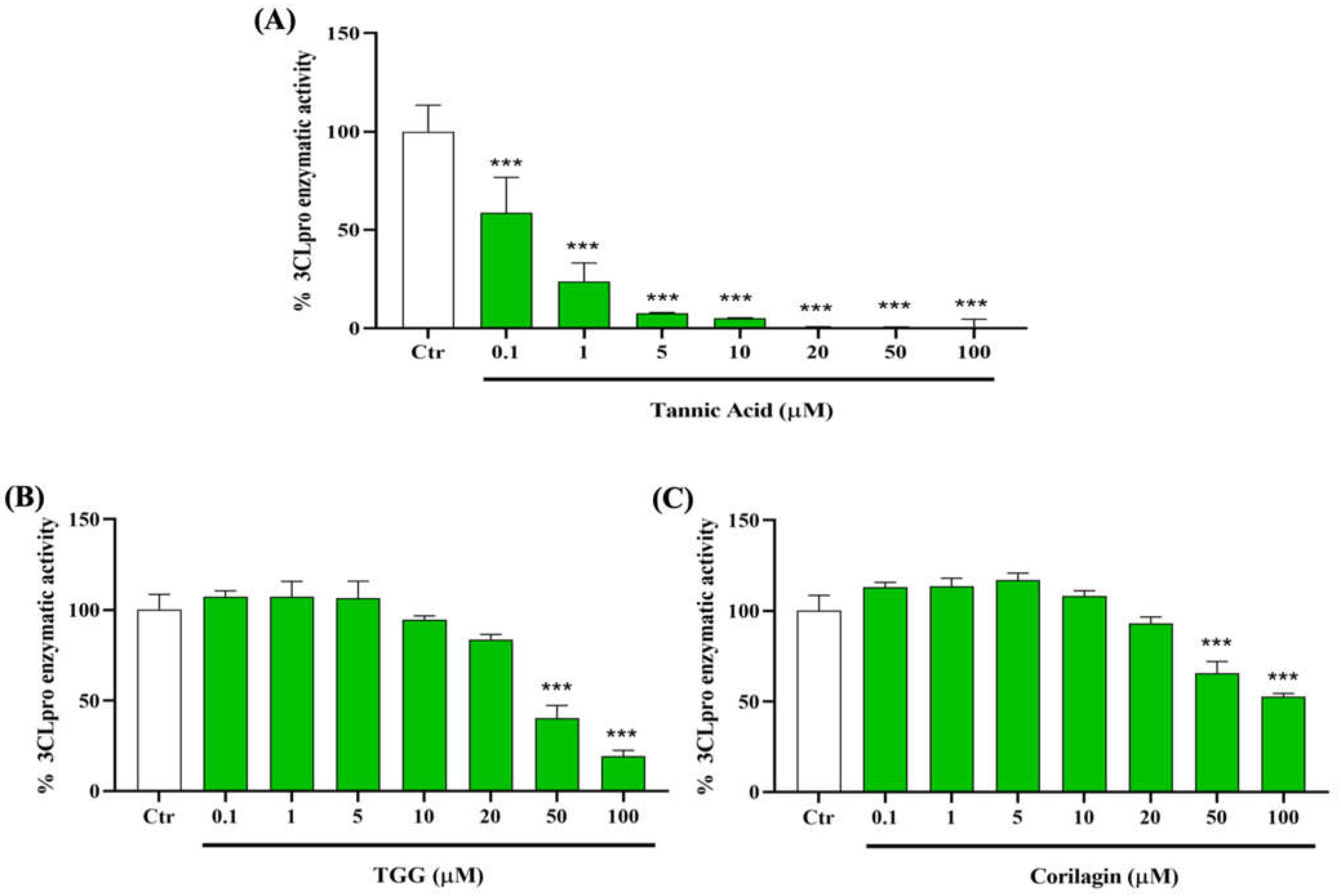
2.2.1. Effect of TA, TGG, and Corilagin on TMPRSS2 Activity
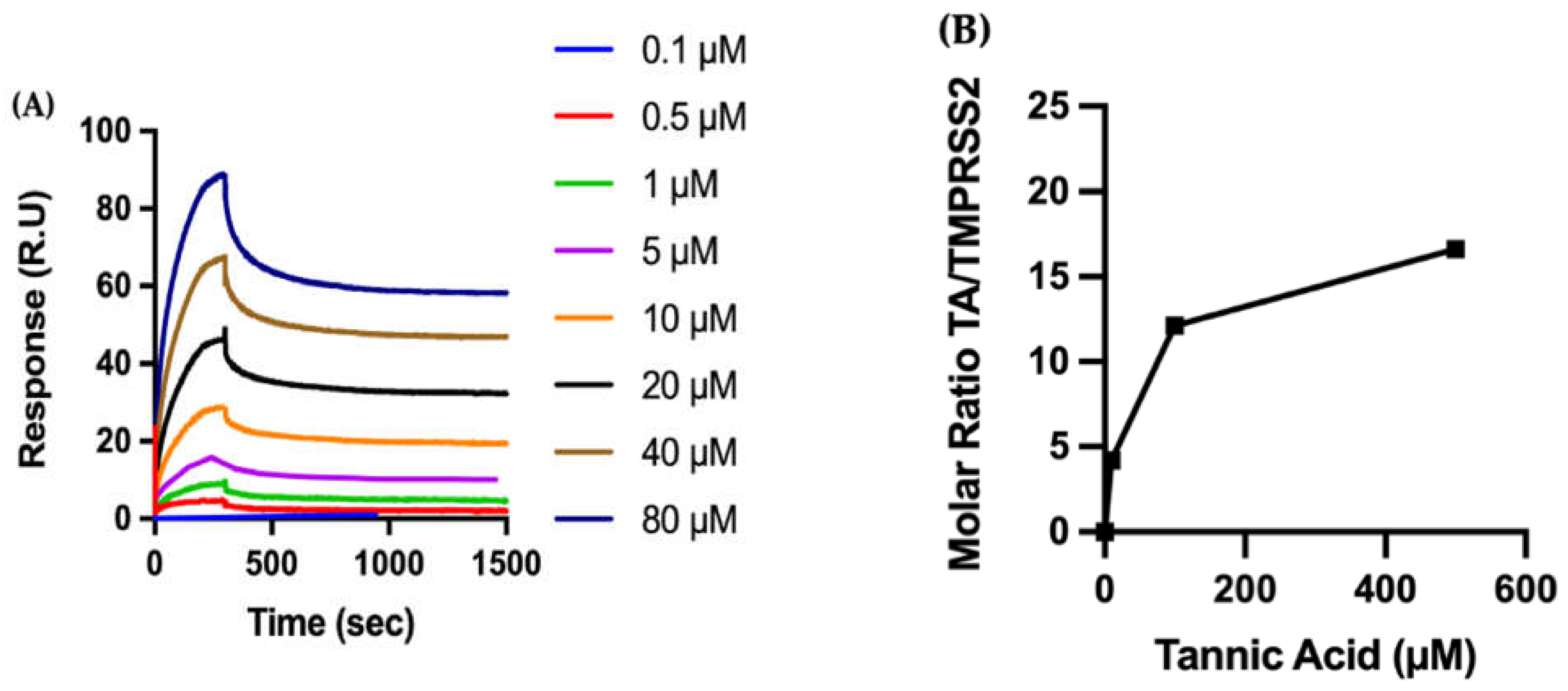
2.2.2. Biophysical Characterization of TA/TMPRSS2 Interactions
2.3. Polyphenols and 3CLpro Enzyme
2.3.1. Effect of TA, TGG, and Corilagin on 3CLpro Activity
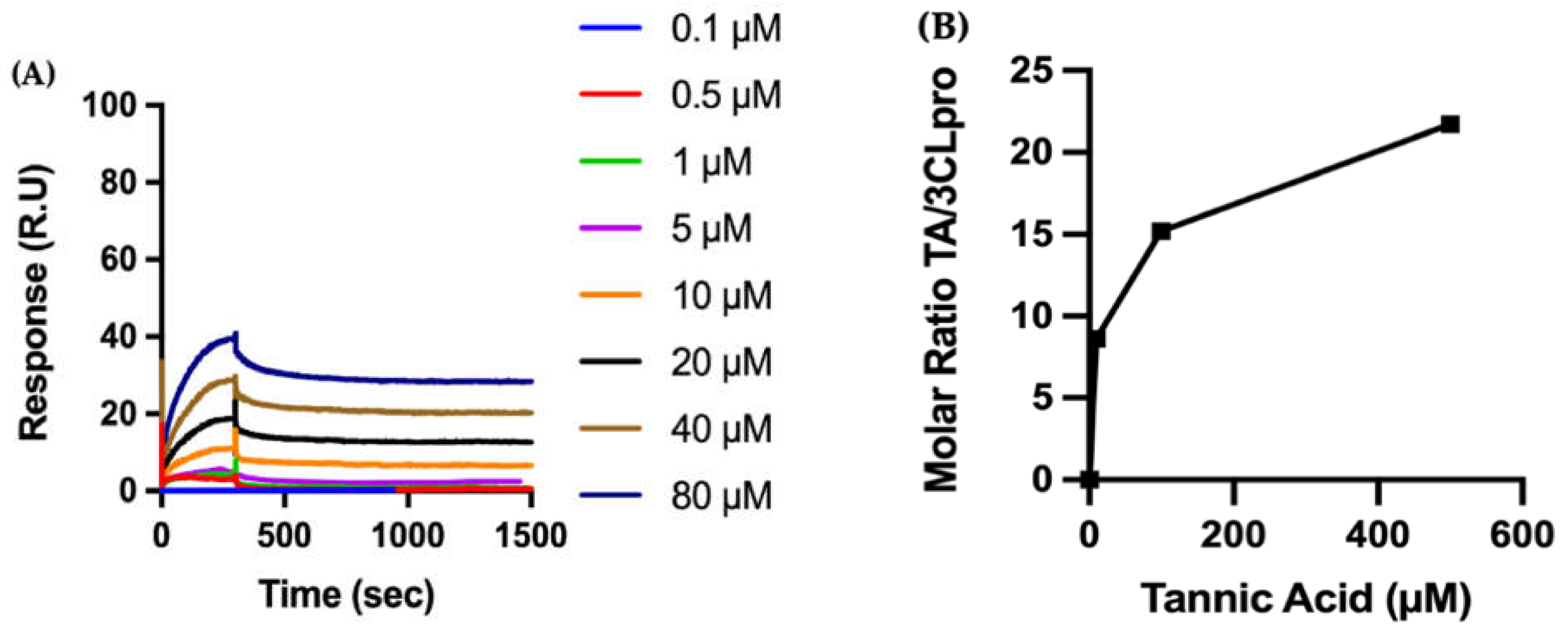
2.3.2. Biophysical Characterization of TA/3CLpro Interactions
2.4. Molecular Modelling
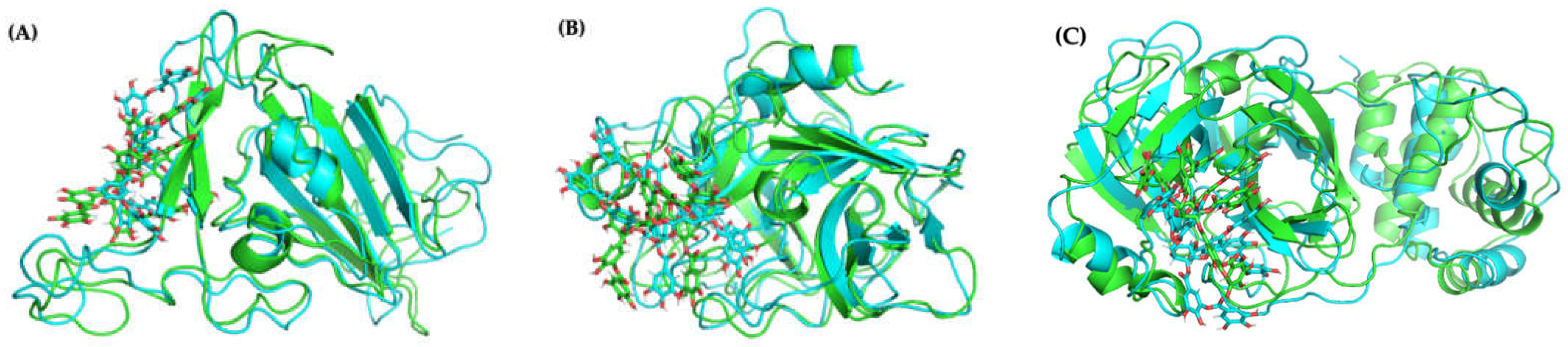
2.4.1. Molecular Docking and Dynamics of TA/RBD Complex
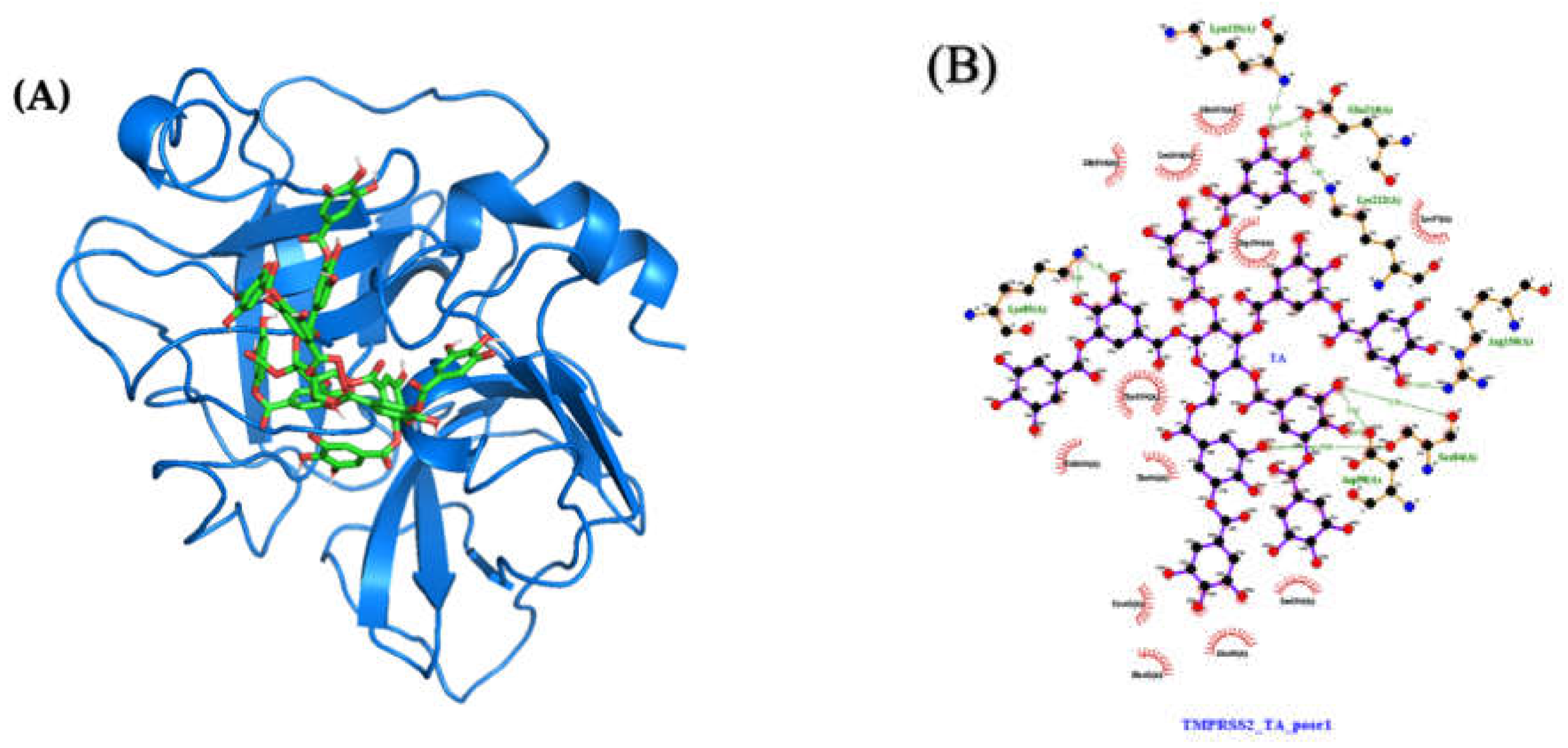
2.4.2. Molecular Docking and Dynamics of TA/TMPRSS2 Complex
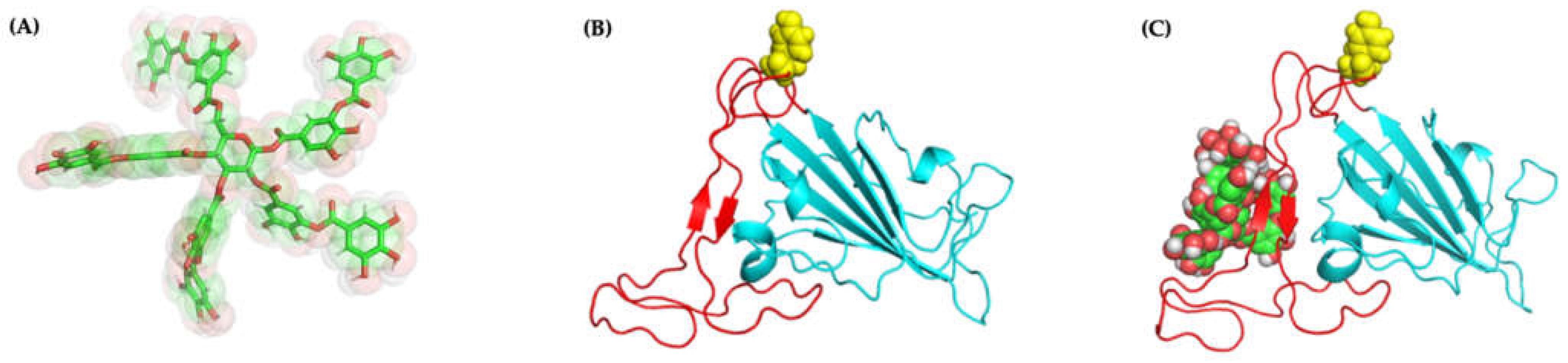
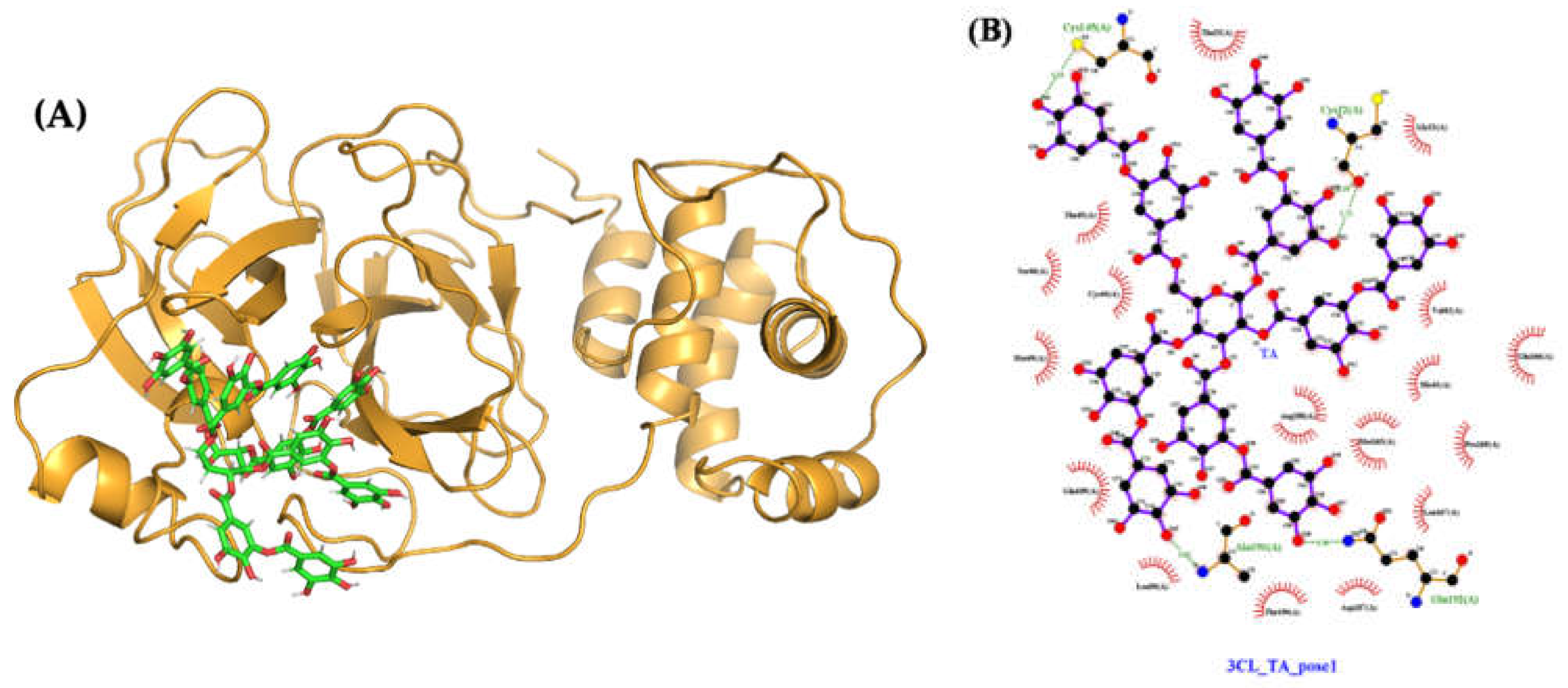
2.4.3. Molecular Docking and Dynamics of TA/3CLpro Complex
3. Discussion
Some Limitations of this Study
4. Materials and Methods
4.1. Products
4.2. Biochemical Assays
4.2.1. SARS-CoV-2 RBD (N501Y) Spike Protein and Human ACE2 Binding Inhibitor Assay
4.2.2. TMPRSS2 Enzymatic Assay
4.2.3. CLpro Enzymatic Assay
4.2.4. Statistical Analysis
4.3. Surface Plasmon Resonance (SPR)
4.4. Quartz Crystal Microbalance with Dissipation Monitoring (QCMD)
4.4.1. QCMD Procedure
4.4.2. Water Content in the Protein Layer Adsorbed on the QCMD Sensor
4.4.3. Molar Ratio of TA/Protein on the Sensor Surface
4.5. Molecular Modeling
4.5.1. Molecular Docking
4.5.2. MD Simulations
4.5.3. Protein–Ligand Simulations
4.5.4. Analysis
4.5.5. Binding Free Energy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, Y.; Liu, J.; Plante, K.S.; Plante, J.A.; Xie, X.; Zhang, X.; Ku, Z.; An, Z.; Scharton, D.; Schindewolf, C. The N501Y spike substitution enhances SARS-CoV-2 infection and transmission. Nature 2021, 602, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Mlcochova, P.; Kemp, S.; Dhar, M.S.; Papa, G.; Meng, B.; Ferreira, I.A.; Datir, R.; Collier, D.A.; Albecka, A.; Singh, S. SARS-CoV-2 B. 1.617. 2 Delta variant replication and immune evasion. Nature 2021, 599, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Dejnirattisai, W.; Huo, J.; Zhou, D.; Zahradnik, J.; Supasa, P.; Liu, C.; Duyvesteyn, H.M.E.; Ginn, H.M.; Mentzer, H.M.; Tuekprakhon, A.; et al. SARS-CoV-2 Omicron-B.1.1.529 leads to widespread escape from neutralizing antibody responses. Cell 2022, 185, 467–484. [Google Scholar] [CrossRef]
- Sztain, T.; Ahn, S.-H.; Bogetti, A.T.; Casalino, L.; Goldsmith, J.A.; Seitz, E.; McCool, R.S.; Kearns, F.L.; Acosta-Reyes, F.; Maji, S. A glycan gate controls opening of the SARS-CoV-2 spike protein. Nat. Chem. 2021, 13, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Shen, H.-M. Targeting the endocytic pathway and autophagy process as a novel therapeutic strategy in COVID-19. Int. J. Biol. Sci. 2020, 16, 1724. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Zhu, Y.; Liu, M.; Lan, Q.; Xu, W.; Wu, Y.; Ying, T.; Liu, S.; Shi, Z.; Jiang, S. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell. Mol. Immunol. 2020, 17, 765–767. [Google Scholar] [CrossRef]
- Rakowska, P.D.; Tiddia, M.; Faruqui, N.; Bankier, C.; Pei, Y.; Pollard, A.J.; Zhang, J.; Gilmore, I.S. Antiviral surfaces and coatings and their mechanisms of action. Commun. Mater. 2021, 2, 1–19. [Google Scholar]
