
Single Cell Analysis of Reversibility of the Cell Death Program in Ethanol-Treated Neuronal PC12 Cells

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
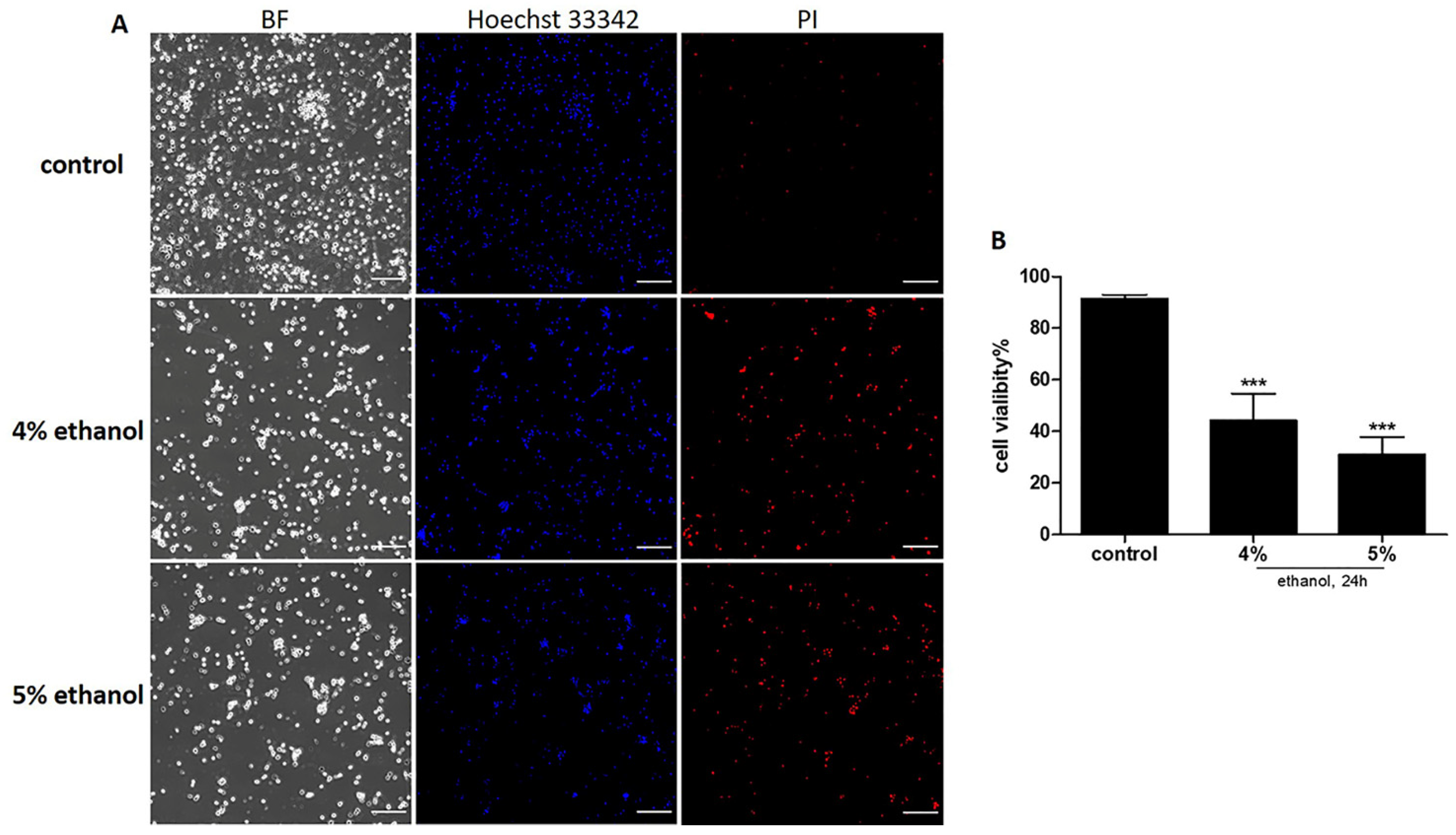
2.1. Ethanol Induced Cell Death on Neuronal PC12 Cells
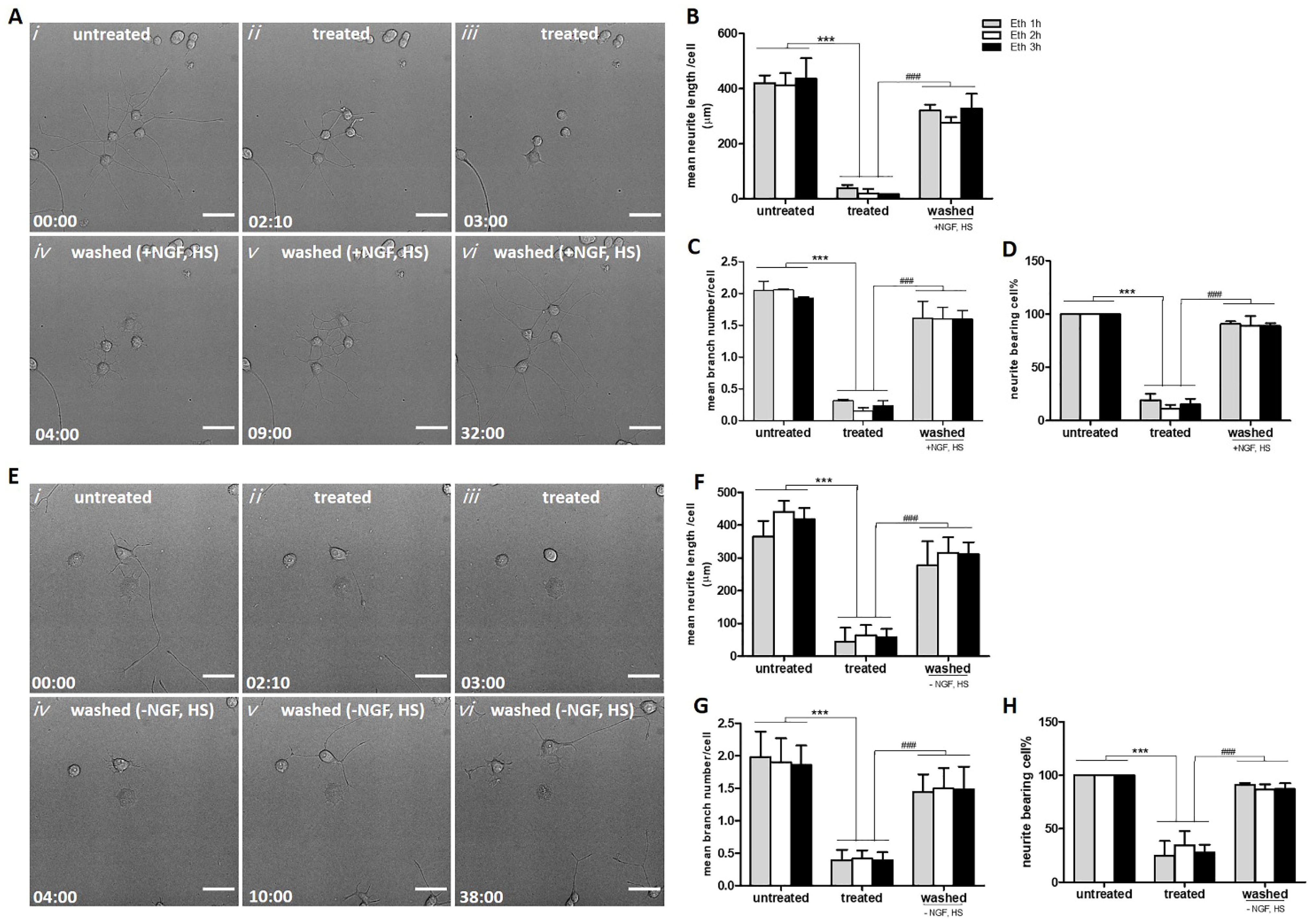
2.2. Neurites Retraction and Regeneration Analysis
2.2.1. Reversible Loss of Neurites Induced by Ethanol in PC12 Cells
2.2.2. Neurite Regrowth Does Not Depend on NGF and HS in the Culture Medium
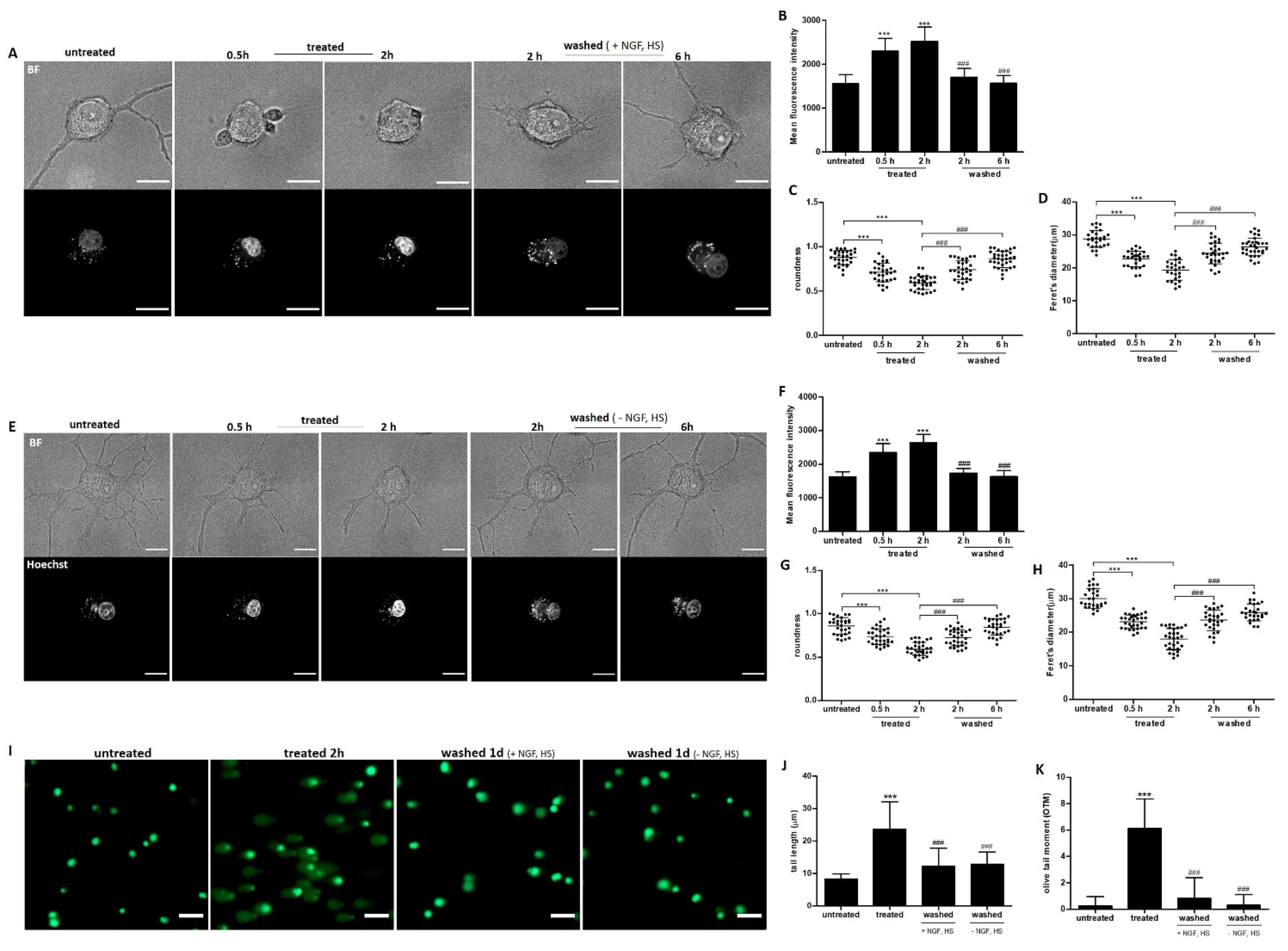
2.3. Elevation of Intracellular ROS and Ca2+ Induced by Ethanol in Neuronal PC12 Cells
2.4. Recovery of Nuclear Shrinkage and DNA Damage with or without Exogenous NGF
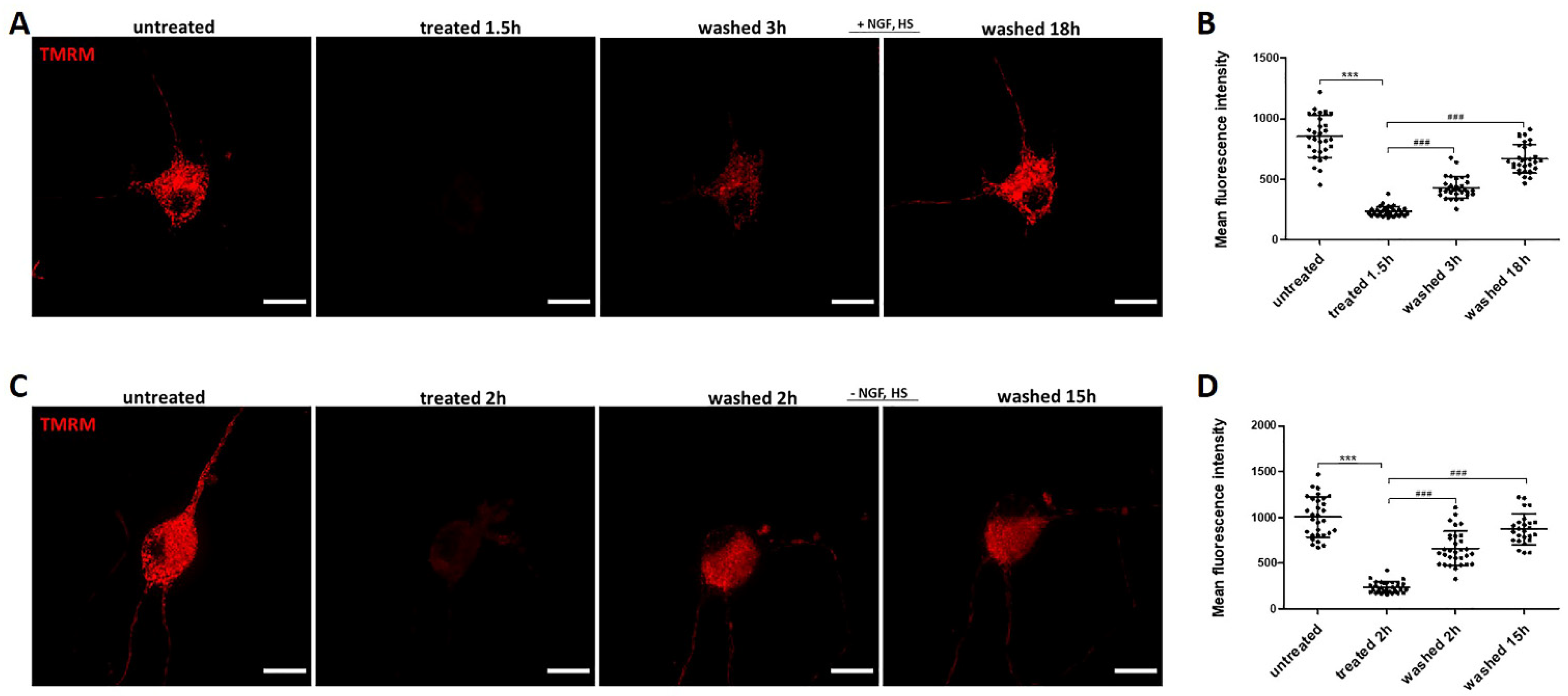
2.5. Recovery of Mitochondrial Fragmentation and Membrane Potential Loss with or without Exogenous NGF
2.6. Reversible PS Exposure in Ethanol Treated Neuronal PC12 Cells
2.7. Cytochrome C Distribution in Ethanol Treated Neuronal PC12 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Viability Assay
4.3. Live Cell Imaging and Neurite Analysis
4.4. Detection of ROS Generation and Intracellular Ca2+ Level
4.5. Assessment of Nuclear Morphology
4.6. Mitochondrial Membrane Potential and Mitochondrial Morphology Analysis
4.7. Detection of Phosphatidylserine (PS) Exposure
4.8. Single-Cell Gel Electrophoresis (Comet) Assay
4.9. Immunofluorescence
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saxena, S.; Caroni, P. Selective neuronal vulnerability in neurodegenerative diseases: From stressor thresholds to degeneration. Neuron 2011, 71, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Troulinaki, K.; Tavernarakis, N. Neurodegenerative conditions associated with ageing: A molecular interplay? Mech. Ageing Dev. 2005, 126, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Roselli, F.; Caroni, P. From intrinsic firing properties to selective neuronal vulnerability in neurodegenerative diseases. Neuron 2015, 85, 901–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, D.A.; Schneider, J.A.; Arvanitakis, Z.; Kelly, J.F.; Aggarwal, N.T.; Shah, R.C.; Wilson, R.S. