
Impact of Air Pollution in Airway Diseases: Role of the Epithelial Cells (Cell Models and Biomarkers)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Environmental Pollution and Health of Respiratory System
2. Role of Bronchial Epithelial Cells in Respiratory Diseases
3. Effects of Air Contaminants on Epithelial Cells of the Lung
4. Cell Systems to Study the Effects of Environmental Contaminants in Respiratory Diseases
5. Exposome, Omics Technologies, and New Biomarkers (miRNA) in Toxicology
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| FVC | forced volume capacity |
| COPD | chronic obstructive pulmonary disease |
| CO | carbon monoxide |
| NOx | nitrogen oxides |
| SO2 | sulfur dioxide |
| IPA | polycyclic aromatic hydrocarbons |
| PM | particulate matter |
| VOC | volatile organic compounds |
| COHb | carboxyhemoglobin |
| NO2 | nitrogen dioxide |
| B[a]P | benzo-[a]-pyrene |
| IARC | International Agency for Research on Cancer |
| ESCAPE | European study of cohorts for air pollution effects |
| SIN | sites of national interest |
| CISAS | International Center for Advanced Studies in Environment, Ecosystem and Human Health |
| OMS | World Health Organization |
| POP | persistent organic pollutants |
| PCB | polychlorinated biphenyls |
| PBDE | polybrominated diphenyl ethers |
| EFSA | European Food Safety Authority |
| TLR4/NF-kB | Toll-like receptor 4/nuclear factor kappa-light-chain-enhancer of activated B cells |
| UE | European Union |
| AM | alveolar macrophages |
| TNF-α | tumor necrosis factor |
| IL- | interleukin- |
| DAMP | damage-associated molecular pattern |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| MCP-1 | monocyte chemoattractant protein 1 |
| TSLP | timic stromal lymphopoietin |
| O3 | ozone |
| C6H6 | benzene |
| As | arsenic |
| Cd | cadmium |
| Ni | nickel |
| ROS | reactive oxygen species |
| DUOX1 | duox ossidasi 1 |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| Nrf2 | erythroid 2-like 2 |
| ALI | air–liquid interface |
| LOC | lung-on-a-chip |
| IPSC | induced pluripotent stem cells |
| NP | nanoparticles |
| TEER | transepithelial electrical resistance |
| NAC | N-acetyl cysteine |
| Hg | mercury |
| COX-2 | cyclooxygenase-2 |
| RNA | ribonucleic acid |
| mRNA | messenger RNA |
| Zonula Occludens-1 | ZO-1 |
References
- Manisalidis, I.; Stavropoulou, E.; Stavropoulos, A.; Bezirtzoglou, E. Environmental and health im-pacts of air pollution: A review. Front. Public Health. 2020, 20, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Ashfaq, A.; Sharma, P. Environmental effects of air pollution and application of engineered methods to combat the problem. J. Indust. Pollut. Control. 2012, 29, 25–28. [Google Scholar]
- Guan, W.J.; Zheng, X.Y.; Chung, K.F.; Zhong, N.S. Impact of air pollution on the burden of chronic res-piratory diseases in China: Time for urgent action. Lancet 2016, 388, 1939–1951. [Google Scholar] [CrossRef]
- Szyszkowicz, M.; Kousha, T.; Valacchi, G. Ambient air pollution and emergency department visits for skin conditions. Glob. Dermatol. 2016, 3, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Kampa, M.; Castanas, E. Human health effects of air pollution. Environ. Pollut. 2008, 151, 362–367. [Google Scholar] [CrossRef]
- Peters, J.M.; Avol, E.; Gauderman, W.J.; Linn, W.S.; Navidi, W.; London, S.J.; Margolis, H.; Rappaport, E.; Vora, H.; Gong, H., Jr.; et al. A study of twelve Southern California communities with differing levels types of air pollution: II. Effects on pulmonary function. Am. J. Respir. Crit. Care Med. 1999, 159, 768–775. [Google Scholar] [CrossRef]
- Doiron, D.; de Hoogh, K.; Probst-Hensch, N.; Fortier, I.; Cai, Y.; De Matteis, S.; Hansell, A.L. Air pollution, lung function and COPD: Results from the population-based UK Biobank study. Eur. Respir. J. 2019, 54, 1802140. [Google Scholar] [CrossRef]
- Gauderman, W.J.; Avol, E.; Gilliland, F.; Vora, H.; Thomas, D.; Berhane, K.; McConnell, R.; Kuenzli, N.; Lurmann, F.; Rappaport, E.; et al. The effect of air pollution on lung development from 10 to 18 years of age. N. Engl. J. Med. 2004, 351, 1057–1067, Erratum in 2005, 352, 1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inquinamento e Salute. Dal Traffico al Fumo, Dalla Chimica all’attività Lavorativa: Come l’ambiente Influenza il Rischio di Ammalarsi di Tumore. Fondazioneveronesi.it. 2018. Available online: https://www.fondazioneveronesi.it/magazine/tools-della-salute/download/i-manuali/inquinamento-e-salute (accessed on 24 January 2022).
- Proietti, M. Inquinamento e Malattie. Edizioni Minerva Medica 2018. Available online: https://www.minervamedica.it/it/volumi/specialitadiche/igiene/scheda.php?cod=L10091 (accessed on 24 January 2022).
- Berend, N. Contribution of air pollution to COPD and small airway dysfunction. Respirology. 2016, 21, 237–244. [Google Scholar] [CrossRef]
- Holland, W.W.; Reid, D.D. The urban factor in chronic bronchitis. Lancet 1965, 1, 445–448. [Google Scholar] [CrossRef]
- Schwartz, J. Lung function and chronic exposure to air pollution: A cross-sectional analysis of NHANES II. Environ. Res. 1989, 50, 309–321. [Google Scholar] [CrossRef]
- Robertson, D.S. Health effects of increase in concentration of carbon dioxide in the atmosphere. Curr. Sci. 2006, 90, 1607–1609. [Google Scholar]
- Zhao, Y.; Hu, J.; Tan, Z.; Liu, T.; Zeng, W.; Li, X.; Huang, C.; Wang, S.; Huang, Z.; Ma, W. Ambient carbon monoxide and increased risk of daily hospital out-patient visits for respiratory diseases in Dongguan, China. Sci. Total Environ. 2019, 668, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Hamra, G.B.; Laden, F.; Cohen, A.J.; Raaschou-Nielsen, O.; Brauer, M.; Loomis, D. Lung cancer and expo-sure to nitrogen dioxide and traffic: A systematic review and meta-analysis. Environ. Health Perspect. 2015, 123, 1107–1112. [Google Scholar] [CrossRef]
- Faustini, A.; Rapp, R.; Forastiere, F. Nitrogen dioxide and mortality: Review andmeta-analysis of long-termstudies. Eur. Respir. J. 2014, 44, 744–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nriagu, J.O. Air pollution from solid fuels. In Encyclopedia of Environmental Health; Elsevier: Amsterdam, The Netherlands, 2011; pp. 46–52. [Google Scholar]
- Abdel-Shafy, H.I.; Mansour, M.S.M. A review on polycyclic aromatic hydrocarbons: Source, envi-ronmental impact, effect on human health and remediation. Egypt J. Pet. 2016, 25, 107–123. [Google Scholar] [CrossRef] [Green Version]
- Arpalombardia. 2018. Available online: https://www.arpalombardia.it/Pages/Aria/Inquinanti/Metalli.aspx?firstlevel=Inquinanti (accessed on 24 January 2022).
