
Current Advances in RNA Therapeutics for Human Diseases



Abstract
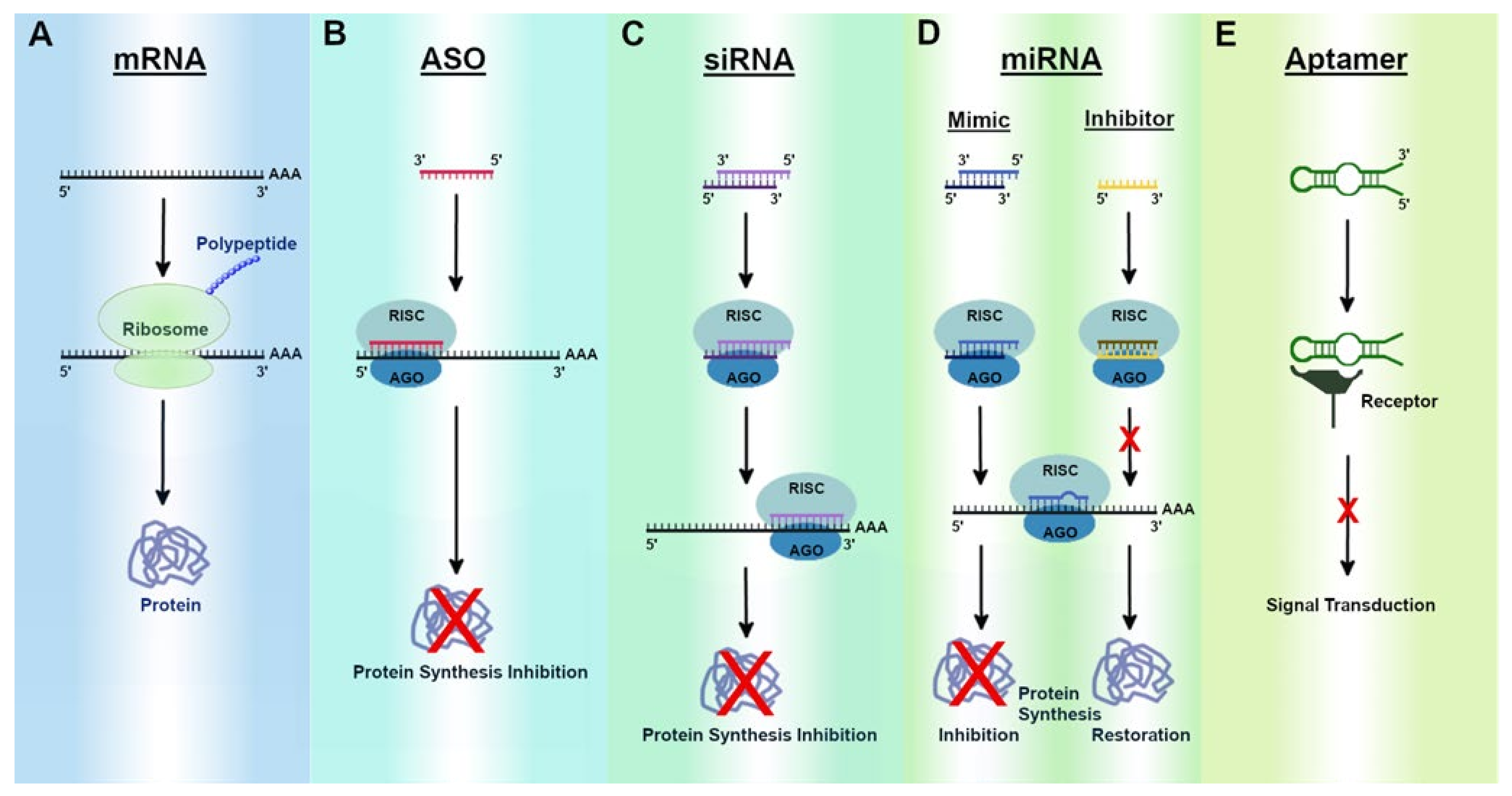
:1. Introduction
2. RNA Therapeutics
2.1. mRNA Therapeutics and Functional Implications
mRNA Vaccines
2.2. ASO Therapeutics and Functional Implications
2.2.1. FDA-Approved ASOs
2.2.2. ASOs in Clinical Trials
2.3. siRNA Therapeutics and Functional Implications
2.3.1. FDA-Approved siRNAs
2.3.2. siRNAs in Clinical Trials
2.4. miRNA Therapeutics and Functional Implications
miRNAs in Clinical Trials
2.5. Aptamer Therapeutics and Functional Implications
2.5.1. FDA-Approved Aptamers
2.5.2. Aptamers in Clinical Trials
3. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Winkle, M.; El-Daly, S.M.; Fabbri, M.; Calin, G.A. Noncoding RNA therapeutics—Challenges and potential solutions. Nat. Rev. Drug Discov. 2021, 20, 629–651. [Google Scholar] [CrossRef] [PubMed]
- Rand, T.A.; Ginalski, K.; Grishin, N.V.; Wang, X. Biochemical identification of Argonaute 2 as the sole protein required for RNA-induced silencing complex activity. Proc. Natl. Acad. Sci. USA 2004, 101, 14385–14389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Zogg, H.; Ro, S. Role of microRNAs in Disorders of Gut-Brain Interactions: Clinical Insights and Therapeutic Alternatives. J. Pers. Med. 2021, 11, 1021. [Google Scholar] [CrossRef]
- Matranga, C.; Tomari, Y.; Shin, C.; Bartel, D.P.; Zamore, P.D. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell 2005, 123, 607–620. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent advances in the development of protein-protein interactions modulators: Mechanisms and clinical trials. Signal Transduct. Target. Ther. 2020, 5, 213. [Google Scholar] [CrossRef]
- Dimitrov, D.S. Therapeutic proteins. Methods Mol. Biol. 2012, 899, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Bruno, B.J.; Miller, G.D.; Lim, C.S. Basics and recent advances in peptide and protein drug delivery. Ther. Deliv. 2013, 4, 1443–1467. [Google Scholar] [CrossRef] [Green Version]
- Bajan, S.; Hutvagner, G. RNA-Based Therapeutics: From Antisense Oligonucleotides to miRNAs. Cells 2020, 9, 137. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, H. Innovative Therapeutic Approaches for Huntington’s Disease: From Nucleic Acids to GPCR-Targeting Small Molecules. Front. Cell Neurosci. 2021, 15, 785703. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.; Hossain, G.S.; Kocerha, J. The Potential for microRNA Therapeutics and Clinical Research. Front. Genet. 2019, 10, 478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Ha, S.E.; Wei, L.; Jin, B.; Zogg, H.; Poudrier, S.M.; Jorgensen, B.G.; Park, C.; Ronkon, C.F.; Bartlett, A.; et al. miR-10b-5p Rescues Diabetes and Gastrointestinal Dysmotility. Gastroenterology 2021, 160, 1662–1678.e18. [Google Scholar] [CrossRef]
- Vitravene Study, G. A randomized controlled clinical trial of intravitreous fomivirsen for treatment of newly diagnosed peripheral cytomegalovirus retinitis in patients with AIDS. Am. J. Ophthalmol. 2002, 133, 467–474. [Google Scholar] [CrossRef]
- Santos, R.D.; Raal, F.J.; Donovan, J.M.; Cromwell, W.C. Mipomersen preferentially reduces small low-density lipoprotein particle number in patients with hypercholesterolemia. J. Clin. Lipidol. 2015, 9, 201–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, S.; Kota, J.; et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Plante-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef]
- Scaglioni, D.; Catapano, F.; Ellis, M.; Torelli, S.; Chambers, D.; Feng, L.; Beck, M.; Sewry, C.; Monforte, M.; Harriman, S.; et al. The administration of antisense oligonucleotide golodirsen reduces pathological regeneration in patients with Duchenne muscular dystrophy. Acta Neuropathol. Commun. 2021, 9, 7. [Google Scholar] [CrossRef]
- Kim, J.; Hu, C.; Moufawad El Achkar, C.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Kuniholm, A.; et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N. Engl. J. Med. 2019, 381, 1644–1652. [Google Scholar] [CrossRef]
- Shirley, M. Casimersen: First Approval. Drugs 2021, 81, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.R.; Kuntz, N.L.; Koenig, E.; East, L.; Upadhyay, S.; Han, B.; Shieh, P.B. Safety, tolerability, and pharmacokinetics of casimersen in patients with Duchenne muscular dystrophy amenable to exon 45 skipping: A randomized, double-blind, placebo-controlled, dose-titration trial. Muscle Nerve 2021, 64, 285–292. [Google Scholar] [CrossRef]
- Clemens, P.R.; Rao, V.K.; Connolly, A.M.; Harper, A.D.; Mah, J.K.; Smith, E.C.; McDonald, C.M.; Zaidman, C.M.; Morgenroth, L.P.; Osaki, H.; et al. Safety, Tolerability, and Efficacy of Viltolarsen in Boys With Duchenne Muscular Dystrophy Amenable to Exon 53 Skipping: A Phase 2 Randomized Clinical Trial. JAMA Neurol. 2020, 77, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Balwani, M.; Sardh, E.; Ventura, P.; Peiro, P.A.; Rees, D.C.; Stolzel, U.; Bissell, D.M.; Bonkovsky, H.L.; Windyga, J.; Anderson, K.E.; et al. Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N. Engl. J. Med. 2020, 382, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Liebow, A.; Li, X.; Racie, T.; Hettinger, J.; Bettencourt, B.R.; Najafian, N.; Haslett, P.; Fitzgerald, K.; Holmes, R.P.; Erbe, D.; et al. An Investigational RNAi Therapeutic Targeting Glycolate Oxidase Reduces Oxalate Production in Models of Primary Hyperoxaluria. J. Am. Soc. Nephrol. 2017, 28, 494–503. [Google Scholar] [CrossRef] [Green Version]
- Lamb, Y.N. Inclisiran: First Approval. Drugs 2021, 81, 389–395. [Google Scholar] [CrossRef]
- Gragoudas, E.S.; Adamis, A.P.; Cunningham, E.T., Jr.; Feinsod, M.; Guyer, D.R.; Group, V.I.S.i.O.N.C.T. Pegaptanib for neovascular age-related macular degeneration. N. Engl. J. Med. 2004, 351, 2805–2816. [Google Scholar] [CrossRef] [Green Version]