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 2006, 66, 1837–1844. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, M.; Sahashi, K.; Iguchi, Y.; Hashizume, A. Preclinical progression of neurodegenerative diseases. Nagoya J. Med. Sci. 2018, 80, 289–298. [Google Scholar] [PubMed]
- Driscoll, I.; Troncoso, J. Asymptomatic Alzheimer’s Disease: A Prodrome or a State of Resilience? Curr. Alzheimer Res. 2011, 8, 330–335. [Google Scholar] [CrossRef]
- Kanazawa, I. How do neurons die in neurodegenerative diseases? Trends Mol. Med. 2001, 7, 339–344. [Google Scholar] [CrossRef]
- Fry, L.E.; Fahy, E.; Chrysostomou, V.; Hui, F.; Tang, J.; van Wijngaarden, P.; Petrou, S.; Crowston, J.G. The coma in glaucoma: Retinal ganglion cell dysfunction and recovery. Prog. Retin. Eye Res. 2018, 65, 77–92. [Google Scholar] [CrossRef]
- Tang, H.L.; Tang, H.M.; Mak, K.H.; Shaomin Hub, S.S.W.; Wong, K.M.; Wong, C.S.T.; Wu, H.Y.; Law, H.T.; Liu, K.; Talbot, C.C., Jr.; et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol. Biol. Cell 2012, 23, 2240–2252. [Google Scholar] [CrossRef]
- Sun, G.; Guzman, E.; Balasanyan, V.; Conner, C.M.; Wong, K.; Zhou, H.R.; Kosik, K.S.; Montell, D.J. A molecular signature for anastasis, recovery from the brink of apoptotic cell death. J. Cell Biol. 2017, 216, 3355–3368. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.L.; Yuen, K.L.; Tang, H.M.; Fung, M.C. Reversibility of apoptosis in cancer cells. Br. J. Cancer 2009, 100, 118–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tait, S.W.; Parsons, M.J.; Llambi, F.; Bouchier-Hayes, L.; Connell, S.; Munoz-Pinedo, C.; Green, D.R. Resistance to caspase-independent cell death requires persistence of intact mitochondria. Dev. Cell 2010, 18, 802–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boya, P.; de la Rosa, E.J. Cell death in early neural life. Birth Defects Res. Part C Embryo Today Rev. 2005, 75, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, R.W. Cell death during development of the nervous system. Annu. Rev. Neurosci. 1991, 14, 453–501. [Google Scholar] [CrossRef]