- Zhao, M.; Yu, Y.; Sun, L.-M.; Xing, J.-Q.; Li, T.; Zhu, Y.; Wang, M.; Yu, Y.; Xue, W.; Xia, T. GCG inhibits SARS-CoV-2 replication by disrupting the liquid phase condensation of its nucleocapsid protein. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Gao, S.; Huang, T.; Song, L.; Xu, S.; Cheng, Y.; Cherukupalli, S.; Kang, D.; Zhao, T.; Sun, L.; Zhang, J.; et al. Medicinal chemistry strategies towards the development of effective SARS-CoV-2 inhibitors. Acta Pharm. Sin. B 2022, 12, 581–599. [Google Scholar] [CrossRef]
- Paraiso, I.L.; Revel, J.S.; Stevens, J.F. Potential use of polyphenols in the battle against COVID-19. Curr. Opin. Food Sci. 2020, 32, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.Y.; Jeon, S.; Jang, Y.; Gotina, L.; Won, J.; Ju, Y.H.; Kim, S.; Jang, M.W.; Won, W.; Park, M.G. Platycodin D, a natural component of Platycodon grandiflorum, prevents both lysosome-and TMPRSS2-driven SARS-CoV-2 infection by hindering membrane fusion. Exp. Mol. Med. 2021, 53, 956–972. [Google Scholar] [CrossRef] [PubMed]
- Chiu, S.-K.; Tsai, K.-W.; Wu, C.-C.; Zheng, C.-M.; Yang, C.-H.; Hu, W.-C.; Hou, Y.-C.; Lu, K.-C.; Chao, Y.-C. Putative Role of Vitamin D for COVID-19 Vaccination. Int. J. Mol. Sci. 2021, 22, 8988. [Google Scholar] [CrossRef]
- Li, Q.; Yi, D.; Lei, X.; Zhao, J.; Zhang, Y.; Cui, X.; Xiao, X.; Jiao, T.; Dong, X.; Zhao, X.; et al. Corilagin inhibits SARS-CoV-2 replication by targeting viral RNA-dependent RNA polymerase. Acta Pharm. Sin. B 2021, 11, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Gao, F. An overview of potential inhibitors targeting non-structural proteins 3 (PLpro and Mac1) and 5 (3CLpro/Mpro) of SARS-CoV-2. Comput. Struct. Biotechnol. J. 2021, 19, 4868. [Google Scholar] [CrossRef]
- Liao, Q.; Chen, Z.; Tao, Y.; Zhang, B.; Wu, X.; Wang, Z.; Yang, L.; Wang, Q. An integrated method for optimized identification of effective natural inhibitors against SARS-CoV-2 3CLpro. Sci. Rep. 2021, 11, 22796. [Google Scholar] [CrossRef] [PubMed]
- Stille, J.; Tjutrins, J.; Wang, G.; Venegas, F.A.; Hennecker, C.; Rueda, A.M.; Sharon, I.; Blaine, N.; Miron, C.E.; Pinus, S. Design, Synthesis and Biological Evaluation of Novel SARS-CoV-2 3CLpro Covalent Inhibitors. Eur. J. Med. Chem. 2021, 229, 114046. [Google Scholar] [CrossRef]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef] [Green Version]
- Silva, R.C.; Freitas, H.F.; Campos, J.M.; Kimani, N.M.; Silva, C.H.; Borges, R.S.; Pita, S.S.; Santos, C.B. Natural Products-Based Drug Design against SARS-CoV-2 Mpro 3CLpro. Int. J. Mol. Sci. 2021, 22, 11739. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Yeo, S.-G.; Song, J.H.; Hong, E.-H.; Lee, B.-R.; Kwon, Y.S.; Chang, S.-Y.; Kim, S.H.; won Lee, S.; Park, J.-H.; Ko, H.-J. Antiviral effects of Phyllanthus urinaria containing corilagin against human enterovirus 71 and Coxsackievirus A16 in vitro. Arch. Pharmacal. Res. 2015, 38, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Li, Z.; Yuan, K.; Qu, X.; Chen, J.; Wang, G.; Zhang, H.; Luo, H.; Zhu, L.; Jiang, P.; et al. Small molecules blocking the entry of severe acute respiratory syndrome coronavirus into host cells. J. Virol. 