- Richardson, P.G.; Smith, A.R.; Triplett, B.M.; Kernan, N.A.; Grupp, S.A.; Antin, J.H.; Lehmann, L.; Shore, T.; Iacobelli, M.; Miloslavsky, M.; et al. Defibrotide for Patients with Hepatic Veno-Occlusive Disease/Sinusoidal Obstruction Syndrome: Interim Results from a Treatment IND Study. Biol. Blood Marrow Transplant. 2017, 23, 997–1004. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.B.; Yang, K.; Ko, H.J.; Shim, J.R.; Choi, B.H.; Lee, J.H.; Ryu, J.H. Successful defibrotide treatment of a patient with veno-occlusive disease after living-donor liver transplantation: A case report. Medicine 2021, 100, e26463. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Perez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, J.W.; Kim, H.; McCauley, M.; Hessel, E.M.; Sims, P.; Fisher, D.C.; Nadler, L.M.; Coffman, R.L.; Freedman, A.S. Combination immunotherapy with a CpG oligonucleotide (1018 ISS) and rituximab in patients with non-Hodgkin lymphoma: Increased interferon-alpha/beta-inducible gene expression, without significant toxicity. Blood 2005, 105, 489–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedberg, J.W.; Kelly, J.L.; Neuberg, D.; Peterson, D.R.; Kutok, J.L.; Salloum, R.; Brenn, T.; Fisher, D.C.; Ronan, E.; Dalton, V.; et al. Phase II study of a TLR-9 agonist (1018 ISS) with rituximab in patients with relapsed or refractory follicular lymphoma. Br. J. Haematol. 2009, 146, 282–291. [Google Scholar] [CrossRef] [Green Version]
- Chi, K.N.; Yu, E.Y.; Jacobs, C.; Bazov, J.; Kollmannsberger, C.; Higano, C.S.; Mukherjee, S.D.; Gleave, M.E.; Stewart, P.S.; Hotte, S.J. A phase I dose-escalation study of apatorsen (OGX-427), an antisense inhibitor targeting heat shock protein 27 (Hsp27), in patients with castration-resistant prostate cancer and other advanced cancers. Ann. Oncol. 2016, 27, 1116–1122. [Google Scholar] [CrossRef]
- Cortes, J.; Kantarjian, H.; Ball, E.D.; Dipersio, J.; Kolitz, J.E.; Fernandez, H.F.; Goodman, M.; Borthakur, G.; Baer, M.R.; Wetzler, M. Phase 2 randomized study of p53 antisense oligonucleotide (cenersen) plus idarubicin with or without cytarabine in refractory and relapsed acute myeloid leukemia. Cancer 2012, 118, 418–427. [Google Scholar] [CrossRef]
- De Velasco, M.A.; Kura, Y.; Sakai, K.; Hatanaka, Y.; Davies, B.R.; Campbell, H.; Klein, S.; Kim, Y.; MacLeod, A.R.; Sugimoto, K.; et al. Targeting castration-resistant prostate cancer with androgen receptor antisense oligonucleotide therapy. JCI Insight 2019, 4, e122688. [Google Scholar] [CrossRef]
- Laskin, J.J.; Nicholas, G.; Lee, C.; Gitlitz, B.; Vincent, M.; Cormier, Y.; Stephenson, J.; Ung, Y.; Sanborn, R.; Pressnail, B.; et al. Phase I/II trial of custirsen (OGX-011), an inhibitor of clusterin, in combination with a gemcitabine and platinum regimen in patients with previously untreated advanced non-small cell lung cancer. J. Thorac. Oncol. 2012, 7, 579–586. [Google Scholar] [CrossRef] [Green Version]
- Chi, K.N.; Eisenhauer, E.; Fazli, L.; Jones, E.C.; Goldenberg, S.L.; Powers, J.; Tu, D.; Gleave, M.E. A phase I pharmacokinetic and pharmacodynamic study of OGX-011, a 2′-methoxyethyl antisense oligonucleotide to clusterin, in patients with localized prostate cancer. J. Natl. Cancer Inst. 2005, 97, 1287–1296. [Google Scholar] [CrossRef]
- El Dika, I.; Lim, H.Y.; Yong, W.P.; Lin, C.C.; Yoon, J.H.; Modiano, M.; Freilich, B.; Choi, H.J.; Chao, T.Y.; Kelley, R.K.; et al. An Open-Label, Multicenter, Phase I, Dose Escalation Study with Phase II Expansion Cohort to Determine the Safety, Pharmacokinetics, and Preliminary Antitumor Activity of Intravenous TKM-080301 in Subjects with Advanced Hepatocellular Carcinoma. Oncologist 2019, 24, 747–e218. [Google Scholar] [CrossRef] [Green Version]
- Steegmaier, M.; Hoffmann, M.; Baum, A.; Lenart, P.; Petronczki, M.; Krssak, M.; Gurtler, U.; Garin-Chesa, P.; Lieb, S.; Quant, J.; et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol. 2007, 17, 316–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultheis, B.; Strumberg, D.; Santel, A.; Vank, C.; Gebhardt, F.; Keil, O.; Lange, C.; Giese, K.; Kaufmann, J.; Khan, M.; et al. First-in-human phase I study of the liposomal RNA interference therapeutic Atu027 in patients with advanced solid tumors. J. Clin. Oncol. 2014, 32, 4141–4148. [Google Scholar] [CrossRef] [PubMed]
- Schultheis, B.; Strumberg, D.; Kuhlmann, J.; Wolf, M.; Link, K.; Seufferlein, T.; Kaufmann, J.; Feist, M.; Gebhardt, F.; Khan, M.; et al. Safety, Efficacy and Pharcacokinetics of Targeted Therapy with The Liposomal RNA Interference Therapeutic Atu027 Combined with Gemcitabine in Patients with Pancreatic Adenocarcinoma. A Randomized Phase Ib/IIa Study. Cancers 2020, 12, 3130. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Khvalevsky, E.Z.; Hubert, A.; Gabai, R.M.; Hen, N.; Segal, A.; Domb, A.; Harari, G.; David, E.B.; Raskin, S.; et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget 2015, 6, 24560–24570. [Google Scholar] [CrossRef] [Green Version]
- Zorde Khvalevsky, E.; Gabai, R.; Rachmut, I.H.; Horwitz, E.; Brunschwig, Z.; Orbach, A.; Shemi, A.; Golan, T.; Domb, A.J.; Yavin, E.; et al. Mutant KRAS is a druggable target for pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20723–20728. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Kaelin, W.G. Targeting HIF2 in Clear Cell Renal Cell Carcinoma. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Triozzi, P.; Kooshki, M.; Alistar, A.; Bitting, R.; Neal, A.; Lametschwandtner, G.; Loibner, H. Phase I clinical trial of adoptive cellular immunotherapy with APN401 in patients with solid tumors. J. ImmunoTherapy Cancer 2015, 3, 175. [Google Scholar] [CrossRef] [Green Version]
- Sekijima, Y.; Wiseman, R.L.; Matteson, J.; Hammarstrom, P.; Miller, S.R.; Sawkar, A.R.; Balch, W.E.; Kelly, J.W. The biological and chemical basis for tissue-selective amyloid disease. Cell 2005, 121, 73–85. [Google Scholar] [CrossRef] [Green Version]
- Habtemariam, B.A.; Karsten, V.; Attarwala, H.; Goel, V.; Melch, M.; Clausen, V.A.; Garg, P.; Vaishnaw, A.K.; Sweetser, M.T.; Robbie, G.J.; et al. Single-Dose Pharmacokinetics and Pharmacodynamics of Transthyretin Targeting N-acetylgalactosamine-Small Interfering Ribonucleic Acid Conjugate, Vutrisiran, in Healthy Subjects. Clin. Pharmacol. Ther. 2021, 109, 372–382. [Google Scholar] [CrossRef]
- Park, E.J.; Choi, J.; Lee, K.C.; Na, D.H. Emerging PEGylated non-biologic drugs. Expert Opin. Emerg. Drugs 2019, 24, 107–119. [Google Scholar] [CrossRef]
- Menne, J.; Eulberg, D.; Beyer, D.; Baumann, M.; Saudek, F.; Valkusz, Z.; Wiecek, A.; Haller, H.; Emapticap Study, G. C-C motif-ligand 2 inhibition with emapticap pegol (NOX-E36) in type 2 diabetic patients with albuminuria. Nephrol. Dial. Transplant. 2017, 32, 307–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kremsner, P.G.; Ahuad Guerrero, R.A.; Arana-Arri, E.; Aroca Martinez, G.J.; Bonten, M.; Chandler, R.; Corral, G.; De Block, E.J.L.; Ecker, L.; Gabor, J.J.; et al. Efficacy and safety of the CVnCoV SARS-CoV-2 mRNA vaccine candidate in ten countries in Europe and Latin America (HERALD): A randomised, observer-blinded, placebo-controlled, phase 2b/3 trial. Lancet Infect. Dis. 2021, 22, 329–340. [Google Scholar] [CrossRef]