- Kole, A.J.; Annis, R.P.; Deshmukh, M. Mature neurons: Equipped for survival. Cell Death Dis. 2013, 4, e689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benn, S.C.; Woolf, C.J. Adult neuron survival strategies--slamming on the brakes. Nat. Rev. Neurosci. 2004, 5, 686–700. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.M.; Deshmukh, M. Restricting apoptosis for postmitotic cell survival and its relevance to cancer. Cell Cycle 2006, 5, 1616–1620. [Google Scholar] [CrossRef] [Green Version]
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef]
- Golde, T.E. The therapeutic importance of understanding mechanisms of neuronal cell death in neurodegenerative disease. Mol. Neurodegener. 2009, 4, 8. [Google Scholar] [CrossRef] [Green Version]
- Young, C.; Klocke, B.J.; Tenkova, T.; Choi, J.; Labruyere, J.; Qin, Y.Q.; Holtzman, D.M.; Roth, K.A. Ethanol-induced neuronal apoptosis in vivo requires BAX in the developing mouse brain. Cell Death Differ. 2003, 10, 1148–1155. [Google Scholar] [CrossRef] [Green Version]
- Mishra, V.; Agas, A.; Schuetz, H.; Kalluru, J.; Haorah, J. Alcohol induces programmed death receptor-1 and programmed death-ligand-1 differentially in neuroimmune cells. Alcohol 2020, 86, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Greene, L.; Tischler, A. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. USA 1976, 73, 2424–2428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Cao, J.X.; Sun, B.; Li, H.L.; Xia, Y.; Wu, Z.; Tang, C.L.; Hu, J. Mesenchymal stem cells inhibition of chronic ethanol-induced oxidative damage via upregulation of phosphatidylinositol-3-kinase/Akt and modulation of extracellular signal-regulated kinase 1/2 activation in PC12 cells and neurons. Neuroscience 2010, 167, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Ermak, G.; Davies, K.J. Calcium and oxidative stress: From cell signaling to cell death. Mol. Immunol. 2002, 38, 713–721. [Google Scholar] [CrossRef]