2004, 78, 11334–11339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, L.; He, Y.; Zhou, Y.; Liu, S.; Zheng, B.J.; Jiang, S. The spike protein of SARS-CoV—A target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009, 7, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-C.; Chen, Y.; Wang, Y.-C.; Wang, W.-J.; Yang, C.-S.; Tsai, C.-L.; Hou, M.-H.; Chen, H.-F.; Shen, Y.-C.; Hung, M.-C. Tannic acid suppresses SARS-CoV-2 as a dual inhibitor of the viral main protease and the cellular TMPRSS2 protease. Am. J. Cancer Res. 2020, 10, 4538. [Google Scholar]
- Swain, S.S.; Panda, S.K.; Luyten, W. Phytochemicals against SARS-CoV as potential drug leads. Biomed. J. 2020, 44, 74–85. [Google Scholar] [CrossRef]
- Pandey, P.; Rane, J.S.; Chatterjee, A.; Kumar, A.; Khan, R.; Prakash, A.; Ray, S. Targeting SARS-CoV-2 spike protein of COVID-19 with naturally occurring phytochemicals: An in silico study for drug development. J. Biomol. Struct. Dyn. 2020, 39, 6306–6316. [Google Scholar] [CrossRef]
- Yang, L.J.; Chen, R.H.; Hamdoun, S.; Coghi, P.; Ng, J.P.; Zhang, D.W.; Guo, X.; Xia, C.; Law, B.Y.K.; Wong, V.K.W. Corilagin prevents SARS-CoV-2 infection by targeting RBD-ACE2 binding. Phytomedicine 2021, 87, 153591. [Google Scholar] [CrossRef]
- Binette, V.; Côté, S.; Haddad, M.; Nguyen, P.T.; Bélanger, S.; Bourgault, S.; Ramassamy, C.; Gaudreault, R.; Mousseau, N. Corilagin and 1,3,6-Tri-O-galloyl-β-d-glucose: Potential inhibitors of SARS-CoV-2 variants. Phys. Chem. Chem. Phys. 2021, 23, 14873–14888. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2021, 23, 3–20. [Google Scholar] [CrossRef]
- Ferreira, J.C.; Rabeh, W.M. Biochemical and biophysical characterization of the main protease, 3-chymotrypsin-like protease (3CLpro) from the novel coronavirus SARS-CoV 2. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Tahir ul Qamar, M.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Pitsillou, E.; Liang, J.; Karagiannis, C.; Ververis, K.; Darmawan, K.K.; Ng, K.; Hung, A.; Karagiannis, T.C. Interaction of small molecules with the SARS-CoV-2 main protease in silico and in vitro validation of potential lead compounds using an enzyme-linked immunosorbent assay. Comput. Biol. Chem. 2020, 89, 107408. [Google Scholar] [CrossRef]
- Loschwitz, J.; Jackering, A.; Keutmann, M.; Olagunju, M.; Eberle, R.J.; Coronado, M.A.; Olubiyi, O.O.; Strodel, B. Novel inhibitors of the main protease enzyme of SARS-CoV-2 identified via molecular dynamics simulation-guided in vitro assay. Bioorg. Chem. 2021, 111, 104862. [Google Scholar] [CrossRef] [PubMed]