- Anttila, V.; Saraste, A.; Knuuti, J.; Jaakkola, P.; Hedman, M.; Svedlund, S.; Lagerstrom-Fermer, M.; Kjaer, M.; Jeppsson, A.; Gan, L.M. Synthetic mRNA Encoding VEGF-A in Patients Undergoing Coronary Artery Bypass Grafting: Design of a Phase 2a Clinical Trial. Mol. Ther. Methods Clin. Dev. 2020, 18, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; McCray, P.B., Jr.; Engelhardt, J.F. Advances in gene therapy for cystic fibrosis lung disease. Hum. Mol. Genet 2019, 28, R88–R94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandler, R.J.; Venditti, C.P. Gene Therapy for Methylmalonic Acidemia: Past, Present, and Future. Hum. Gene Ther. 2019, 30, 1236–1244. [Google Scholar] [CrossRef]
- An, D.; Schneller, J.L.; Frassetto, A.; Liang, S.; Zhu, X.; Park, J.S.; Theisen, M.; Hong, S.J.; Zhou, J.; Rajendran, R.; et al. Systemic Messenger RNA Therapy as a Treatment for Methylmalonic Acidemia. Cell Rep. 2017, 21, 3548–3558. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef]
- Janssen, H.L.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [Green Version]
- Gomez, I.G.; MacKenna, D.A.; Johnson, B.G.; Kaimal, V.; Roach, A.M.; Ren, S.; Nakagawa, N.; Xin, C.; Newitt, R.; Pandya, S.; et al. Anti-microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J. Clin. Investig. 2015, 125, 141–156. [Google Scholar] [CrossRef]
- Bedewy, A.M.L.; Elmaghraby, S.M.; Shehata, A.A.; Kandil, N.S. Prognostic Value of miRNA-155 Expression in B-Cell Non-Hodgkin Lymphoma. Turk. J. Haematol. 2017, 34, 207–212. [Google Scholar] [CrossRef]
- Bonauer, A.; Carmona, G.; Iwasaki, M.; Mione, M.; Koyanagi, M.; Fischer, A.; Burchfield, J.; Fox, H.; Doebele, C.; Ohtani, K.; et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science 2009, 324, 1710–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trajkovski, M.; Hausser, J.; Soutschek, J.; Bhat, B.; Akin, A.; Zavolan, M.; Heim, M.H.; Stoffel, M. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature 2011, 474, 649–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.C.; Valencia, T.; Allerson, C.; Schairer, A.; Flaten, A.; Yheskel, M.; Kersjes, K.; Li, J.; Gatto, S.; Takhar, M.; et al. Discovery and preclinical evaluation of anti-miR-17 oligonucleotide RGLS4326 for the treatment of polycystic kidney disease. Nat. Commun. 2019, 10, 4148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taubel, J.; Hauke, W.; Rump, S.; Viereck, J.; Batkai, S.; Poetzsch, J.; Rode, L.; Weigt, H.; Genschel, C.; Lorch, U.; et al. Novel antisense therapy targeting microRNA-132 in patients with heart failure: Results of a first-in-human Phase 1b randomized, double-blind, placebo-controlled study. Eur. Heart J. 2021, 42, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Cushing, L.; Kuang, P.P.; Qian, J.; Shao, F.; Wu, J.; Little, F.; Thannickal, V.J.; Cardoso, W.V.; Lu, J. miR-29 is a major regulator of genes associated with pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2011, 45, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 180–188. [Google Scholar] [CrossRef]
- Mignone, F.; Gissi, C.; Liuni, S.; Pesole, G. Untranslated regions of mRNAs. Genome Biol. 2002, 3, REVIEWS0004. [Google Scholar] [CrossRef]
- Leppek, K.; Das, R.; Barna, M. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 2018, 19, 158–174. [Google Scholar] [CrossRef]
- Da Silva Sanchez, A.; Paunovska, K.; Cristian, A.; Dahlman, J.E. Treating Cystic Fibrosis with mRNA and CRISPR. Hum. Gene Ther. 2020, 31, 940–955. [Google Scholar] [CrossRef]
- Musunuru, K.; Chadwick, A.C.; Mizoguchi, T.; Garcia, S.P.; DeNizio, J.E.; Reiss, C.W.; Wang, K.; Iyer, S.; Dutta, C.; Clendaniel, V.; et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature 2021, 593, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- Jirikowski, G.F.; Sanna, P.P.; Maciejewski-Lenoir, D.; Bloom, F.E. Reversal of diabetes insipidus in Brattleboro rats: Intrahypothalamic injection of vasopressin mRNA. Science 1992, 255, 996–998. [Google Scholar] [CrossRef]
- Martinon, F.; Krishnan, S.; Lenzen, G.; Magne, R.; Gomard, E.; Guillet, J.G.; Levy, J.P.; Meulien, P. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur. J. Immunol. 1993, 23, 1719–1722. [Google Scholar] [CrossRef] [PubMed]
- Aldrich, C.; Leroux-Roels, I.; Huang, K.B.; Bica, M.A.; Loeliger, E.; Schoenborn-Kellenberger, O.; Walz, L.; Leroux-Roels, G.; von Sonnenburg, F.; Oostvogels, L. Proof-of-concept of a low-dose unmodified mRNA-based rabies vaccine formulated with lipid nanoparticles in human volunteers: A phase 1 trial. Vaccine 2021, 39, 1310–1318. [Google Scholar] [CrossRef]
- Petsch, B.; Schnee, M.; Vogel, A.B.; Lange, E.; Hoffmann, B.; Voss, D.; Schlake, T.; Thess, A.; Kallen, K.J.; Stitz, L.; et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat. Biotechnol. 2012, 30, 1210–1216. [Google Scholar] [CrossRef]
- Aliprantis, A.O.; Shaw, C.A.; Griffin, P.; Farinola, N.; Railkar, R.A.; Cao, X.; Liu, W.; Sachs, J.R.; Swenson, C.J.; Lee, H.; et al. A phase 1, randomized, placebo-controlled study to evaluate the safety and immunogenicity of an mRNA-based RSV prefusion F protein vaccine in healthy younger and older adults. Hum. Vaccin Immunother. 2021, 17, 1248–1261. [Google Scholar] [CrossRef]
- Schnee, M.; Vogel, A.B.; Voss, D.; Petsch, B.; Baumhof, P.; Kramps, T.; Stitz, L. An mRNA Vaccine Encoding Rabies Virus Glycoprotein Induces Protection against Lethal Infection in Mice and Correlates of Protection in Adult and Newborn Pigs. PLoS Negl. Trop. Dis. 2016, 10, e0004746. [Google Scholar] [CrossRef]
- Medina-Magues, L.G.; Gergen, J.; Jasny, E.; Petsch, B.; Lopera-Madrid, J.; Medina-Magues, E.S.; Salas-Quinchucua, C.; Osorio, J.E. mRNA Vaccine Protects against Zika Virus. Vaccines 2021, 9, 1464. [Google Scholar] [CrossRef]
- John, S.; Yuzhakov, O.; Woods, A.; Deterling, J.; Hassett, K.; Shaw, C.A.; Ciaramella, G. Multi-antigenic human cytomegalovirus mRNA vaccines that elicit potent humoral and cell-mediated immunity. Vaccine 2018, 36, 1689–1699. [Google Scholar] [CrossRef]