- Takadera, T.; Ohyashiki, T. Glycogen synthase kinase-3 inhibitors prevent caspase-dependent apoptosis induced by ethanol in cultured rat cortical neurons. Eur. J. Pharmacol. 2004, 499, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Gutierrez, E.I.; Hernandez-Garza, F.; Garcia-Perez, J.O.; Davila-Rodriguez, M.I.; Aguado-Barrera, M.E.; Cerda-Flores, R.M. Evaluation of DNA single and double strand breaks in women with cervical neoplasia based on alkaline and neutral comet assay techniques. J. Biomed. Biotechnol. 2012, 2012, 385245. [Google Scholar] [CrossRef] [PubMed]
- Brookes, P.S.; Yoon, Y.S.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. J. Physiol.-Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Iannetti, E.F.; Smeitink, J.A.; Beyrath, J.; Willems, P.H.; Koopman, W.J. Multiplexed high-content analysis of mitochondrial morphofunction using live-cell microscopy. Nat. Protoc. 2016, 11, 1693–1710. [Google Scholar] [CrossRef]
- Xiao, B.; Deng, X.; Zhou, W.; Tan, E.K. Flow Cytometry-Based Assessment of Mitophagy Using MitoTracker. Front. Cell. Neurosci. 2016, 10, 76. [Google Scholar] [CrossRef] [Green Version]
- Rimon, G.; Bazenet, C.E.; Philpott, K.L.; Rubin, L.L. Increased surface phosphatidylserine is an early marker of neuronal apoptosis. J. Neurosci. Res. 1997, 48, 563–570. [Google Scholar] [CrossRef]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal Cell Death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef]
- Das, K.P.; Freudenrich, T.M.; Mundy, W.R. Assessment of PC12 cell differentiation and neurite growth: A comparison of morphological and neurochemical measures. Neurotoxicology Teratol. 2004, 26, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Jeon, C.Y.; Jin, J.K.; Koh, Y.H.; Chun, W.; Choi, I.G.; Kown, H.J.; Kim, Y.S.; Park, J.B. Neurites from PC12 cells are connected to each other by synapse-like structures. Synapse 2010, 64, 765–772. [Google Scholar] [CrossRef]