- Du, A.; Zheng, R.; Disoma, C.; Li, S.; Chen, Z.; Li, S.; Liu, P.; Zhou, Y.; Shen, Y.; Liu, S. Epigallocatechin-3-gallate, an active ingredient of Traditional Chinese Medicines, inhibits the 3CLpro activity of SARS-CoV-2. Int. J. Biol. Macromol. 2021, 176, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Coelho, C.; Gallo, G.; Campos, C.B.; Hardy, L.; Würtele, M. Biochemical screening for SARS-CoV-2 main protease inhibitors. PLoS ONE 2020, 15, e0240079. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-N.; Lin, C.P.; Huang, K.-K.; Chen, W.-C.; Hsieh, H.-P.; Liang, P.-H.; Hsu, J.T.-A. Inhibition of SARS-CoV 3C-like protease activity by theaflavin-3, 3’-digallate (TF3). Evid. -Based Complementary Altern. Med. 2005, 2, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.T.H.; Jung, J.-H.; Kim, M.-K.; Lim, S.; Choi, J.-M.; Chung, B.; Kim, D.-W.; Kim, D. The Inhibitory Effects of Plant Derivate Polyphenols on the Main Protease of SARS Coronavirus 2 and Their Structure–Activity Relationship. Molecules 2021, 26, 1924. [Google Scholar] [CrossRef]
- Gaudreault, R.; van de Ven, T.G.; Whitehead, M.A. Molecular modeling of poly (ethylene oxide) model cofactors; 1, 3, 6-tri-O-galloyl-β-D-glucose and corilagin. Mol. Modeling Annu. 2002, 8, 73–80. [Google Scholar] [CrossRef]
- Gaudreault, R.; Whitehead, M.A.; van de Ven, T.G. Molecular orbital studies of gas-phase interactions between complex molecules. J. Phys. Chem. A 2006, 110, 3692–3702. [Google Scholar] [CrossRef]
- Gaudreault, R.; Mousseau, N. Mitigating Alzheimer’s disease with natural polyphenols: A review. Curr. Alzheimer Res. 2019, 16, 529–543. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Deng, Y.; Zheng, Z.; Huang, W.; Chen, L.; Tong, Q.; Ming, Y. Corilagin, a promising medicinal herbal agent. Biomed. Pharmacother. 2018, 99, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.U.; Mullick, R.; Kumar, A.; Sharma, G.; Bag, P.; Roy, C.L.; Sudha, G.; Tandon, H.; Dave, P.; Shukla, A.; et al. A natural small molecule inhibitor corilagin blocks HCV replication and modulates oxidative stress to reduce liver damage. Antivir. Res. 2018, 150, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Jin, F.; Cheng, D.; Tao, J.-Y.; Zhang, S.-L.; Pang, R.; Guo, Y.-J.; Ye, P.; Dong, J.-H.; Zhao, L. Anti-inflammatory and anti-oxidative effects of corilagin in a rat model of acute cholestasis. BMC Gastroenterol. 2013, 13, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.-T.; Lin, T.-C.; Hsu, F.-L. Antihypertensive effect of corilagin in the rat. Can. J. Physiol. Pharmacol. 1995, 73, 1425–1429. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.-f.; Ju, H.-q.; Li, S.; Zhang, Y.-j.; Yang, C.-r.; Wang, Y.-f. Effects of 1, 2, 4, 6-tetra-O-galloyl-β-D-glucose from P. emblica on HBsAg and HBeAg secretion in HepG2. 2.15 cell culture. Virol. Sin. 2010, 25, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, O.; Gouzi, H.; El-Hoshoudy, A.N.; Benaceur, F.; Patel, C.; Goswami, D.; Boukerouis, D.; Bendahou, M. Berries anthocyanins as potential SARS-CoV-2 inhibitors targeting the viral attachment and replication; molecular docking simulation. Egypt. J. Pet. 2021, 30, 33–43. [Google Scholar] [CrossRef]
- Godinho, P.I.; Soengas, R.G.; Silva, V.L. Therapeutic Potential of Glycosyl Flavonoids as Anti-Coronaviral Agents. Pharmaceuticals 2021, 14, 546. [Google Scholar] [CrossRef] [PubMed]