- Webster, H.; Valencia, S.; Kumar, A.; Chan, C.; Dennis, M.; Roark, H.; Woods, A.; John, S.; Carfi, A.; Permar, S.R. Pre-existing immunity to cytomegalovirus in macaques influences human CMV vaccine responses in preclinical models. Vaccine 2021, 39, 5358–5367. [Google Scholar] [CrossRef]
- Conklin, L.D.; McAninch, R.E.; Schulz, D.; Kaluza, G.L.; LeMaire, S.A.; Coselli, J.S.; Raizner, A.E.; Sutton, R.E. HIV-based vectors and angiogenesis following rabbit hindlimb ischemia. J. Surg. Res. 2005, 123, 55–66. [Google Scholar] [CrossRef]
- Giacca, M.; Zacchigna, S. VEGF gene therapy: Therapeutic angiogenesis in the clinic and beyond. Gene Ther. 2012, 19, 622–629. [Google Scholar] [CrossRef]
- Gaffney, M.M.; Hynes, S.O.; Barry, F.; O’Brien, T. Cardiovascular gene therapy: Current status and therapeutic potential. Br. J. Pharmacol. 2007, 152, 175–188. [Google Scholar] [CrossRef] [Green Version]
- Butnariu, L.I.; Tarca, E.; Cojocaru, E.; Rusu, C.; Moisa, S.M.; Leon Constantin, M.M.; Gorduza, E.V.; Trandafir, L.M. Genetic Modifying Factors of Cystic Fibrosis Phenotype: A Challenge for Modern Medicine. J. Clin. Med. 2021, 10, 5821. [Google Scholar] [CrossRef]
- Fraser, J.L.; Venditti, C.P. Methylmalonic and propionic acidemias: Clinical management update. Curr. Opin. Pediatr. 2016, 28, 682–693. [Google Scholar] [CrossRef]
- Connolly, B.; Isaacs, C.; Cheng, L.; Asrani, K.H.; Subramanian, R.R. SERPINA1 mRNA as a Treatment for Alpha-1 Antitrypsin Deficiency. J. Nucleic Acids 2018, 2018, 8247935. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; An, D.; Galduroz, M.; Zhuo, J.; Liang, S.; Eybye, M.; Frassetto, A.; Kuroda, E.; Funahashi, A.; Santana, J.; et al. mRNA Therapy Improves Metabolic and Behavioral Abnormalities in a Murine Model of Citrin Deficiency. Mol. Ther. 2019, 27, 1242–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porciuncula, A.; Morgado, M.; Gupta, R.; Syrigos, K.; Meehan, R.; Zacharek, S.J.; Frederick, J.P.; Schalper, K.A. Spatial Mapping and Immunomodulatory Role of the OX40/OX40L Pathway in Human Non-Small Cell Lung Cancer. Clin. Cancer Res. 2021, 27, 6174–6183. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, S.L.; Bailey, D.; Zielinski, J.; Apte, A.; Musenge, F.; Karp, R.; Burke, S.; Garcon, F.; Mishra, A.; Gurumurthy, S.; et al. Intratumoral IL12 mRNA Therapy Promotes TH1 Transformation of the Tumor Microenvironment. Clin. Cancer Res. 2020, 26, 6284–6298. [Google Scholar] [CrossRef]
- Zhu, X.; Yin, L.; Theisen, M.; Zhuo, J.; Siddiqui, S.; Levy, B.; Presnyak, V.; Frassetto, A.; Milton, J.; Salerno, T.; et al. Systemic mRNA Therapy for the Treatment of Fabry Disease: Preclinical Studies in Wild-Type Mice, Fabry Mouse Model, and Wild-Type Non-human Primates. Am. J. Hum. Genet 2019, 104, 625–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evers, M.M.; Toonen, L.J.; van Roon-Mom, W.M. Antisense oligonucleotides in therapy for neurodegenerative disorders. Adv. Drug Deliv. Rev. 2015, 87, 90–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lima, W.F.; Zhang, H.; Fan, A.; Sun, H.; Crooke, S.T. Determination of the role of the human RNase H1 in the pharmacology of DNA-like antisense drugs. J. Biol. Chem. 2004, 279, 17181–17189. [Google Scholar] [CrossRef] [Green Version]
- Wan, L.; Dreyfuss, G. Splicing-Correcting Therapy for SMA. Cell 2017, 170, 5. [Google Scholar] [CrossRef]
- Lim, K.H.; Han, Z.; Jeon, H.Y.; Kach, J.; Jing, E.; Weyn-Vanhentenryck, S.; Downs, M.; Corrionero, A.; Oh, R.; Scharner, J.; et al. Antisense oligonucleotide modulation of non-productive alternative splicing upregulates gene expression. Nat. Commun. 2020, 11, 3501. [Google Scholar] [CrossRef]
- Ward, A.J.; Norrbom, M.; Chun, S.; Bennett, C.F.; Rigo, F. Nonsense-mediated decay as a terminating mechanism for antisense oligonucleotides. Nucleic Acids Res. 2014, 42, 5871–5879. [Google Scholar] [CrossRef] [Green Version]
- Boiziau, C.; Kurfurst, R.; Cazenave, C.; Roig, V.; Thuong, N.T.; Toulme, J.J. Inhibition of translation initiation by antisense oligonucleotides via an RNase-H independent mechanism. Nucleic Acids Res. 1991, 19, 1113–1119. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.H.; Shen, W.; Sun, H.; Migawa, M.T.; Vickers, T.A.; Crooke, S.T. Translation efficiency of mRNAs is increased by antisense oligonucleotides targeting upstream open reading frames. Nat. Biotechnol. 2016, 34, 875–880. [Google Scholar] [CrossRef]
- Vickers, T.A.; Wyatt, J.R.; Burckin, T.; Bennett, C.F.; Freier, S.M. Fully modified 2′ MOE oligonucleotides redirect polyadenylation. Nucleic Acids Res. 2001, 29, 1293–1299. [Google Scholar] [CrossRef] [Green Version]
- Piascik, P. Fomiversen sodium approved to treat CMV retinitis. J. Am. Pharm. Assoc. 1999, 39, 84–85. [Google Scholar] [CrossRef]
- Jabs, D.A.; Griffiths, P.D. Fomivirsen for the treatment of cytomegalovirus retinitis. Am. J. Ophthalmol. 2002, 133, 552–556. [Google Scholar] [CrossRef]
- Won, J.I.; Zhang, J.; Tecson, K.M.; McCullough, P.A. Balancing Low-density Lipoprotein Cholesterol Reduction and Hepatotoxicity With Lomitapide Mesylate and Mipomersen in Patients With Homozygous Familial Hypercholesterolemia. Rev. Cardiovasc. Med. 2017, 18, 21–28. [Google Scholar] [PubMed]
- Cesaro, A.; Fimiani, F.; Gragnano, F.; Moscarella, E.; Schiavo, A.; Vergara, A.; Akioyamen, L.; D’Erasmo, L.; Averna, M.; Arca, M.; et al. New Frontiers in the Treatment of Homozygous Familial Hypercholesterolemia. Heart Fail Clin. 2022, 18, 177–188. [Google Scholar] [CrossRef]
- Gouni-Berthold, I.; Berthold, H.K. Mipomersen and lomitapide: Two new drugs for the treatment of homozygous familial hypercholesterolemia. Atheroscler. Suppl. 2015, 18, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A. FDA Approval of Nusinersen for Spinal Muscular Atrophy Makes 2016 the Year of Splice Modulating Oligonucleotides. Nucleic Acid. Ther. 2017, 27, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Burr, P.; Reddivari, A.K.R. Spinal Muscle Atrophy; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 2017, 27, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, J.R.; Chamberlain, J.S. Progress toward Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther. 2017, 25, 1125–1131. [Google Scholar] [CrossRef] [Green Version]