- Brunekreef, B.; Beelen, R.; Hoek, G.; Schouten, L.; Bausch-Goldbohm, S.; Fischer, P.; Armstrong, B.; Hughes, E.; Jerrett, M.; van den Brandt, P. Effects of long-term exposure to traffic-related air pol-lution on respiratory and cardiovascular mortality in the Netherlands: The NLCS-AIR study. Res. Rep. 2009, 139, 5–71. [Google Scholar]
- Colais, P.; Faustini, A.; Stafoggia, M.; Berti, G.; Bisanti, L.; Cadum, E.; Cernigliaro, A.; Mallone, S.; Pacelli, B.; Serinelli, M.; et al. EPIAIR Collaborative Group. Particulate air pollution and hospital admissions for cardiac diseases in potentially sensitive subgroups. Epidemiology 2012, 23, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Beelen, R.; Hoek, G.; van den Brandt, P.A.; Goldbohm, R.A.; Fischer, P.; Schouten, L.J.; Armstrong, B.; Brunekreef, B. Long-term exposure to traffic-related air pollution and lung cancer risk. Epidemiology 2008, 19, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Vineis, P.; Hoek, G.; Krzyzanowski, M.; Vigna-Taglianti, F.; Veglia, F.; Airoldi, L.; Autrup, H.; Dunning, A.; Garte, S.; Hainaut, P.; et al. Air pollution and risk of lung cancer in a prospective study in Europe. Int. J. Cancer 2006, 119, 169–174. [Google Scholar] [CrossRef]
- Palli, D.; Saieva, C.; Munnia, A.; Peluso, M.; Grechi, D.; Zanna, I.; Caini, S.; Decarli, A.; Sera, F.; Masala, G. DNA adducts and PM10 exposure in traffic-exposed workers and urban residents from the EPIC-Florence city study. Sci. Total Environ. 2008, 403, 105–112. [Google Scholar] [CrossRef]
- Heinrich, J.; Thiering, E.; Rzehak, P.; Krämer, U.; Hochadel, M.; Rauchfuss, K.M.; Gehring, U.; Wichmann, H.E. Long-term exposure to NO2 and PM10 and all-cause and cause-specific mortality in a prospective cohort of women. Occup. Environ. Med. 2013, 70, 179–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raaschou-Nielsen, O.; Andersen, Z.J.; Beelen, R.; Samoli, E.; Stafoggia, M.; Weinmayr, G.; Hoffmann, B.; Fischer, P.; Nieuwenhuijsen, M.J.; Brunekreef, B.; et al. Air pollution and lung cancer incidence in 17 european cohorts: Prospective analyses from the European Study of Cohorts for Air Pollution Effects (ESCAPE). Lancet Oncol. 2013, 14, 813–822. [Google Scholar] [CrossRef]
- Schmid, O.; Stoeger, T. Surface area is the biologically most effective dose metric for acute nanoparticle toxicity in the lung. J. Aerosol. Sci. 2016, 99, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Teeguarden, J.G.; Hinderliter, P.M.; Orr, G.; Thrall, B.D.; Pounds, J.G. Particokinetics in vitro: Dosimetry considerations for in vitro nanoparticle toxicity assessments. Toxicol. Sci. 2007, 95, 300–312, Erratum in Toxicol. Sci. 2007, 97, 614. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, K.E.; Palmberg, L.; Witasp, E.; Kupczyk, M.; Feliu, N.; Gerde, P.; Seisenbaeva, G.A.; Fadeel, B.; Dahlen, S.E.; Kessler, V.G. Solution engineered palladium nanoparticles: Model for health effect studies of automotive particulate pollution. ACS Nano 2011, 5, 5312–5324. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer (IARC). Air Pollution and Cancer; Straif, K., Cohen, A., Samet, J., Eds.; IARC Scientific Publication: Lyon, France, 2013; ISBN1 13978-92-832-2166-1. ISBN2 13978-92-832-2161-6. [Google Scholar]
- Piscitelli, P.; Valenzano, B.; Rizzo, E.; Maggiotto, G.; Rivezzi, M.; Esposito Corcione, F.; Miani, A. Air pollution and estimated health costs related to road transportations of goods in Italy: A first healthcare burden assessment. Int. J. Environ. Res. Public Health 2019, 16, 2876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Air Pollution—European Environment Agency. 2021. Available online: https://www.eea.europa.eu/themes/air/intro (accessed on 24 January 2022).
- GBD 2015 Chronic Respiratory Disease Collaborators. Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Respir. Med. 2017, 5, 691–706, Erratum in Lancet Respir. Med. 2017, 5, e30. [Google Scholar] [CrossRef] [Green Version]
- International Agency for Research on Cancer (IARC). Outdoor Air Pollution 2016—Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer (IARC): Lyon, France, 2016; Volume 109, ISBN1 13-978-92-832-0147-2. ISBN2 13-978-92-832-0175-5. [Google Scholar]
- Martin, P.J.; Héliot, A.; Tremolet, G.; Landkocz, Y.; Dewaele, D.; Cazier, F.; Ledoux, F.; Courcot, D. Cellular response and extracellular vesicles characteri-zation of human macrophages exposed to fine atmospheric particulate matter. Environ. Pollut. 2019, 254 Pt A, 112933. [Google Scholar] [CrossRef]
- Guarnieri, M.; Balmes, J.R. Outdoor air pollution and asthma. Lancet 2014, 383, 1581–1592. [Google Scholar] [CrossRef] [Green Version]
- Galasso, R.; Gruppo di lavoro Sentieri. SENTIERI/Quinto Rapporto—Studio Epidemiologico Na-zionale dei Territori e degli Insediamenti Esposti a Rischio da Inquinamento. Valutazione della evidenza epidemiologica. Epidemiol. Prev. 2019, 43 (Suppl. 2–3), 1–208. [Google Scholar] [CrossRef]
- Ko, F.W.S.; Hui, D.S.C. Effects of air pollution on lung health. Clin. Pulm. Med. 2010, 17, 300–304. [Google Scholar] [CrossRef]
- Pirastu, R.; Ancona, A.; Iavarone, I.; Mitis, F.; Zona, A.; Comba, P. SENTIERI/Quinto Rapporto—Studio Epidemiologico Nazionale dei Territori e degli Insediamenti Esposti a Rischio da Inqui-namento. Valutazione della evidenza epidemiologica. Epidemiol. Prev. 2010, 34 (Suppl. 3), 1–96. [Google Scholar] [PubMed]
- Degrendele, C.; Wilson, J.; Kukučka, P.; Klánová, J.; Lemmel, G. Are atmospheric PBDE levels declin-ing in central Europe? Examination of the seasonal and semi-long-term variations, gas—particle partitioning and implications for long-range atmospheric transport. Atmos. Chem. Phys. 2018, 18, 12877–12890. [Google Scholar] [CrossRef] [Green Version]