- Cochran, M.; Black, M. PC12 Neurite Regeneration and Long-Term Maintenance in the Absence of Exogenous Nerve Growth Factor in Response to Contact with Schwann Cells. Dev. Brain Res. 1985, 17, 105–116. [Google Scholar] [CrossRef]
- Chiaramello, M.U.A. Expression profiling upon Nex1/MATH-2-mediated neuritogenesis in PC12 cells and its implication in regeneration. J. Neurochem. 2004, 91, 1332–1343. [Google Scholar]
- Madreiter-Sokolowski, C.T.; Thomas, C.; Ristow, M. Interrelation between ROS and Ca2+ in aging and age-related diseases. Redox Biol. 2020, 36, 101678. [Google Scholar] [CrossRef]
- Su, L.J.; Zhang, J.H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxidative Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef] [Green Version]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [Green Version]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2018, 592, 728–742. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bano, D.; Nicotera, P. Ca2+ signals and neuronal death in brain ischemia. Stroke 2007, 38, 674–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, V.; Menzies, K.J.; Auwerx, J. Repairing Mitochondrial Dysfunction in Disease. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 353–389. [Google Scholar] [CrossRef] [PubMed]
- McElnea, E.M.; Quill, B.; Docherty, N.G.; Irnaten, M.; Siah, W.F.; Clark, A.F.; O’Brien, C.J.; Wallace, D.M. Oxidative stress, mitochondrial dysfunction and calcium overload in human lamina cribrosa cells from glaucoma donors. Mol. Vis. 2011, 17, 1182–1191. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3102025/pdf/mv-v17-1182.pdf (accessed on 9 January 2022). [PubMed]
- Knott, A.B.; Perkins, G.; Schwarzenbacher, R.; Bossy-Wetzel, E. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 505–518. [Google Scholar] [CrossRef] [Green Version]