- De Angelis, F.G.; Sthandier, O.; Berarducci, B.; Toso, S.; Galluzzi, G.; Ricci, E.; Cossu, G.; Bozzoni, I. Chimeric snRNA molecules carrying antisense sequences against the splice junctions of exon 51 of the dystrophin pre-mRNA induce exon skipping and restoration of a dystrophin synthesis in Delta 48-50 DMD cells. Proc. Natl. Acad. Sci. USA 2002, 99, 9456–9461. [Google Scholar] [CrossRef] [Green Version]
- Sekijima, Y. Hereditary Transthyretin Amyloidosis; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; GeneReviews: Seattle, WA, USA, 1993. [Google Scholar]
- Mahfouz, M.; Maruyama, R.; Yokota, T. Inotersen for the Treatment of Hereditary Transthyretin Amyloidosis. Methods Mol. Biol. 2020, 2176, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Iftikhar, M.; Frey, J.; Shohan, M.J.; Malek, S.; Mousa, S.A. Current and emerging therapies for Duchenne muscular dystrophy and spinal muscular atrophy. Pharmacol. Ther. 2021, 220, 107719. [Google Scholar] [CrossRef]
- Brudvig, J.J.; Weimer, J.M. On the cusp of cures: Breakthroughs in Batten disease research. Curr. Opin. Neurobiol. 2021, 72, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Feuerhake, I.; Frentzel-Beyme, R.; Wolff, R. Non-Hodgkin lymphomas and ionizing radiation: Case report and review of the literature. Ann. Hematol. 2022, 101, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, N.C.; Lozier, A.; Flament, C.; Ricciardi-Castagnoli, P.; Bellet, D.; Suter, M.; Perricaudet, M.; Tursz, T.; Maraskovsky, E.; Zitvogel, L. Dendritic cells directly trigger NK cell functions: Cross-talk relevant in innate anti-tumor immune responses in vivo. Nat. Med. 1999, 5, 405–411. [Google Scholar] [CrossRef]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Loriot, Y.; Beraldi, E.; Zhang, F.; Wyatt, A.W.; Al Nakouzi, N.; Mo, F.; Zhou, T.; Kim, Y.; Monia, B.P.; et al. Generation 2.5 antisense oligonucleotides targeting the androgen receptor and its splice variants suppress enzalutamide-resistant prostate cancer cell growth. Clin. Cancer Res. 2015, 21, 1675–1687. [Google Scholar] [CrossRef] [Green Version]
- Fakhr, E.; Zare, F.; Teimoori-Toolabi, L. Precise and efficient siRNA design: A key point in competent gene silencing. Cancer Gene Ther. 2016, 23, 73–82. [Google Scholar] [CrossRef]
- Song, M.S.; Rossi, J.J. Molecular mechanisms of Dicer: Endonuclease and enzymatic activity. Biochem. J. 2017, 474, 1603–1618. [Google Scholar] [CrossRef] [Green Version]
- Iwakawa, H.O.; Tomari, Y. Life of RISC: Formation, action, and degradation of RNA-induced silencing complex. Mol. Cell 2022, 82, 30–43. [Google Scholar] [CrossRef]
- Niaz, S. The AGO proteins: An overview. Biol. Chem. 2018, 399, 525–547. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Patisiran: First Global Approval. Drugs 2018, 78, 1625–1631. [Google Scholar] [CrossRef] [PubMed]
- Kaku, M.; Berk, J.L. Neuropathy Associated with Systemic Amyloidosis. Semin Neurol. 2019, 39, 578–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Paula Brandao, P.R.; Titze-de-Almeida, S.S.; Titze-de-Almeida, R. Leading RNA Interference Therapeutics Part 2: Silencing Delta-Aminolevulinic Acid Synthase 1, with a Focus on Givosiran. Mol. Diagn. Ther. 2020, 24, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Givosiran: A Review in Acute Hepatic Porphyria. Drugs 2021, 81, 841–848. [Google Scholar] [CrossRef]
- Scott, L.J.; Keam, S.J. Lumasiran: First Approval. Drugs 2021, 81, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Andrisani, O.M.; Studach, L.; Merle, P. Gene signatures in hepatocellular carcinoma (HCC). Semin. Cancer Biol. 2011, 21, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef]
- Wolf, M.M.; Kimryn Rathmell, W.; Beckermann, K.E. Modeling clear cell renal cell carcinoma and therapeutic implications. Oncogene 2020, 39, 3413–3426. [Google Scholar] [CrossRef] [Green Version]
- Krieg, M.; Haas, R.; Brauch, H.; Acker, T.; Flamme, I.; Plate, K.H. Up-regulation of hypoxia-inducible factors HIF-1alpha and HIF-2alpha under normoxic conditions in renal carcinoma cells by von Hippel-Lindau tumor suppressor gene loss of function. Oncogene 2000, 19, 5435–5443. [Google Scholar] [CrossRef] [Green Version]
- Bachmaier, K.; Krawczyk, C.; Kozieradzki, I.; Kong, Y.Y.; Sasaki, T.; Oliveira-dos-Santos, A.; Mariathasan, S.; Bouchard, D.; Wakeham, A.; Itie, A.; et al. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature 2000, 403, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Paolino, M.; Choidas, A.; Wallner, S.; Pranjic, B.; Uribesalgo, I.; Loeser, S.; Jamieson, A.M.; Langdon, W.Y.; Ikeda, F.; Fededa, J.P.; et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 2014, 507, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Fabian, M.R.; Sonenberg, N. The mechanics of miRNA-mediated gene silencing: A look under the hood of miRISC. Nat. Struct. Mol. Biol. 2012, 19, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Schirle, N.T.; Sheu-Gruttadauria, J.; MacRae, I.J. Structural basis for microRNA targeting. Science 2014, 346, 608–613. [Google Scholar] [CrossRef]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet 2015, 16, 421–433. [Google Scholar] [CrossRef]
- Panigrahi, M.; Thibault, P.A.; Wilson, J.A. miR-122 affects both the initiation and maintenance of Hepatitis C Virus infections. J. Virol. 2021, 96, JVI-01903. [Google Scholar] [CrossRef]
- Kashtan, C.E. Alport Syndrome: Achieving Early Diagnosis and Treatment. Am. J. Kidney Dis. 2021, 77, 272–279. [Google Scholar] [CrossRef]
- Witten, L.; Slack, F.J. miR-155 as a novel clinical target for hematological malignancies. Carcinogenesis 2020, 41, 2–7. [Google Scholar] [CrossRef]
- Seto, A.G.; Beatty, X.; Lynch, J.M.; Hermreck, M.; Tetzlaff, M.; Duvic, M.; Jackson, A.L. Cobomarsen, an oligonucleotide inhibitor of miR-155, co-ordinately regulates multiple survival pathways to reduce cellular proliferation and survival in cutaneous T-cell lymphoma. Br. J. Haematol. 2018, 183, 428–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastasiadou, E.; Seto, A.G.; Beatty, X.; Hermreck, M.; Gilles, M.E.; Stroopinsky, D.; Pinter-Brown, L.C.; Pestano, L.; Marchese, C.; Avigan, D.; et al. Cobomarsen, an Oligonucleotide Inhibitor of miR-155, Slows DLBCL Tumor Cell Growth In Vitro and In Vivo. Clin. Cancer Res. 2021, 27, 1139–1149. [Google Scholar] [CrossRef] [PubMed]