- Besis, A.; Lammel, G.; Kukučka, P.; Samara, C.; Sofuoglu, A.; Dumanoglu, Y.; Eleftheriadis, K.; Kouvarakis, G.; Sofuoglu, S.C.; Vassilatou, V.; et al. Polybrominated diphenyl ethers (PBDEs) in background air around the Aegean: Implications for phase partitioning and size distribution. Environ. Sci. Pollut. Res. Int. 2017, 24, 28102–28120. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Meeker, J.D.; Ferguson, K.K. Serum polybrominated diphenyl ether (PBDE) concentrations in relation to biomarkers of oxidative stress and inflammation: The National Health and Nutrition Examination Survey 2003–2004. Sci. Total Environ. 2017, 575, 400–405. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Klösener, J.; Flor, S.; Peters, T.M.; Ludewig, G.; Thorne, P.S.; Robertson, L.W.; Luthe, G. Toxicity assessment of air-delivered particle-bound polybro-minated diphenyl ethers. Toxicology 2014, 317, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Montalbano, A.M.; Albano, G.D.; Anzalone, G.; Moscato, M.; Gagliardo, R.; Di Sano, C.; Bonanno, A.; Ruggieri, S.; Cibella, F.; Profita, M. Cytotoxic and genotoxic effects of the flame retardants (PBDE-47, PBDE-99 and PBDE-209) in human bronchial epithelial cells. Chemosphere 2020, 245, 125600. [Google Scholar] [CrossRef]
- Pozo, K.; Palmeri, M.; Palmeri, V.; Estellano, V.H.; Mulder, M.D.; Efstathiou, C.I.; Sará, G.L.; Romeo, T.; Lammel, G.; Focardi, S. Assessing persistent organic pollutants (POPs) in the Sicily island atmosphere, mediterranean, using PUF disk passive air samplers. Environ. Sci. Pollut. Res. Int. 2016, 23, 20796–20804. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Ferrante, M.C.; Di Guida, F.; Pirozzi, C.; Lama, A.; Simeoli, R.; Clausi, M.T.; Monnolo, A.; Mollica, M.P.; Mattace Raso, G.; et al. Polychlorinated Biphenyls (PCB 101, 153, and 180) Impair Murine Macrophage Responsiveness to Lipopolysaccharide: Involvement of NF-κB Pathway. Toxicol Sci. 2015, 147, 255–269. [Google Scholar] [CrossRef]
- Frederiksen, M.; Vorkamp, K.; Mathiesen, L.; Mose, T.; Knudsen, L.E. Placental transfer of the polybrominated di-phenyl ethers BDE-47, BDE-99 and BDE-209 in a human placenta perfusion system: An exper-imental study. Environ. Health 2010, 9, 32. [Google Scholar] [CrossRef] [Green Version]
- La Guardia, M.J.; Hale, R.C.; Harvey, E. Detailed polybrominated diphenyl ether (PBDE) congener composition of the widely used penta-, octa-, and deca-PBDE technical flame-retardant mix-tures. Environ. Sci. Technol. 2006, 40, 6247–6254. [Google Scholar] [CrossRef]
- ECD. Official Journal of the European Union Commission Decision 2005/717/EC—Exemption of DecaBDE from the Prohibition on Use, C 116 (9 May 2008). 2009. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=OJ:C:2008:116:FULL&from=PL (accessed on 24 January 2022).
- Crystal, R.G.; Randell, S.H.; Engelhardt, J.F.; Voynow, J.; Sunday, M.E. Airway epithelial cells: Current concepts and challenges. Proc. Am Thorac. Soc. 2008, 5, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Shih, L.; Wu, R. Pulmonary epithelium: Cell types and functions. In The Pulmonary Epithelium in Health and Disease, 3rd ed.; Proud, D., Ed.; John Wiley and Sons Ltd.: Hoboken, NJ, USA, 2008. [Google Scholar]
- Tam, A.; Wadsworth, S.; Dorscheid, D.; Man, S.F.; Sin, D.D. The airway epithelium: More than just a structural barrier. Ther. Adv. Respir. Dis. 2011, 5, 255–273. [Google Scholar] [CrossRef] [PubMed]
- Schleimer, R.P.; Kato, A.; Kern, R.; Kuperman, D.; Avila, P.C. Epithelium: At the interface of innate and adaptive immune responses. J. Allergy Clin. Immunol. 2007, 120, 1279–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, D.A.; Holgate, S.T. The airway epithelium: Structural and functional properties in health and disease. Respirology 2003, 8, 432–446. [Google Scholar] [CrossRef] [PubMed]
- Strengert, M.; Knaus, U.G. Analysis of epithelial barrier integrity in polarized lung epithelial cells. Methods Mol. Biol. 2011, 763, 195–206. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Pollard, J.W. Trophic macrophages in development and disease. Nat. Rev. Immunol. 2009, 9, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Miyata, R.; Van Eeden, S.F. The innate and adaptive immune response induced by alveolar macrophages exposed to ambient particulate matter. Toxicol. Appl. Pharmacol. 2011, 257, 209–226. [Google Scholar] [CrossRef]
- Lanyu, Z.; Feilong, H. Emerging role of extracellular vesicles in lung injury and inflammation. Biomed. Pharmacother. 2019, 113, 108748. [Google Scholar] [CrossRef]
- Luyts, K.; Napierska, D.; Dinsdale, D.; Klein, S.G.; Serchi, T.; Hoet, P.H. A coculture model of the lung–blood barrier: The role of activated phagocytic cells. Toxicol. In Vitro 2015, 29, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Hiemstra, P.S.; McCray, P.B., Jr.; Bals, R. The innate immune function of airway epithelial cells in inflammatory lung disease. Eur. Respir. J. 2015, 45, 1150–1162. [Google Scholar] [CrossRef] [Green Version]
- Cromwell, O.; Hamid, Q.; Corrigan, C.J.; Barkans, J.; Meng, Q.; Collins, P.D.; Kay, A.B. Expression and generation of interleukin-8, IL-6 and granulocyte-macrophage colony-stimulating factor by bronchial epithelial cells and enhance-ment by IL-1 beta and tumour necrosis factor-alpha. Immunology 1992, 77, 330–337. [Google Scholar] [PubMed]
- Chow, A.W.; Liang, J.F.; Wong, J.S.; Fu, Y.; Tang, N.L.; Ko, W.H. Polarized secretion of interleukin (IL)-6 and IL-8 by human airway epithelia 16HBE14o- cells in response to cationic polypeptide chal-lenge. PLoS ONE 2010, 5, e12091. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dong, C.; Wang, G.; Zheng, H.; Wang, X.; Bai, C. Pulmonary epithelial CCR3 promotes LPS-induced lung inflammation by mediating release of IL-8. J. Cell Physiol. 2011, 226, 2398–2405. [Google Scholar] [CrossRef]
- Piper, S.C.; Ferguson, J.; Kay, L.; Parker, L.C.; Sabroe, I.; Sleeman, M.A.; Briend, E.; Finch, D.K. The role of Interleukin-1 and Interleukin-18 in pro-inflammatory and anti-viral responses to rhinovirus in primary bronchial epithelial cells. PLoS ONE 2013, 8, e63365. [Google Scholar] [CrossRef] [Green Version]
- Standiford, T.J.; Kunkel, S.L.; Basha, M.A.; Chensue, S.W.; Lynch, J.P., 3rd; Toews, G.B.; Westwick, J.; Strieter, R.M. Interleukin-8 gene expression by a pulmonary epithelial cell line. A model for cytokine networks in the lung. J. Clin. Investig. 1990, 86, 1945–1953. [Google Scholar] [CrossRef] [Green Version]
- Wissel, H.; Schulz, C.; Koehne, P.; Richter, E.; Maass, M.; Rüdiger, M. Chlamydophila pneumoniae in-duces expression of toll-like receptor 4 and release of TNF-alpha and MIP-2 via an NF-kappaB pathway in rat type II pneumocytes. Respir. Res. 2005, 6, 51. [Google Scholar] [CrossRef] [Green Version]