- Salmaso, V.; Moro, S. Bridging Molecular Docking to Molecular Dynamics in Exploring Ligand-Protein Recognition Process: An Overview. Front. Pharm. 2018, 9, 923. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery, C.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Fratev, F. N501Y and K417N Mutations in the Spike Protein of SARS-CoV-2 Alter the Interactions with Both hACE2 and Human-Derived Antibody: A Free Energy of Perturbation Retrospective Study. J. Chem. Inf. Modeling 2021, 61, 6079–6084. [Google Scholar] [CrossRef]
- Willyard, C. How antiviral pill molnupiravir shot ahead in the COVID drug hunt. Nature 2021. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, B.; Batool, M.; Kim, M.S.; Choi, S. Exploring the binding mechanism of PF-07321332 SARS-CoV-2 protease inhibitor through molecular dynamics and binding free energy simulations. Int. J. Mol. Sci. 2021, 22, 9124. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Thao, T.T.N.; Hoffmann, D.; Taddeo, A.; Ebert, N.; Labroussaa, F.; Pohlmann, A.; King, J.; Steiner, S.; Kelly, J.N. SARS-CoV-2 spike D614G change enhances replication and transmission. Nature 2021, 592, 122–127. [Google Scholar] [CrossRef]
- Lange, N.W.; Salerno, D.M.; Jennings, D.L.; Choe, J.; Hedvat, J.; Kovac, D.B.; Scheffert, J.; Shertel, T.; Ratner, L.E.; Brown, R.S., Jr. Nirmatrelvir/ritonavir use: Managing clinically significant drug-drug interactions with transplant immunosuppressants. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2022. [Google Scholar] [CrossRef]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [Green Version]
- Matsuyama, S.; Nao, N.; Shirato, K.; Kawase, M.; Saito, S.; Takayama, I.; Nagata, N.; Sekizuka, T.; Katoh, H.; Kato, F. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc. Natl. Acad. Sci. USA 2020, 117, 7001–7003. [Google Scholar] [CrossRef] [Green Version]
- Elmezayen, A.D.; Al-Obaidi, A.; Şahin, A.T.; Yelekçi, K. Drug repurposing for coronavirus (COVID-19): In silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. J. Biomol. Struct. Dyn. 2021, 39, 2980–2992. [Google Scholar] [CrossRef] [Green Version]
- Pinto, A.F.; Nascimento, J.M.d.; Sobral, R.V.d.S.; Amorim, E.L.C.d.; Silva, R.O.; Leite, A.C.L. Tannic acid as a precipitating agent of human plasma proteins. Eur. J. Pharm. Sci. 2019, 138, 105018. [Google Scholar] [CrossRef]
- Hagerman, A. Chemistry of tannin-protein complexation. In Chemistry and Significance of Condensed Tannins; Hemingway, R.W., Karchesy, J.J., Eds.; Plenum Press: New York, NY, USA, 1989. [Google Scholar]
- Solin, K.; Beaumont, M.; Rosenfeldt, S.; Orelma, H.; Borghei, M.; Bacher, M.; Opietnik, M.; Rojas, O.J. Self-Assembly of Soft Cellulose Nanospheres into Colloidal Gel Layers with Enhanced Protein Adsorption Capability for Next-Generation Immunoassays. Small 2020, 16, 2004702. [Google Scholar] [CrossRef] [PubMed]
- Mauriz, E.; Lechuga, L.M. Current Trends in SPR Biosensing of SARS-CoV-2 Entry Inhibitors. Chemosensors 2021, 9, 330. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Z. Recent progress of surface plasmon resonance in the development of coronavirus disease-2019 drug candidates. Eur. J. Med. Chem. Rep. 2021, 1, 100003. [Google Scholar] [CrossRef]