- Abplanalp, W.T.; Fischer, A.; John, D.; Zeiher, A.M.; Gosgnach, W.; Darville, H.; Montgomery, R.; Pestano, L.; Allee, G.; Paty, I.; et al. Efficiency and Target Derepression of Anti-miR-92a: Results of a First in Human Study. Nucleic Acid Ther. 2020, 30, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Gallant-Behm, C.L.; Piper, J.; Dickinson, B.A.; Dalby, C.M.; Pestano, L.A.; Jackson, A.L. A synthetic microRNA-92a inhibitor (MRG-110) accelerates angiogenesis and wound healing in diabetic and nondiabetic wounds. Wound Repair Regen. 2018, 26, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Rottiers, V.; Naar, A.M. MicroRNAs in metabolism and metabolic disorders. Nat. Rev. Mol. Cell Biol. 2012, 13, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Park, J.H. Genetic Mechanisms of ADPKD. Adv. Exp. Med. Biol. 2016, 933, 13–22. [Google Scholar] [CrossRef]
- Ucar, A.; Gupta, S.K.; Fiedler, J.; Erikci, E.; Kardasinski, M.; Batkai, S.; Dangwal, S.; Kumarswamy, R.; Bang, C.; Holzmann, A.; et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat. Commun. 2012, 3, 1078. [Google Scholar] [CrossRef]
- Foinquinos, A.; Batkai, S.; Genschel, C.; Viereck, J.; Rump, S.; Gyongyosi, M.; Traxler, D.; Riesenhuber, M.; Spannbauer, A.; Lukovic, D.; et al. Preclinical development of a miR-132 inhibitor for heart failure treatment. Nat. Commun. 2020, 11, 633. [Google Scholar] [CrossRef]
- Batkai, S.; Genschel, C.; Viereck, J.; Rump, S.; Bar, C.; Borchert, T.; Traxler, D.; Riesenhuber, M.; Spannbauer, A.; Lukovic, D.; et al. CDR132L improves systolic and diastolic function in a large animal model of chronic heart failure. Eur. Heart J. 2021, 42, 192–201. [Google Scholar] [CrossRef]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [Green Version]
- Bandi, N.; Zbinden, S.; Gugger, M.; Arnold, M.; Kocher, V.; Hasan, L.; Kappeler, A.; Brunner, T.; Vassella, E. miR-15a and miR-16 are implicated in cell cycle regulation in a Rb-dependent manner and are frequently deleted or down-regulated in non-small cell lung cancer. Cancer Res. 2009, 69, 5553–5559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonci, D.; Coppola, V.; Musumeci, M.; Addario, A.; Giuffrida, R.; Memeo, L.; D’Urso, L.; Pagliuca, A.; Biffoni, M.; Labbaye, C.; et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat. Med. 2008, 14, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Reid, G.; Pel, M.E.; Kirschner, M.B.; Cheng, Y.Y.; Mugridge, N.; Weiss, J.; Williams, M.; Wright, C.; Edelman, J.J.; Vallely, M.P.; et al. Restoring expression of miR-16: A novel approach to therapy for malignant pleural mesothelioma. Ann. Oncol. 2013, 24, 3128–3135. [Google Scholar] [CrossRef] [PubMed]
- Chava, S.; Reynolds, C.P.; Pathania, A.S.; Gorantla, S.; Poluektova, L.Y.; Coulter, D.W.; Gupta, S.C.; Pandey, M.K.; Challagundla, K.B. miR-15a-5p, miR-15b-5p, and miR-16-5p inhibit tumor progression by directly targeting MYCN in neuroblastoma. Mol. Oncol. 2020, 14, 180–196. [Google Scholar] [CrossRef]
- Grabowski, G.; Pacana, M.J.; Chen, E. Keloid and Hypertrophic Scar Formation, Prevention, and Management: Standard Review of Abnormal Scarring in Orthopaedic Surgery. J. Am. Acad. Orthop. Surg. 2020, 28, e408–e414. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Wang, Y.; Wang, D.; Wu, Y. Identification of collagen 1 as a post-transcriptional target of miR-29b in skin fibroblasts: Therapeutic implication for scar reduction. Am. J. Med. Sci. 2013, 346, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, Z.; Wang, Y.; Li, L.; Wang, D.; Zhang, W.; Liu, L.; Jiang, H.; Yang, J.; Cheng, J. Overexpression of miR-29b reduces collagen biosynthesis by inhibiting heat shock protein 47 during skin wound healing. Transl. Res. 2016, 178, 38–53.e36. [Google Scholar] [CrossRef]
- Montgomery, R.L.; Yu, G.; Latimer, P.A.; Stack, C.; Robinson, K.; Dalby, C.M.; Kaminski, N.; van Rooij, E. MicroRNA mimicry blocks pulmonary fibrosis. EMBO Mol. Med. 2014, 6, 1347–1356. [Google Scholar] [CrossRef]
- Gallant-Behm, C.L.; Piper, J.; Lynch, J.M.; Seto, A.G.; Hong, S.J.; Mustoe, T.A.; Maari, C.; Pestano, L.A.; Dalby, C.M.; Jackson, A.L.; et al. A MicroRNA-29 Mimic (Remlarsen) Represses Extracellular Matrix Expression and Fibroplasia in the Skin. J. Investig. Dermatol. 2019, 139, 1073–1081. [Google Scholar] [CrossRef]
- Liu, C.; Kelnar, K.; Liu, B.; Chen, X.; Calhoun-Davis, T.; Li, H.; Patrawala, L.; Yan, H.; Jeter, C.; Honorio, S.; et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat. Med. 2011, 17, 211–215. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Hermeking, H. miR-34a and miR-34b/c Suppress Intestinal Tumorigenesis. Cancer Res. 2017, 77, 2746–2758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Q.; Hao, X.; Zhang, M.; Tang, W.; Yang, M.; Li, L.; Xiang, D.; Desano, J.T.; Bommer, G.T.; Fan, D.; et al. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS ONE 2009, 4, e6816. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Wu, Y.; Zhang, S.; Han, Y.; Shen, J.; Zheng, W.; Wei, L.; Liu, Y.; Ren, L.; Gu, Z.; et al. Distinct roles of miR-34 family members on suppression of lung squamous cell carcinoma. Biomed. Pharmacother. 2021, 142, 111967. [Google Scholar] [CrossRef]
- Schmid, G.; Notaro, S.; Reimer, D.; Abdel-Azim, S.; Duggan-Peer, M.; Holly, J.; Fiegl, H.; Rossler, J.; Wiedemair, A.; Concin, N.; et al. Expression and promotor hypermethylation of miR-34a in the various histological subtypes of ovarian cancer. BMC Cancer 2016, 16, 102. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Huang, Y.; Yang, J.; Wu, J.; Luo, C. miR-34a is downregulated in human osteosarcoma stem-like cells and promotes invasion, tumorigenic ability and self-renewal capacity. Mol. Med. Rep. 2017, 15, 1631–1637. [Google Scholar] [CrossRef] [Green Version]
- Raver-Shapira, N.; Marciano, E.; Meiri, E.; Spector, Y.; Rosenfeld, N.; Moskovits, N.; Bentwich, Z.; Oren, M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol. Cell 2007, 26, 731–743. [Google Scholar] [CrossRef]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The biology and role of CD44 in cancer progression: Therapeutic implications. J. Hematol. Oncol. 2018, 11, 64. [Google Scholar] [CrossRef] [Green Version]
- Tarasov, V.; Jung, P.; Verdoodt, B.; Lodygin, D.; Epanchintsev, A.; Menssen, A.; Meister, G.; Hermeking, H. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle 2007, 6, 1586–1593. [Google Scholar] [CrossRef] [Green Version]