- Asokananthan, N.; Graham, P.T.; Fink, J.; Knight, D.A.; Bakker, A.J.; McWilliam, A.S.; Thompson, P.J.; Stewart, G.A. Activation of protease-activated receptor (PAR)-1, PAR-2, and PAR-4 stimulates IL-6, IL-8, and prostaglandin E2 release from human respiratory epithelial cells. J. Immunol. 2002, 168, 3577–3585. [Google Scholar] [CrossRef] [Green Version]
- Day, S.B.; Ledford, J.R.; Zhou, P.; Lewkowich, I.P.; Pagina, K. German cockroach proteases and prote-ase-activated receptor-2 regulate chemokine production and dendritic cell recruitment. J. Innate Immun. 2012, 4, 100–110. [Google Scholar] [CrossRef] [Green Version]
- Jairaman, A.; Maguire, C.H.; Schleimer, R.P.; Prakriya, M. Allergens stimulate store-operated calci-um entry and cytokine production in airway epithelial cells. Sci. Rep. 2016, 6, 32311. [Google Scholar] [CrossRef] [Green Version]
- Jairaman, A.; Yamashita, M.; Schleimer, R.P.; Prakriya, M. Store-operated Ca2+ release-activated Ca2+ channels regulate PAR2-activated Ca2+ signaling and cytokine production in airway epi-thelial cells. J. Immunol. 2015, 195, 2122–2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, C.; Brennan, S.; Thompson, P.J.; Stewart, G.A. Dust Mite Proteolytic Allergens Induce Cytokine Re-lease from Cultured Airway Epithelium. J. Immunol. 1998, 161, 3645–3651. [Google Scholar] [PubMed]
- Sun, Y.; Wu, F.; Sun, F.; Huang, P. Adenosine promotes IL-6 release in airway epithelia. J. Immunol. 2008, 180, 4173–4181. [Google Scholar] [CrossRef]
- Osterlund, C.; Grönlund, H.; Polovic, N.; Sundström, S.; Gafvelin, G.; Bucht, A. The Non-Proteolytic House Dust Mite Allergen Der p 2 Induce NF-kappaB and MAPK Dependent Activation of Bronchial Epithelial Cells. Clin. Exp. Allergy 2009, 39, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Usmani, S.M.; von Einem, J.; Frick, M.; Miklavc, P.; Mayenburg, M.; Husmann, M.; Dietl, P.; Wittekindt, O.H. Molecular basis of early epithelial response to strep-tococcal exotoxin: Role of STIM1 and Orai1 proteins. Cell Microbiol. 2012, 14, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Kanda, A.; Ueki, S.; Ito, W.; Kamada, Y.; Oyamada, H.; Saito, N.; Kayaba, H.; Chihara, J. Prostaglandin D2 induces IL-8 and GM-CSF by bronchial epithe-lial cells in a CRTH2-independent pathway. Int. Arch Allergy Immunol. 2006, 141, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, H.; Ohtoshi, T.; Kikutani, T.; Okazaki, H.; Akiyama, N.; Sato, M.; Shoji, S.; Ito, K. Histamine activates bronchial epithelial cells to re-lease inflammatory cytokines in vitro. Int Arch. Allergy Immunol. 1995, 108, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Li, L.; Wang, Y.; Zhang, S.; Adcock, I.M.; Barnes, P.J.; Huang, M.; Yao, X. Bronchial epithelial cells: The key effector cells in the pathogenesis of chronic obstructive pulmonary disease? Respirology 2015, 20, 722–729. [Google Scholar] [CrossRef]
- Farias, R.; Rousseau, S. The TAK1 → IKKβ → TPL2 → MKK1/MKK2 signaling cascade regulates IL-33 expression in cystic fibrosis airway epithelial cells following infection by Pseudomonas Aeruginosa. Front. Cell Dev. Biol. 2016, 3, 87. [Google Scholar] [CrossRef] [Green Version]
- Heyen, L.; Müller, U.; Siegemund, S.; Schulze, B.; Protschka, M.; Alber, G.; Piehler, D. Lung epithelium is the major source of IL-33 and is regulated by IL-33-dependent and IL-33-independent mechanisms in pulmonary cryptococ-cosis. Pathog. Dis. 2016, 74, ftw086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, S.; Wang, W.; Meng, Q.; Fang, C.; Lv, Z.; An, Y.Q.; Wang, Y.H.; Liu, Y.J.; Caballero, R.; Lee, T.H.; et al. Allergen-induced expression of IL-25 and IL-25 receptor in atopic asthmatic airways and late-phase cutaneous responses. J. Allergy Clin. Immunol. 2011, 128, 116–124. [Google Scholar] [CrossRef]
- Hristova, M.; Habibovic, A.; Veith, C.; Janssen-Heininger, Y.M.; Dixon, A.E.; Geiszt, M.; van der Vliet, A. Airway epithelial dual oxidase 1 mediates allergen-induced IL-33 secretion and activation of type 2 immune responses. J. Allergy Clin. Immunol. 2016, 137, 1545–1556.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khosravi, A.R.; Erle, D.J. Chitin-induced airway epithelial cell innate immune responses are inhib-ited by Carvacrol/thymol. PLoS ONE 2016, 11, e0159459. [Google Scholar] [CrossRef] [PubMed]
- Nabe, T.; Wakamori, H.; Yano, C.; Nishiguchi, A.; Yuasa, R.; Kido, H.; Tomiyama, Y.; Tomoda, A.; Kida, H.; Takiguchi, A.; et al. Production of interleukin (IL)-33 in the lungs during multi-ple antigen challenge-induced airway inflammation in mice, and its modulation by a glucocor-ticoid. Eur. J. Pharmacol. 2015, 757, 34–41. [Google Scholar] [CrossRef]
- Oh, K.; Seo, M.W.; Lee, G.Y.; Byoun, O.J.; Kang, H.R.; Cho, S.H.; Lee, D.S. Airway epithelial cells initiate the allergen response through transglutaminase 2 by inducing IL-33 expression and a subsequent Th2 response. Respir. Res. 2013, 14, 35. [Google Scholar] [CrossRef] [Green Version]
- Préfontaine, D.; Nadigel, J.; Chouiali, F.; Audusseau, S.; Semlali, A.; Chakir, J.; Martin, J.G.; Hamid, Q. Increased IL-33 expression by epithelial cells in bronchial asthma. J. Allergy Clin. Immunol. 2010, 125, 752–754. [Google Scholar] [CrossRef]
- Hellermann, G.R.; Nagy, S.B.; Kong, X.; Lockey, R.F.; Mohapatra, S.S. Mechanism of cigarette smoke condensate-induced acute inflammatory response in human bronchial epithelial cells. Respir. Res. 2002, 3, 22. [Google Scholar] [CrossRef] [Green Version]
- Beale, J.; Jayaraman, A.; Jackson, D.J.; Macintyre, J.D.; Edwards, M.R.; Walton, R.P.; Zhu, J.; Ching, Y.M.; Shamji, B.; Edwards, M.; et al. Rhinovirus-induced IL-25 in asthma exacerbation drives type 2 immunity and allergic pulmonary inflammation. Sci. Transl. Med. 2014, 6, 256ra134. [Google Scholar] [CrossRef] [Green Version]
- Takizawa, H.; Ohtoshi, T.; Kawasaki, S.; Abe, S.; Sugawara, I.; Nakahara, K.; Matsushima, K.; Kudoh, S. Diesel exhaust particles activate human bronchial epithelial cells to express inflammatory mediators in the airways: A review. Respirology 2000, 5, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Puchelle, E.; Zahm, J.M.; Tournier, J.M.; Coraux, C. Airway epithelial repair, regeneration, and re-modeling after injury in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2006, 3, 726–733. [Google Scholar] [CrossRef] [Green Version]