- Singh, R.; Gautam, A.; Chandel, S.; Ghosh, A.; Dey, D.; Roy, S.; Ravichandiran, V.; Ghosh, D. Protease inhibitory effect of natural polyphenolic compounds on SARS-CoV-2: An in silico study. Molecules 2020, 25, 4604. [Google Scholar] [CrossRef]
- Patil, R.; Chikhale, R.; Khanal, P.; Gurav, N.; Ayyanar, M.; Sinha, S.; Prasad, S.; Dey, Y.N.; Wanjari, M.; Gurav, S.S. Computational and network pharmacology analysis of bioflavonoids as possible natural antiviral compounds in COVID-19. Inform. Med. Unlocked 2021, 22, 100504. [Google Scholar] [CrossRef]
- Albohy, A.; Zahran, E.M.; Abdelmohsen, U.R.; Salem, M.A.; Al-Warhi, T.; Al-Sanea, M.M.; Abelyan, N.; Khalil, H.E.; Desoukey, S.Y.; Fouad, M.A. Multitarget in silico studies of Ocimum menthiifolium, family Lamiaceae against SARS-CoV-2 supported by molecular dynamics simulation. J. Biomol. Struct. Dyn. 2020, 1–11. [Google Scholar] [CrossRef]
- Gogoi, B.; Chowdhury, P.; Goswami, N.; Gogoi, N.; Naiya, T.; Chetia, P.; Mahanta, S.; Chetia, D.; Tanti, B.; Borah, P. Identification of potential plant-based inhibitor against viral proteases of SARS-CoV-2 through molecular docking, MM-PBSA binding energy calculations and molecular dynamics simulation. Mol. Divers. 2021, 25, 1963–1977. [Google Scholar] [CrossRef]
- Andrade, R.G., Jr.; Dalvi, L.T.; Silva, J.M.C., Jr.; Lopes, G.K.; Alonso, A.; Hermes-Lima, M. The antioxidant effect of tannic acid on the in vitro copper-mediated formation of free radicals. Arch. Biochem. Biophys. 2005, 437, 1–9. [Google Scholar] [CrossRef]
- Wu, Y.; Zhong, L.; Yu, Z.; Qi, J. Anti-neuroinflammatory effects of tannic acid against lipopolysaccharide-induced BV2 microglial cells via inhibition of NF-κB activation. Drug Dev. Res. 2019, 80, 262–268. [Google Scholar] [CrossRef]
- Karkhanei, B.; Ghane, E.T.; Mehri, F. Evaluation of oxidative stress level: Total antioxidant capacity, total oxidant status and glutathione activity in patients with Covid-19. New Microbes New Infect. 2021, 42, 100897. [Google Scholar] [CrossRef]
- Pincemail, J.; Cavalier, E.; Charlier, C.; Cheramy–Bien, J.-P.; Brevers, E.; Courtois, A.; Fadeur, M.; Meziane, S.; Goff, C.L.; Misset, B. Oxidative stress status in COVID-19 patients hospitalized in intensive care unit for severe pneumonia. A pilot study. Antioxidants 2021, 10, 257. [Google Scholar] [CrossRef]
- Alam, M.S.; Czajkowsky, D.M. SARS-CoV-2 infection and oxidative stress: Pathophysiological insight into thrombosis and therapeutic opportunities. Cytokine Growth Factor Rev. 2021. [Google Scholar] [CrossRef] [PubMed]
- Yeo, J.; Lee, J.; Yoon, S.; Kim, W.J. Tannic acid-based nanogel as an efficient anti-inflammatory agent. Biomater. Sci. 2020, 8, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xu, F.; Liu, C.; Cai, A.; Dain, J.A.; Li, D.; Seeram, N.P.; Cho, B.P.; Ma, H. Inhibitory Effects and Surface Plasmon Resonance-Based Binding Affinities of Dietary Hydrolyzable Tannins and Their Gut Microbial Metabolites on SARS-CoV-2 Main Protease. J. Agric. Food Chem. 2021, 69, 12197–12208. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Yao, S.; Zhao, W.; Zhang, Y.; Liu, J.; Shao, Q.; Wang, Q.; Li, M.; Xie, H.; Shang, W.; et al. Identification of pyrogallol as a warhead in design of covalent inhibitors for the SARS-CoV-2 3CL protease. Nat. Commun. 2021, 12, 3623. [Google Scholar] [CrossRef] [PubMed]