- Kislin, M.; Sword, J.; Fomitcheva, I.V.; Croom, D.; Pryazhnikov, E.; Lihavainen, E.; Toptunov, D.; Rauvala, H.; Ribeiro, A.S.; Khiroug, L.; et al. Reversible Disruption of Neuronal Mitochondria by Ischemic and Traumatic Injury Revealed by Quantitative Two-Photon Imaging in the Neocortex of Anesthetized Mice. J. Neurosci. 2017, 37, 333–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnoult, D. Mitochondrial fragmentation in apoptosis. Trends Cell Biol. 2007, 17, 6–12. [Google Scholar] [CrossRef]
- Deshmukh, M.; Johnson, E.M. Evidence of a Novel Event during Neuronal Death: Development of Competence-to-Die in Response to Cytoplasmic Cytochrome c. Neuron 1998, 21, 695–705. [Google Scholar] [CrossRef] [Green Version]
- Deshmukh, M.; Kuida, K.; Johnson, E.M., Jr. Caspase inhibition extends the commitment to neuronal death beyond cytochrome c release to the point of mitochondrial depolarization. J. Cell Biol. 2000, 150, 131–143. [Google Scholar] [CrossRef]
- Martinou, I.; Desagher, S.; Eskes, R.; Antonsson, B.; Andre, E.; Fakan, S.; Martinou, J.C. The release of cytochrome c from mitochondria during apoptosis of NGF-deprived sympathetic neurons is a reversible event. J. Cell Biol. 1999, 144, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Chen, J.; Chan, J.R.; Langen, R. Engineering a polarity-sensitive biosensor for time-lapse imaging of apoptotic processes and degeneration. Nat. Methods 2010, 7, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.L.; Tang, H.M.; Hardwick, J.M.; Fung, M.C. Strategies for tracking anastasis, a cell survival phenomenon that reverses apoptosis. J. Vis. Exp. 2015, 16, e51964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Eijnde, S.M.; van den Hoff, M.J.B.; Reutelingsperger, C.P.M.; van Heerde, W.L.; Henfling, M.E.R.; Vermeij-Keers, C.; Schutte, B.; Borgers, M.; Ramaekers, F.C.S. Transient expression of phosphatidylserine at cell-cell contact areas is required for myotube formation. J. Cell Sci. 2001, 114, 3631–3642. [Google Scholar] [CrossRef]
- Madabhushi, R.; Pan, L.; Tsai, L.H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef] [Green Version]