- Aghasafari, P.; George, U.; Pidaparti, R. A review of inflammatory mechanism in airway diseases. Inflamm. Res. 2019, 68, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Frank, J.K.; Julia, C.F. Air pollution and public health: Emerging hazards and im-proved understanding of risk. Environ. Geochem. Health 2015, 37, 631–649. [Google Scholar] [CrossRef] [Green Version]
- Murrison, L.B.; Brandt, E.B.; Myers, J.B.; Hershey, G.K.K. Environmental exposures and mechanisms in allergy and asthma development. J. Clin. Investig. 2019, 129, 1504–1515. [Google Scholar] [CrossRef] [Green Version]
- Naclerio, R.; Ansotegui, I.J.; Bousquet, J.; Canonica, G.W.; d’Amato, G.; Rosario, N.; Pawankar, R.; Peden, D.; Bergmann, K.C.; Bielory, L.; et al. International expert consensus on the management of allergic rhinitis (AR) aggravated by air pollutants: Impact of air pollution on patients with AR: Current knowledge and future strategies. World Allergy Organ. J. 2020, 13, 100106. [Google Scholar] [CrossRef]
- Andreau, K.; Leroux, M.; Bouharrur, A. Health and cellular impacts of air pollutants: From cyto-protection to cytotoxicity. Biochem. Res. Int. 2012, 2012, 493894. [Google Scholar] [CrossRef] [Green Version]
- Cooper, D.M.; Loxham, M. Particulate matter and the airway epithelium: The special case of the underground? Eur. Respir. Rev. 2019, 28, 190066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Zhou, Y.; Liu, S.; Chen, X.; Zou, W.; Zhao, D.; Li, X.; Pu, J.; Huang, L.; Chen, J.; et al. Association between exposure to ambient particulate matter and chronic obstructive pulmonary disease: Results from a cross-sectional study in China. Thorax 2017, 72, 788–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winterbottom, C.J.; Shah, R.J.; Patterson, K.C.; Kreider, M.E.; Panettieri, R.A., Jr.; Rivera-Lebron, B.; Miller, W.T.; Litzky, L.A.; Penning, T.M.; Heinlen, K.; et al. Exposure to ambient particulate matter is asso-ciated with accelerated functional decline in idiopathic pulmonary fibrosis. Chest 2018, 153, 1221–1228. [Google Scholar] [CrossRef]
- Hamra, G.B.; Guha, N.; Cohen, A.; Laden, F.; Raaschou-Nielsen, O.; Samet, J.M.; Vineis, P.; Forastiere, F.; Saldiva, P.; Yorifuji, T.; et al. Outdoor particulate matter exposure and lung cancer: A systematic review and meta-analysis. Environ. Health Perspect. 2014, 122, 906–991. [Google Scholar] [CrossRef] [Green Version]
- Kelly, F.J. Oxidative stress: Its role in air pollution and adverse health effects. Occup. Environ. Med. 2003, 60, 612–616. [Google Scholar] [CrossRef] [Green Version]
- Øvrevik, J.; Refsnes, M.; Låg, M.; Holme, J.A.; Schwarze, P.E. Activation of proinflammatory responses in cells of the airway mucosa by particulate matter: Oxidant- and non-oxidant-mediated triggering mechanisms. Biomolecules 2015, 5, 1399–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Klaveren, R.J.; Nemery, B. Role of reactive oxygen species in occupational and environmen-tal obstructive pulmonary diseases. Curr. Opin. Pulm. Med. 1999, 5, 118–123. [Google Scholar] [CrossRef]
- Kelly, F.J.; Fussell, J.C. Size, source and chemical composition as determinants of toxicity attribut-able to ambient particulate matter. Atmos. Environ. 2012, 60, 504–526. [Google Scholar] [CrossRef]
- Wong, L.N.; Aung, H.H.; Lamé, M.W.; Wegesser, T.C.; Wilson, D.W. Fine particulate matter from urban ambient and wildfire sources from California’s San Joaquin Valley initiate differential inflammatory, oxidative stress, and xenobiotic responses in human bronchial epithelial cells. Toxicol. In Vitro 2011, 25, 1895–1905. [Google Scholar] [CrossRef]
- Becher, R.; Bucht, A.; Øvrevik, J.; Hongslo, J.K.; Dahlman, H.J.; Samuelsen, J.T.; Schwarze, P.E. Involvement of NADPH oxidase and iNOS in rodent pulmo-nary cytokine responses to urban air and mineral particles. Inhal. Toxicol. 2007, 19, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Øvrevik, J.; Refsnes, M.; Totlandsdal, A.I.; Holme, J.A.; Schwarze, P.E.; Låg, M. TACE/TGF-α/EGFR regulates CXCL8 in bronchial epithelial cells exposed to particulate matter components. Eur. Respir. J. 2011, 38, 1189–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leclercq, B.; Kluza, J.; Antherieu, S.; Sotty, J.; Alleman, L.Y.; Perdrix, E.; Loyens, A.; Coddeville, P.; Guidice, J.M.; Marchetti, P.; et al. Air pollution-derived PM2.5 impairs mitochondrial func-tion in healthy and chronic obstructive pulmonary diseased human bronchial epithelial cells. Environ. Pollut. 2018, 243, 1434–1449. [Google Scholar] [CrossRef]
- Lavrich, K.S.; Corteselli, E.M.; Wages, P.A.; Bromberg, P.A.; Simmons, S.O.; Gibbs-Flournoy, E.A.; Samet, J.M. Investigating mitochondrial dysfunction in human lung cells exposed to redox-active PM components. Toxicol. Appl. Pharmacol. 2018, 342, 99–107. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, M.; Li, Z.; Yue, J.; Xu, M.; Zhang, Y.; Yung, K.K.; Li, R. Fine particulate matter induces mitochondrial dysfunction and oxidative stress in human SH-SY5Y cells. Chemosphere 2019, 218, 577–588. [Google Scholar] [CrossRef]
- Wang, J.; Huang, J.; Wang, L.; Chen, C.; Yang, D.; Jin, M.; Bai, C.; Song, Y. Urban particulate matter triggers lung inflammation via the ROS-MAPK-NF-κB signaling pathway. J. Thorac. Dis. 2017, 9, 4398–4412. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Li, D.; Li, X.; Ma, L.; Bai, X.; Wen, Z.; Zhang, X.; Chen, D.; Peng, L. Exposure to PM2.5 induces aberrant activation of NF-κB in human air-way epithelial cells by downregulating miR-331 expression. Environ. Toxicol. Pharmacol. 2017, 50, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Berman, R.; Downey, G.P.; Dakhama, A.; Day, B.J.; Chu, H.W. Afghanistan particulate matter enhances pro-inflammatory responses in IL-13-exposed human airway epithelium via TLR2 signaling. Toxicol. Sci. 2018, 166, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, H.L.; Holgersson, A.; Möller, L. Mechanisms related to the genotoxicity of particles in the subway and from other sources. Chem. Res. Toxicol. 2008, 21, 726–731. [Google Scholar] [CrossRef]