- Shrimp, J.H.; Kales, S.C.; Sanderson, P.E.; Simeonov, A.; Shen, M.; Hall, M.D. An enzymatic TMPRSS2 assay for assessment of clinical candidates and discovery of inhibitors as potential treatment of COVID-19. ACS Pharmacol. Transl. Sci. 2020, 3, 997–1007. [Google Scholar] [CrossRef]
- Mody, V.; Ho, J.; Wills, S.; Mawri, A.; Lawson, L.; Ebert, M.C.C.J.C.; Fortin, G.M.; Rayalam, S.; Taval, S. Identification of 3-chymotrypsin like protease (3CLPro) inhibitors as potential anti-SARS-CoV-2 agents. Commun. Biol. 2021, 4, 93. [Google Scholar] [CrossRef]
- Sauerbrey, G. Verwendung von Schwingquarzen zur Wägung dünner Schichten und zur Mikrowägung. Z. Für Phys. 1959, 155, 206–222. [Google Scholar] [CrossRef]
- Quevedo, I.R.; Olsson, A.L.; Clark, R.J.; Veinot, J.G.; Tufenkji, N. Interpreting deposition behavior of polydisperse surface-modified nanoparticles using QCM-D and sand-packed columns. Environ. Eng. Sci. 2014, 31, 326–337. [Google Scholar] [CrossRef] [Green Version]
- Lapointe, M.; Farner, J.M.; Hernandez, L.M.; Tufenkji, N. Understanding and improving microplastic removal during water treatment: Impact of coagulation and flocculation. Environ. Sci. Technol. 2020, 54, 8719–8727. [Google Scholar] [CrossRef]
- Olsson, A.L.; Wargenau, A.; Tufenkji, N. Optimizing bacteriophage surface densities for bacterial capture and sensing in quartz crystal microbalance with dissipation monitoring. ACS Appl. Mater. Interfaces 2016, 8, 13698–13706. [Google Scholar] [CrossRef] [PubMed]
- Dabros, T.; Van De Ven, T. Collision-induced dispersion of droplets attached to solid particles. J. Colloid Interface Sci. 1994, 163, 28–36. [Google Scholar] [CrossRef]
- Gaudreault, R.; Van de Ven, T.; Whitehead, M. A theoretical study of the interactions of water with gallic acid and a PEO/TGG complex. Mol. Simul. 2006, 32, 17–27. [Google Scholar] [CrossRef]
- Martinez, N.; Junquera, E.; Aicart, E. Ultrasonic, density, and potentiometric characterization of the interaction of gentisic and gallic acids with an apolar cavity in aqueous solution. Phys. Chem. Chem. Phys. 1999, 1, 4811–4817. [Google Scholar] [CrossRef]
- Stewart, J.J. Optimization of parameters for semi-empirical methods I. Method. J. Comput. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.J. Optimization of parameters for semiempirical methods II. Applications. J. Comput. Chem. 1989, 10, 221–264. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Huggins, D.J. Structural analysis of experimental drugs binding to the SARS-CoV-2 target TMPRSS2. J. Mol. Graph. Model. 2020, 100, 107710. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Snijder, E.J. Viral cysteine proteinases. Perspect. Drug Discov. Des. 1996, 6, 64–86. [Google Scholar] [CrossRef]