- Siu, L.; Brody, J.; Gupta, S.; Marabelle, A.; Jimeno, A.; Munster, P.; Grilley-Olson, J.; Rook, A.H.; Hollebecque, A.; Wong, R.K.S.; et al. Safety and clinical activity of intratumoral MEDI9197 alone and in combination with durvalumab and/or palliative radiation therapy in patients with advanced solid tumors. J. Immunother. Cancer 2020, 8, e001095. [Google Scholar] [CrossRef]
- Tao, L.; Xu, C.; Shen, W.; Tan, J.; Li, L.; Fan, M.; Sun, D.; Lai, Y.; Cheng, H. HIPK3 Inhibition by Exosomal hsa-miR-101-3p Is Related to Metabolic Reprogramming in Colorectal Cancer. Front. Oncol. 2021, 11, 758336. [Google Scholar] [CrossRef]
- Chang, L.; Yin, L.; Zhang, D.; Wang, C.; Li, G.; Tan, C.; Zhang, X.; Su, J. MicroRNA-221 Promotes Tumor Progression by Targeting HHIP in Human Glioblastoma. Transl. Cancer Res. 2021, 10, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yan, P.; Wang, J.; Zhang, Y.; Zhang, M.; Wang, Z.; Fu, Q.; Liang, W. Clinical Significance of Tumor miR-21, miR-221, miR-143, and miR-106a as Biomarkers in Patients with Osteosarcoma. Int. J. Biol. Markers 2019, 2, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Iida, M.; Hazama, S.; Tsunedomi, R.; Tanaka, H.; Takenouchi, H.; Kanekiyo, S.; Tokumitsu, Y.; Tomochika, S.; Tokuhisa, Y.; Sakamoto, K.; et al. Overexpression of miR-221 and miR-222 in the Cancer Stroma Is Associated with Malignant Potential in Colorectal Cancer. Oncol. Rep. 2018, 40, 1621–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef]
- Li-Ping, Z.; Ge, Y.; Xiao-Min, Z.; Feng, Q. Development of Aptamer Screening against Proteins and Its Applications. Chin. J. Anal. Chem. 2020, 48, 560–572. [Google Scholar] [CrossRef]
- Ricci, F.; Bandello, F.; Navarra, P.; Staurenghi, G.; Stumpp, M.; Zarbin, M. Neovascular Age-Related Macular Degeneration: Therapeutic Management and New-Upcoming Approaches. Int. J. Mol. Sci. 2020, 21, 8242. [Google Scholar] [CrossRef]
- Jager, R.D.; Mieler, W.F.; Miller, J.W. Age-related macular degeneration. N. Engl. J. Med. 2008, 358, 2606–2617. [Google Scholar] [CrossRef] [Green Version]
- Kernan, N.A.; Grupp, S.; Smith, A.R.; Arai, S.; Triplett, B.; Antin, J.H.; Lehmann, L.; Shore, T.; Ho, V.T.; Bunin, N.; et al. Final results from a defibrotide treatment-IND study for patients with hepatic veno-occlusive disease/sinusoidal obstruction syndrome. Br. J. Haematol. 2018, 181, 816–827. [Google Scholar] [CrossRef]
- Richardson, P.G.; Riches, M.L.; Kernan, N.A.; Brochstein, J.A.; Mineishi, S.; Termuhlen, A.M.; Arai, S.; Grupp, S.A.; Guinan, E.C.; Martin, P.L.; et al. Phase 3 trial of defibrotide for the treatment of severe veno-occlusive disease and multi-organ failure. Blood 2016, 127, 1656–1665. [Google Scholar] [CrossRef] [Green Version]
- Steurer, M.; Montillo, M.; Scarfo, L.; Mauro, F.R.; Andel, J.; Wildner, S.; Trentin, L.; Janssens, A.; Burgstaller, S.; Fromming, A.; et al. Olaptesed pegol (NOX-A12) with bendamustine and rituximab: A phase IIa study in patients with relapsed/refractory chronic lymphocytic leukemia. Haematologica 2019, 104, 2053–2060. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Carmona, M.; Williams, A.; Schreiber, J.; Hohmann, N.; Pruefer, U.; Krauss, J.; Jager, D.; Fromming, A.; Beyer, D.; Eulberg, D.; et al. Combined inhibition of CXCL12 and PD-1 in MSS colorectal and pancreatic cancer: Modulation of the microenvironment and clinical effects. J. Immunother. Cancer 2021, 9, e002505. [Google Scholar] [CrossRef] [PubMed]
- Zboralski, D.; Hoehlig, K.; Eulberg, D.; Fromming, A.; Vater, A. Increasing Tumor-Infiltrating T Cells through Inhibition of CXCL12 with NOX-A12 Synergizes with PD-1 Blockade. Cancer Immunol. Res. 2017, 5, 950–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberthur, D.; Achenbach, J.; Gabdulkhakov, A.; Buchner, K.; Maasch, C.; Falke, S.; Rehders, D.; Klussmann, S.; Betzel, C. Crystal structure of a mirror-image L-RNA aptamer (Spiegelmer) in complex with the natural L-protein target CCL2. Nat. Commun. 2015, 6, 6923. [Google Scholar] [CrossRef] [Green Version]
- Mongelard, F.; Bouvet, P. AS-1411, a guanosine-rich oligonucleotide aptamer targeting nucleolin for the potential treatment of cancer, including acute myeloid leukemia. Curr. Opin. Mol. Ther. 2010, 12, 107–114. [Google Scholar]

{kind=link}
| Product | Route of Delivery | Target | Mechanism of Action | Disease/Clinical Outcome | Company | Approval Status | References |
|---|---|---|---|---|---|---|---|
| ASO | |||||||
| Fomivirsen | IVT | CMV mRNA | Downregulates IE2 | Cytomegalovirus (CMV) retinitis | Ionis Pharmaceutical, Novartis | FDA (1998) | [14] |
| Mipomersen | SC | apo-B-100 mRNA | Downregulates ApoB | Homozygous familial hypercholesterolemia | Kastle Therapeutics, Ionis Pharmaceuticals, Genzyme | FDA (2013) | [15] |
| Nusinersen | ITH | SMN2 pre-mRNA | Splicing modulation | Spinal muscular atrophy | Ionis Pharmaceuticals, Biogen | FDA (2016) | [16] |
| Eteplirsen | IV | Exon 51 of DMD | Splicing modulation | Duchenne muscular dystrophy | Sarepta Therapeutics | FDA (2016) | [17] |
| Inotersen | SC | TTR mRNA | Downregulates transthyretin mRNA | Familial amyloid polyneuropathy | Ionis Pharmaceuticals | FDA (2018) | [18] |
| Golodirsen | IV | Exon 53 of DMD | Splicing modulation | Duchenne muscular dystrophy | Sarepta Therapeutics | FDA (2019) | [19] |
| Milasen | Intrathecal | CLN7 | Splicing modulation | Mila Makovec’s CLN7 gene associated with Batten disease | Boston Children’s Hospital | FDA (2018) | [20] |
| Casimersen | IV | Exon 45 of DMD | Splicing modulation | Duchenne muscular dystrophy | Sarepta Therapeutics | FDA (2021) | [21,22,23] |
| siRNA | |||||||
| Patisiran | IV | TTR mRNA | Downregulation of transthyretin | Polyneuropathy caused by hATTR amyloidosis | Alnylam | FDA (2018) | [24] |
| Givosiran | SC | ALS1 mRNA | Downregulation of ALAS1 | Acute hepatic porphyria | Alnylam | FDA (2020) | [25] |
| Lumasiran | SC | HAO1 mRNA | Downregulation of glycolate oxidase | Primary hyperoxaluria type 1 | Alnylam | FDA (2020) | [26] |
| Inclisiran | SC | PCSK9 | Downregulation of proprotein convertase subtilsin/kexin type 9 | Atherosclerotic cardiovascular disease | Novartis | FDA (2021) | [27] |
| Aptamer | |||||||
| Pegaptanib | Intravitreal | Heparin-binding domain of VEGF-165 | Blocking VEGF-165 | Neovascular age-related macular degeneration | OSI Pharmaceuticals | FDA (2004) | [28] |
| Defibrotide | IV | Adenosine A1/A2receptor | Activating Adenosine A1/A2 receptor | Veno-occlusive disease in liver | Jazz Pharmaceuticals | FDA (2020) | [29,30] |