- Karlsson, H.L.; Ljungman, A.G.; Lindbom, J.; Möller, L. Comparison of genotoxic and inflammatory ef-fects of particles generated by wood combustion, a road simulator and collected from street and subway. Toxicol. Lett. 2006, 165, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Salvi, S.; Blomberg, A.; Rudell, B.; Kelly, F.; Sandstrom, T.; Holgate, S.T.; Frew, A. Acute inflammatory responses in the airways and periph-eral blood after short-term exposure to diesel exhaust in healthy human volunteers. Am. J. Respir. Crit. Care Med. 1999, 159, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef]
- Lippai, M.; Szatmári, Z. Autophagy-from molecular mechanisms to clinical relevance. Cell Biol. Toxicol. 2017, 33, 145–168. [Google Scholar] [CrossRef]
- Bai, R.; Guan, L.; Zhang, W.; Xu, J.; Rui, W.; Zhang, F.; Ding, W. Comparative study of the effects of PM1-induced oxidative stress on autophagy and surfactant protein B and C expressions in lung alveolar type II epithe-lial MLE-12 cells. Biochim. Biophys. Acta 2016, 1860, 2782–2792. [Google Scholar] [CrossRef]
- Long, F.; Jiang, H.; Yi, H.; Su, L.; Sun, J. Particulate matter 2.5 induced bronchial epithelial cell injury via activation of 5′-adenosine monophosphate-activated protein kinase-mediated au-tophagy. J. Cell Biochem. 2019, 120, 3294–3305. [Google Scholar] [CrossRef] [PubMed]
- Frias, D.P.; Gomes, R.L.; Yoshizaki, K.; Carvalho-Oliveira, R.; Matsuda, M.; Junqueira, M.D.; Teodoro, W.R.; Vasconcellos, P.D.; Pereira, D.C.; Conceição, P.R.; et al. Nrf2 positively regulates autophagy antioxidant re-sponse in human bronchial epithelial cells exposed to diesel exhaust particles. Sci. Rep. 2020, 10, 3704. [Google Scholar] [CrossRef] [Green Version]
- Grilli, A.; Bengalli, R.; Longhin, E.; Capasso, L.; Proverbio, M.C.; Forcato, M.; Bicciato, S.; Gualtieri, M.; Battaglia, C.; Camatini, M. Transcriptional profiling of human bronchial epithelial cell BEAS-2B exposed to diesel and biomass ultrafine particles. BMC Genomics 2018, 19, 302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, D.N.; Panda, P.K.; Naik, P.P.; Mukhopadhyay, S.; Sinha, N.; Bhutia, S.K. Phytotherapeutic approach: A new hope for polycyclic aromatic hydrocarbons induced cellular disorders, autophagic and apoptotic cell death. Toxicol. Mech. Methods 2017, 27, 1–17. [Google Scholar] [CrossRef] [PubMed]
- IARC Working Group. ARC Working Group. A review of human carcinogens: Arsenic, metals, fibres, and dusts. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 2012; pp. 121–141. Available online: https://www.ncbi.nlm.nih.gov/books/NBK304375/ (accessed on 24 January 2022).
- Jing, Y.; Liu, L.Z.; Jiang, Y.; Zhu, Y.; Guo, N.L.; Barnett, J.; Rojanasakul, Y.; Agani, F.; Jiang, B.H. Cadmium increases HIF-1 and VEGF expression through ROS, ERK, and AKT signaling pathways and induces malignant transformation of human bronchial epithelial cells. Toxicol. Sci. 2012, 125, 10–19. [Google Scholar] [CrossRef]
- Son, Y.O.; Pratheeshkumar, P.; Roy, R.V.; Hitron, J.A.; Wang, L.; Zhang, Z.; Shi, X. Nrf2/p62 signaling in apoptosis resistance and its role in cadmium-induced carcinogenesis. J. Biol. Chem. 2014, 289, 28660–28675. [Google Scholar] [CrossRef] [Green Version]
- Cartularo, L.; Kluz, T.; Cohen, L.; Shen, S.S.; Costa, M. Molecular mechanisms of malignant transfor-mation by low dose cadmium in normal human bronchial epithelial cells. PLoS ONE 2016, 11, e0155002. [Google Scholar] [CrossRef] [Green Version]
- Fujiki, K.; Inamura, H.; Miyayama, T.; Matsuoka, M. Involvement of Notch1 signaling in malignant progression of A549 cells subjected to prolonged cadmium exposure. J. Biol. Chem. 2017, 292, 7942–7953. [Google Scholar] [CrossRef] [Green Version]
- Park, H.-R.; Kamau, P.W.; Loch-Caruso, R. Involvement of reactive oxygen species in brominat-ed diphenyl ether-47-induced inflammatory cytokine release from human extravillous tropho-blasts in vitro. Toxicol. Appl. Pharmacol. 2014, 274, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Albano, G.D.; Moscato, M.; Montalbano, A.M.; Anzalone, G.; Gagliardo, R.; Bonanno, A.; Giacomazza, D.; Barone, R.; Drago, G.; Cibella, F.; et al. Can PBDEs affect the pathophysiologic com-plex of epithelium in lung diseases? Chemosphere 2020, 241, 125087. [Google Scholar] [CrossRef]
- Rim, K. In vitro Models for Chemical Toxicity: Review of their applications and prospects. Toxicol. Environ. Health Sci. 2019, 11, 94–103. [Google Scholar] [CrossRef]
- Schechtman, L.M. Implementation of the 3Rs (refinement, reduction, and replacement): Vali-dation and regulatory acceptance considerations for alternative toxicological test methods. ILAR J. 2002, 43, S85–S94. [Google Scholar] [CrossRef]
- Belliveau, M.E. The drive for a safer chemicals policy in the United States. New Solut. 2011, 21, 359–386. [Google Scholar] [CrossRef]
- Impinen, A.; Nygaard, U.C.; Carlsen, K.L.; Mowinckel, P.; Carlsen, K.H.; Haug, L.S.; Granum, B. Prenatal exposure to perfluoralkyl sub-stances (PFASs) associated with respiratory tract infections but not allergy- and asthma-related health outcomes in childhood. Environ. Res. 2018, 160, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Tannenbaum, J.; Bennett, B.T. Russell and Burch’s 3Rs then and now: The need for clarity in definition and purpose. J. Am. Assoc. Lab. Anim Sci. 2015, 54, 120–132. [Google Scholar] [PubMed]
- Eagle, H. Nutrition needs of mammalian cells in tissue culture. Science 1955, 122, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Eagle, H. The specific amino acid requirements of a human carcinoma cell (Stain HeLa) in tis-sue culture. J. Exp. Med. 1955, 102, 3748. [Google Scholar] [CrossRef] [Green Version]
- Ritter, D.; Knebel, J.; Niehof, M.; Loinaz, I.; Marradi, M.; Gracia, R.; Te Welscher, Y.; van Nostrum, C.F.; Falciani, C.; Pini, A.; et al. In vitro inhalation cytotoxicity testing of therapeutic nanosystems for pulmonary infection. Toxicol. In Vitro 2020, 63, 104714. [Google Scholar] [CrossRef]