- Kneller, D.W.; Phillips, G.; O’Neill, H.M.; Jedrzejczak, R.; Stols, L.; Langan, P.; Joachimiak, A.; Coates, L.; Kovalevsky, A. Structural plasticity of SARS-CoV-2 3CL M pro active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 2020, 11, 1–6. [Google Scholar] [CrossRef]
- Goodsell, D.S.; Morris, G.M.; Olson, A.J. Automated docking of flexible ligands: Applications of AutoDock. J. Mol. Recognit 1996, 9, 1–5. [Google Scholar] [CrossRef]
- Santos-Martins, D.; Forli, S.; Ramos, M.J.; Olson, A.J. AutoDock4(Zn): An improved AutoDock force field for small-molecule docking to zinc metalloproteins. J. Chem Inf Model. 2014, 54, 2371–2379. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Nosé, S. A unified formulation of the constant temperature molecular-dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A Gen. Phys. 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅ log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. SETTLE: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Kollman, P.A. Application of RESP charges to calculate conformational energies, hydrogen bond energies, and free energies of solvation. J. Am. Chem. Soc. 2002, 115, 9620–9631. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; van Gunsteren, W.F.; Mark, A.E. Peptide folding: When simulation meets experiment. Angew. Chem. Int. Ed. 1999, 38, 236–240. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 1.8; Schrödinger, LLC.: New York, NY, USA, 2010.
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding Free Energy of TA/Protein Complex (kcal/mol) | |
|---|---|
| Protein (Pose) | MD (MMPBSA) |
| RBD (N501Y) (1) | −66 |
| RBD (N501Y) (2) | −44 |
| RBD (N501Y) (3) | −41 |
| RBD (N501Y) (4) | −70 |
| TMPRSS2 (1) | −68 |
| TMPRSS2 (2) | −33 |
| TMPRSS2 (3) | −57 |
| TMPRSS2 (4) | −71 |
| 3CLpro | −65 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haddad, M.; Gaudreault, R.; Sasseville, G.; Nguyen, P.T.; Wiebe, H.; Van De Ven, T.; Bourgault, S.; Mousseau, N.; Ramassamy, C. Molecular Interactions of Tannic Acid with Proteins Associated with SARS-CoV-2 Infectivity. Int. J. Mol. Sci. 2022, 23, 2643. https://doi.org/10.3390/ijms23052643
Haddad M, Gaudreault R, Sasseville G, Nguyen PT, Wiebe H, Van De Ven T, Bourgault S, Mousseau N, Ramassamy C. Molecular Interactions of Tannic Acid with Proteins Associated with SARS-CoV-2 Infectivity. International Journal of Molecular Sciences. 2022; 23(5):2643. https://doi.org/10.3390/ijms23052643
Chicago/Turabian StyleHaddad, Mohamed, Roger Gaudreault, Gabriel Sasseville, Phuong Trang Nguyen, Hannah Wiebe, Theo Van De Ven, Steve Bourgault, Normand Mousseau, and Charles Ramassamy. 2022. "Molecular Interactions of Tannic Acid with Proteins Associated with SARS-CoV-2 Infectivity" International Journal of Molecular Sciences 23, no. 5: 2643. https://doi.org/10.3390/ijms23052643
APA StyleHaddad, M., Gaudreault, R., Sasseville, G., Nguyen, P. T., Wiebe, H., Van De Ven, T., Bourgault, S., Mousseau, N., & Ramassamy, C. (2022). Molecular Interactions of Tannic Acid with Proteins Associated with SARS-CoV-2 Infectivity. International Journal of Molecular Sciences, 23(5), 2643. https://doi.org/10.3390/ijms23052643