- Kasischke, K.; Buchner, M.; Ludolph, A.C.; Riepe, M.W. Nuclear shrinkage in live mouse hippocampal slices. Acta Neuropathol. 2001, 101, 483–490. [Google Scholar] [CrossRef]
- Nouspikel, T. DNA repair in differentiated cells: Some new answers to old questions. Neuroscience 2007, 145, 1213–1221. [Google Scholar] [CrossRef]
- Fishel, M.L.; Vasko, M.R.; Kelley, M.R. DNA repair in neurons: So if they don’t divide what’s to repair? Mutat. Res. 2007, 614, 24–36. [Google Scholar] [CrossRef]
- Mata-Garrido, J.; Tapia, O.; Casafont, I.; Berciano, M.T.; Cuadrado, A.; Lafarga, M. Persistent accumulation of unrepaired DNA damage in rat cortical neurons: Nuclear organization and ChIP-seq analysis of damaged DNA. Acta Neuropathol. Commun. 2018, 6, 68. [Google Scholar] [CrossRef]
- Shanbhag, N.M.; Evans, M.D.; Mao, W.; Nana, A.L.; Seeley, W.W.; Adame, A.; Rissman, R.A.; Masliah, E.; Mucke, L. Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease. Acta Neuropathol. Commun. 2019, 7, 77. [Google Scholar] [CrossRef] [Green Version]
- Brasnjevic, I.; Hof, P.R.; Steinbusch, H.W.; Schmitz, C. Accumulation of nuclear DNA damage or neuron loss: Molecular basis for a new approach to understanding selective neuronal vulnerability in neurodegenerative diseases. DNA Repair 2008, 7, 1087–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutten, B.P.; Schmitz, C.; Gerlach, O.H.; Oyen, H.M.; de Mesquita, E.B.; Steinbusch, H.W.; Korr, H. The aging brain: Accumulation of DNA damage or neuron loss? Neurobiol. Aging 2007, 28, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, I.K.; Taghipour, N.; Hmaidan, S.; Palomba, R.; Scaria, S.; Munoz, A.; Boone, T.B.; Tasciotti, E. Antibody-mediated inhibition of Nogo-A signaling promotes neurite growth in PC-12 cells. J. Tissue Eng. 2016, 7, 9767. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Lan, T.; Liao, W.; Zhao, M.; Yang, H. Genistein inhibits Aβ25–35–induced neurotoxicity in PC12 cells via PKC signaling pathway. Neurochem. Res. 2012, 37, 2787–2794. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, N.M.; Van Opdenbosch, N.; Van Gorp, H.; Parthoens, E.; Lamkanfi, M. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ. 2019, 26, 146–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engeland, M.; Ramaekers, F.C.; Schutte, B.; Reutelingsperger, C.P. A novel assay to measure loss of plasma membrane asymmetry during apoptosis of adherent cells in culture. Cytometry 1996, 24, 131–139. Available online: https://www.ncbi.nlm.nih.gov/pubmed/8725662 (accessed on 9 January 2022). [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, W.; Berendschot, T.T.J.M.; Knoops, K.; van Zandvoort, M.A.M.J.; Webers, C.A.B.; Reutelingsperger, C.P.M.; Gorgels, T.G.M.F. Single Cell Analysis of Reversibility of the Cell Death Program in Ethanol-Treated Neuronal PC12 Cells. Int. J. Mol. Sci. 2022, 23, 2650. https://doi.org/10.3390/ijms23052650
You W, Berendschot TTJM, Knoops K, van Zandvoort MAMJ, Webers CAB, Reutelingsperger CPM, Gorgels TGMF. Single Cell Analysis of Reversibility of the Cell Death Program in Ethanol-Treated Neuronal PC12 Cells. International Journal of Molecular Sciences. 2022; 23(5):2650. https://doi.org/10.3390/ijms23052650
Chicago/Turabian StyleYou, Wenting, Tos T. J. M. Berendschot, Kèvin Knoops, Marc A. M. J. van Zandvoort, Carroll A. B. Webers, Chris P. M. Reutelingsperger, and Theo G. M. F. Gorgels. 2022. "Single Cell Analysis of Reversibility of the Cell Death Program in Ethanol-Treated Neuronal PC12 Cells" International Journal of Molecular Sciences 23, no. 5: 2650. https://doi.org/10.3390/ijms23052650
APA StyleYou, W., Berendschot, T. T. J. M., Knoops, K., van Zandvoort, M. A. M. J., Webers, C. A. B., Reutelingsperger, C. P. M., & Gorgels, T. G. M. F. (2022). Single Cell Analysis of Reversibility of the Cell Death Program in Ethanol-Treated Neuronal PC12 Cells. International Journal of Molecular Sciences, 23(5), 2650. https://doi.org/10.3390/ijms23052650