- BeruBe, K.; Aufderheide, M.; Breheny, D.; Clothier, R.; Combes, R.; Duffin, R.; Forbes, B.; Gaca, M.; Gray, A.; Hall, I.; et al. In vitro models of inhalation toxicity and disease. The report of a FRAME workshop. Altern. Lab. Anim. 2009, 37, 89–141. [Google Scholar]
- Hiemstra, P.S.; Grootaers, G.; van der Does, A.M.; Krul, C.A.M.; Kooter, I.M. Human lung epithelial cell cultures for analysis of inhaled toxicants: Lessons learned and future directions. Toxicol. In Vitro 2018, 47, 137–146. [Google Scholar] [CrossRef]
- Lewinski, N.A.; Liu, N.J.; Asimakopoulou, A.; Papaioannou, E.; Konstandopoulos, A.; Riediker, M. Air-liquid interface cell exposures to nanoparticle aerosols. Methods Mol. Biol. 2017, 1570, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Polk, W.W.; Sharma, M.; Sayes, C.M.; Hotchkiss, J.A.; Clippinger, A.J. Aerosol generation and charac-terization of multi-walled carbon nanotubes exposed to cells cultured at the airliquid interface. Part. Fibre Toxicol. 2016, 13, 20. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Peters, T.M.; O’Shaughnessy, P.T.; Adamcakova-Dodd, A.; Thorne, P.S. Validation of an in vitro exposure system for toxicity assessment of air-delivered nanomaterials. Toxicol. In Vitro 2013, 27, 164–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, J.; Hedelin, A.; Malmlöf, M.; Kessler, V.; Seisenbaeva, G.; Gerde, P.; Palmberg, L. Development of combining of human bronchial mucosa models with XposeALI(R) for exposure of air pollution nanoparticles. PLoS ONE 2017, 12, e0170428. [Google Scholar] [CrossRef]
- Dvorak, A.; Tilley, A.E.; Shaykhiev, R.; Wang, R.; Crystal, R.G. Do airway epithelium air-liquid cultures represent the in vivo airway epithelium transcriptome? Am. J. Respir. Cell Mol. Biol. 2011, 44, 465–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rock, J.R.; Onaitis, M.W.; Rawlins, E.L.; Lu, Y.; Clark, C.P.; Xue, Y.; Randell, S.H.; Hogan, B.L. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 12771–12775. [Google Scholar] [CrossRef] [Green Version]
- Tadokoro, T.; Wang, Y.; Barak, L.S.; Bai, Y.; Randell, S.H.; Hogan, B.L. IL-6/STAT3 promotes regenera-tion of airway ciliated cells from basal stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E3641–E3649. [Google Scholar] [CrossRef] [Green Version]
- Polosukhin, V.V.; Cates, J.M.; Lawson, W.E.; Milstone, A.P.; Matafonov, A.G.; Massion, P.P.; Lee, J.W.; Randell, S.H.; Blackwell, T.S. Hypoxiainducible factor-1 signalling promotes goblet cell hyperplasia in airway epithelium. J. Pathol. 2011, 224, 203–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Bali, A.S.; Randell, S.H.; Hogan, B.L. GRHL2 coordinates regeneration of a polarized mucocil-iary epithelium from basal stem cells. J. Cell Biol. 2015, 211, 669–682. [Google Scholar] [CrossRef]
- Upadhyay, S.; Palmberg, L. Air-Liquid Interface: Relevant in vitro models for investigating air pollutant-induced pulmonary toxicity. Toxicol. Sci. 2018, 164, 21–30. [Google Scholar] [CrossRef] [Green Version]
- King, P.T. Inflammation in chronic obstructive pulmonary disease and its role in cardiovascular disease and lung cancer. Clin. Transl. Med. 2015, 4, 68. [Google Scholar] [CrossRef] [Green Version]
- Faber, S.C.; McCullough, S.D. Through the looking glass: In vitro models for inhalation toxicology and interindividual variability in the airway. Appl. In Vitro Toxicol. 2018, 4, 115–128. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.; Daneshian, M.; Bouwstra, J.; Caloni, F.; Constant, S.; Davies, D.E.; Dandekar, G.; Guzman, C.A.; Fabian, E.; Haltner, E.; et al. Non-animal models of epithelial barriers (skin, in-testine and lung) in research, industrial applications and regulatory toxicology. Altex 2015, 32, 327–378. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Scence 2014, 345, 1247125. [Google Scholar] [CrossRef] [PubMed]
- Fatehullah, A.; Tan, S.H.; Barker, N. Organoids as an in vitro model of human development and disease. Nat. Cell Biol. 2016, 18, 246–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohl, C.; Hofmann, H.; Moisch, M.; Papritz, M.; Hermanns, M.I.; Dei-Anang, J.; Mayer, E.; Kehe, K.; Kirkpatrick, C.J. Acute cytotoxicity and apoptotic effects after l-Pam ex-posure in different cocultures of the proximal and distal respiratory system. J. Biotechnol. 2010, 148, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Papritz, M.; Pohl, C.; Wübbeke, C.; Moisch, M.; Hofmann, H.; Hermanns, M.I.; Thiermann, H.; Kirkpatrick, C.J.; Kehe, K. Side-specific effects by cadmium exposure: Apical and basolateral treatment in a coculture model of the blood-air barrier. Toxicol. Appl. Pharmacol. 2010, 245, 361–369. [Google Scholar] [CrossRef]
- Emmler, J.; Hermanns, M.I.; Steinritz, D.; Kreppel, H.; Kirkpatrick, C.J.; Bloch, W.; Szinicz, L.; Kehe, K. Assessment of alterations in barrier functionality and induction of proinflammatory and cytotoxic effects after sulfur mustard exposure of an in vitro coculture model of the human alveolo-capillary barrier. Inhal. Toxicol. 2007, 19, 657–665. [Google Scholar] [CrossRef]
- Miller, A.J.; Hill, D.R.; Nagy, M.S.; Aoki, Y.; Dye, B.R.; Chin, A.M.; Huang, S.; Zhu, F.; White, E.S.; Lama, V.; et al. In vitro induction and in vivo engraftment of lung bud tip progenitor cells derived from human pluripotent stem cells. Stem Cell Rep. 2018, 10, 101–119. [Google Scholar] [CrossRef] [Green Version]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting organ-level lung functions on a chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H. Modeling development and disease with organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [Green Version]
- Paget, V.; Dekali, S.; Kortulewski, T.; Grall, R.; Gamez, C.; Blazy, K.; Aguerre-Chariol, O.; Chevillard, S.; Braun, A.; Rat, P.; et al. Specific uptake and genotoxicity induced by polystyrene nanobeads with distinct surface chemistry on human lung epithelial cells and macrophages. PLoS ONE 2015, 10, e0123297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalayanda, D.D.; Wang, Q.; Fulton, W.B.; Wang, T.H.; Abdullah, F. Engineering an artificial alveolar-capillary membrane: A novel continuously perfused model within microchannels. J. Pediatr. Surg. 2010, 45, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Stucki, A.O.; Stucki, J.D.; Hall, S.R.; Felder, M.; Mermoud, Y.; Schmid, R.A.; Geiser, T.; Guenat, O.T. A lung-on-a chip array with an integrated bio-inspired respiration mechanism. Lab. Chip 2015, 15, 1302–1310. [Google Scholar] [CrossRef] [Green Version]