| mRNA | |||||||
| BNT162b2 | IM | Immunogenicity and antibody response to SARS-CoV-2 S antigens | SARS-CoV-2 S antigens’ expression | COVID-19 | BioNTech and Pfizer | FDA (2020) | [31] |
| mRNA-1273 | IM | Immunogenicity and antibody response to SARS-CoV-2 S antigens | SARS-CoV-2 S antigens’ expression | COVID-19 | Moderna | FDA (2020) | [32] |
| Oligonucleotide Therapeutics | Route of Delivery | Target | Mechanism of Action | Disease/Clinical Outcome | Company | Clinical Trial Status | References |
|---|---|---|---|---|---|---|---|
| ASO | |||||||
| 1018 ISS | IV | TLR9 | Enhancement of cytotoxic effector mechanisms | Non-Hodgkin’s Lymphoma | Dana-Farber Cancer Institute, Brigham and Women’s Hospital, Massachusetts General Hospital, University of Rochester | NCT00251394 (Phase II) | [33,34] |
| Apatorsen (OGX-427) | IV | HSP27 | Inhibits expression of heat shock protein (Hsp27) | Urologic Cancer, Bladder Cancer, Prostate Cancer, Urothelial Cancer, Non-Small-Cell Lung Cancer | Achieve Life Sciences PRA Health Sciences | NCT00487786, NCT01454089 (Phase I/II) | [35] |
| Cenersen (EL625) | IV | TP53 | Blocks the effects of p53 | Acute Myelogenous Leukemia, Lymphoma | Eleos, Inc. | NCT00074737 (Phase II) | [36] |
| ARRx (AZD5312) | IV | AR | Suppression of human AR expression | Prostate Cancer | AstraZeneca | NCT02144051, (Phase I/II) | [37] |
| Custirsen (OGX-011) | IV | ApoJ | Inhibition of clusterin expression | Prostate Cancer, Breast Cancer, Non-Small-Cell Lung Cancer | NCIC Clinical Trials Group, Achieve Life Sciences | NCT00054106, NCT00138658, (Phase I/II) | [38,39] |
| siRNA | |||||||
| TKM-080301 | Intra-arterial/IV | PLK1 | Inhibition of PLK1 activity | Cancer with hepatic metastases, Hepatocellular Cancer | National Cancer Institute, Arbutus Biopharma Corporation | NCT01437007, NCT02191878, (Phase I/II) | [40,41] |
| Atu027 | IV | PNK3 | Silences expression of PNK3 | Solid Tumors, Pancreatic Cancer | Silence Therapeutics GmbH, Granzer Regulatory Consulting & Services | NCT00938574, NCT01808638 (Phase I/II) | [42,43] |
| siG12D LODER | Locally implanted through EUS biopsy procedure | KRASG12D | Inhibits KRAS expression | Pancreatic Cancer | Silenseed Ltd. | NCT01676259, NCT01188785 (Phase I/II) | [44,45] |
| ARO-HIF2 | IV | HIF2A | Deregulation of HIF2A | Clear Cell Renal Cell Carcinoma | Arrowhead Pharmaceuticals | NCT04169711 (Phase I) | [46] |
| APN401 | IV | CBLB | Inhibition of Cbl-b enhances natural killer cell and T cell mediated antitumor activity | Brain Cancer, Melanoma, Pancreatic Cancer, Renal Cell Cancer | Wake Forest University Health Sciences, National Cancer Institute | NCT03087591, NCT02166255 (Phase I) | [47] |
| Vutrisiran | SQ | TTR | Reduces TTR protein expression | Transthyretin mediated amyloidosis with or without cardiomyopathy | Alnylam Pharmaceuticals | NCT03759379 NCT04153149 (Phase 3) | [48,49] |
| Aptamer | |||||||
| NOX-A12 | IV | CXCL12 | Disrupts CXCR4-CXCL12 interactions | Pancreatic Cancer, Colorectal Cancer, Multiple myeloma | NOXXON Pharma AG, Merck Sharp & Dohme Corp. | NCT01521533, NCT01521533, NCT03168139 (Phase I/II) | [50] |
| NOX-E36 | IV/SQ | CCL2 | Specifically binds and inhibits the pro-inflammatory chemokine CCL2 | Diabetic nephropathy | NOXXON Pharma AG | Phase I | [51] |
| mRNA | |||||||
| CVnCoV | IM | Immunogenicity and antibody response to SARS-CoV-2 S antigens | SARS-CoV-2 S antigens’ expression | COVID-19 | CureVac AG | NCT04652102 (Phase III) | [52] |
| AZD8601 | Epicardial | VEGF-A | Restores VEGF-A expression | Ischemic heart disease | AstraZeneca | NCT03370887 (Phase II) | [53] |
| MRT5005 | Inhalation | CFTR | Restores CFTR expression | Cystic Fibrosis | Translate Bio | NCT03375047 (Phase I/II) | [54] |
| mRNA-3704 | IV | MUT | Restores MUT expression | Methylmalonic aciduria | Moderna | NCT03810690 (Phase I/II) | [55,56] |
| BNT111 | IV | Targets four non-mutated, TAAs (NY-ESO-1, MAGEA3, tyrosinase and TPTE | Induction of immune response against the four selected malignant melanoma-associated antigens (New York-ESO 1 (NY-ESO-1), tyrosinase, Melanoma-associated antigen A3 (MAGE-A3), and Trans-membrane phosphatase with tensin homology (TPTE)) | Advanced Melanoma | BioNTech SE | NCT02410733 (Phase I) | [57] |
| miRNA | |||||||
| Miravirsen | SC | miR-122 | miRNA-inhibitor | HCV | Roche/Santaris | NCT01200420 (Phase II) | [58] |
| RG-012 (lademirsen) | SC | miR-21 | miRNA-inhibitor | Alport Syndrome | Sanofi | NCT03373786 (Phase II) | [59] |
| Cobomarsen | IV/SQ | miR-155 | miRNA-inhibitor | Cutaneous T-Cell Lymphoma/Mycosis Fungoides | miRagen | NCT03713320, NCT02580552 (Phase II) | [60] |
| MRG-110 | Intradermal | miR-92a | miRNA-inhibitor | Wound healing | miRagen | NCT03603431 (Phase I) | [61] |
| AZD4076 | SC | miR-103/107 | miRNA-inhibitor | T2D with NAFLD | AstraZeneca | NCT02826525 (Phase I/IIa) | [62] |
| RGLS4326 | SC | miR-17 | miRNA-inhibitor | Autosomal dominant polycystic kidney disease | Regulus Therapeutics Inc. | NCT04536688 (Phase I) | [63] |
| CDR132L | IV | miR-132 | miRNA-inhibitor | Heart Failure | Cardior Pharmaceuticals GmbH | NCT04045405 (Phase I) | [64] |
| TargomiRs | IV | miR-16 | miRNA-mimic | Malignant Pleural Mesothelioma | EnGeneIC Limited | NCT02369198 (Phase I) | [65] |
| Remlarsen | Intradermal | miR-29 | miRNA-mimic | Keloids, scleroderma | miRagen | NCT03601052 (Phase II) | [66] |
| MRX34 | IV | miR-34a | miRNA-mimic | Melanoma | miRNA Therapeutics, Inc. | NCT01829971 (Phase I) | [67] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zogg, H.; Singh, R.; Ro, S. Current Advances in RNA Therapeutics for Human Diseases. Int. J. Mol. Sci. 2022, 23, 2736. https://doi.org/10.3390/ijms23052736
Zogg H, Singh R, Ro S. Current Advances in RNA Therapeutics for Human Diseases. International Journal of Molecular Sciences. 2022; 23(5):2736. https://doi.org/10.3390/ijms23052736
Chicago/Turabian StyleZogg, Hannah, Rajan Singh, and Seungil Ro. 2022. "Current Advances in RNA Therapeutics for Human Diseases" International Journal of Molecular Sciences 23, no. 5: 2736. https://doi.org/10.3390/ijms23052736
APA StyleZogg, H., Singh, R., & Ro, S. (2022). Current Advances in RNA Therapeutics for Human Diseases. International Journal of Molecular Sciences, 23(5), 2736. https://doi.org/10.3390/ijms23052736