- Huh, D.D. A human breathing lung-on-a-chip. Ann. Am. Thorac. Soc. 2015, 12 (Suppl. 1), S42–S44. [Google Scholar] [CrossRef]
- Fishler, R.; Sznitman, J. A microfluidic model of biomimetically breathing pulmonary acinar airways. J. Vis. Exp. 2016, 9, e53588. [Google Scholar] [CrossRef] [Green Version]
- Kotas, M.E.; Medzhitov, R. Homeostasis, inflammation, and disease susceptibility. Cell 2015, 160, 816–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCullough, S.D.; On, D.M.; Bowers, E.C. Using chromatin immunoprecipitation in toxicology: A Step-by-step guide to increasing efficiency, reducing variability, and expanding applications. Curr. Protoc. Toxicol. 2017, 72, 3.14.1–3.14.28. [Google Scholar] [CrossRef]
- Lenz, A.G.; Karg, E.; Brendel, E.; Hinze-Heyn, H.; Maier, K.L.; Eickelberg, O.; Stoeger, T.; Schmid, O. Inflammatory and oxidative stress responses of an alveolar epithelial cell line to airborne zinc oxide nanoparticles at the air-liquid interface: A comparison with conventional, submerged cell-culture conditions. Biomed. Res. Int. 2013, 2013, 652632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salthammer, T.; Bahadir, M. Occurrence, dynamics and reactions of organic pol-lutants in the indoor environment. Clean 2009, 37, 417–435. [Google Scholar]
- Petry, T.; Vitale, D.; Joachim, F.J.; Smith, B.; Cruse, L.; Mascarenhas, R.; Schneider, S.; Singal, M. Human health risk evaluation of selected VOC, SVOC and particulate emissions from scented candles. Regul. Toxicol. Pharmacol. 2014, 69, 55–70. [Google Scholar] [CrossRef]
- Tsoutsoulopoulos, A.; Möhle, N.; Aufderheide, M.; Schmidt, A.; Thiermann, H.; Steinritz, D. Optimization of the CULTEX(®) radial flow system for in vitro investigation of lung damaging agents. Toxicol. Lett. 2016, 244, 28–34. [Google Scholar] [CrossRef]
- Gminski, R.; Tang, T.; Mersch-Sundermann, V. Cytotoxicity and genotoxicity in human lung epithelial A549 cells caused by airborne volatile organic compounds emitted from pine wood and oriented strand boards. Toxicol. Lett. 2010, 196, 33–41. [Google Scholar] [CrossRef]
- Dwivedi, A.M.; Upadhyay, S.; Johanson, G.; Ernstgård, L.; Palmberg, L. Inflammatory effects of acrolein, crotonaldehyde and hexanal vapors on human primary bronchial epithelial cells cultured at air-liquid interface. Toxicol. In Vitro 2018, 46, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Gostner, J.M.; Zeisler, J.; Alam, M.T.; Gruber, P.; Fuchs, D.; Becker, K.; Neubert, K.; Kleinhappl, M.; Martini, S.; Überall, F. Cellular reactions to long-term volatile organic compound (VOC) exposures. Sci. Rep. 2016, 6, 37842. [Google Scholar] [CrossRef] [PubMed]
- Siroux, V.; Agier, L.; Slama, R. The exposome concept: A challenge and a potential driver for environmental health research. Eur. Respir. Rev. 2016, 25, 124–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braakhuis, H.M.; Kloet, S.K.; Kezic, S.; Kuper, F.; Park, M.V.; Bellmann, S.; van der Zande, M.; Le Gac, S.; Krystek, P.; Peters, R.J.; et al. Progress and future of in vitro models to study translo-cation of nanoparticles. Arch. Toxicol. 2015, 89, 1469–1495. [Google Scholar] [CrossRef] [Green Version]
- Miller, G.W.; Jones, D.P. The nature of nurture: Refining the definition of the exposome. Toxicol. Sci. 2014, 137, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Maitre, L.; De Bont, J.; Casas, M.; Robinson, O.; Aasvang, G.M.; Agier, L.; Andrušaitytė, S.; Ballester, F.; Basagaña, X.; Borràs, E.; et al. Human Early Life Exposome (HELIX) study: A Eu-ropean population-based exposome cohort. BMJ Open 2018, 8, e021311. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Lv, S.; Liu, Y.; Li, Y. Biomarkers for the adverse effects on respiratory system health associated with atmospheric particulate matter exposure. J. Hazard. Mater. 2022, 421, 126760. [Google Scholar] [CrossRef]
- Vrijens, K.; Bollati, V.; Nawrot, T.S. MicroRNAs as potential sig-natures of environmental exposure or effect: A systematic review. Environ. Health Perspect. 2015, 123, 399–411. [Google Scholar] [CrossRef] [Green Version]
- Tseng, C.H. A review on environmental factors regulating arsenic methylation in hu-mans. Toxicol. Appl. Pharmacol. 2009, 235, 338–350. [Google Scholar] [CrossRef]
- Poston, R.G.; Saha, R.N. epigenetic effects of polybrominated diphenyl ethers on human health. Int. J. Environ. Res. Public Health 2019, 16, 2703. [Google Scholar] [CrossRef] [Green Version]
- Tutar, Y. miRNA and cancer; computational and experimental approaches. Curr. Pharm. Biotechnol. 2014, 15, 429. [Google Scholar] [CrossRef]
- Liang, C.; Yu, S.; Luo, J. Adaptive multi-view multi-label learning for identifying dis-ease-associated candidate miRNAs. PLoS Comput. Biol. 2019, 15, e1006931. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Front. Overview of microRNA biogenesis, me-chanisms of actions, and circulation. Endocrinol (Lausanne) 2018, 9, 402. [Google Scholar] [CrossRef] [Green Version]
- Condrat, C.E.; Thompson, D.C.; Barbu, M.G.; Bugnar, O.L.; Boboc, A.; Cretoiu, D.; Suciu, N.; Cretoiu, S.M.; Voinea, S.C. miRNAs as biomarkers in disease: Latest findings regarding their role in diagnosis and prognosis. Cells 2020, 9, E276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izzotti, A.; Calin, G.A.; Arrigo, P.; Steele, V.E.; Croce, C.M.; De Flora, S. Downregulation of microRNA expression in the lungs of rats exposed to cigarette smoke. FASEB J. 2009, 23, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.; Momtaz, S.; Abdollahi, M. The relationship between mercury exposure and epi-genetic alterations regarding human health, risk assessment and diagnostic strategies. J. Trace Elem. Med. Biol. 2019, 52, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Ding, E.; Guo, J.; Bai, Y.; Zhang, H.; Liu, X.; Cai, W.; Zhong, L.; Zhu, B. MiR-92a and miR-486 are potential diagnostic biomarkers for mercury poisoning and jointly sustain NF-κB activity in mercury toxicity. Sci. Rep. 2017, 7, 15980. [Google Scholar] [CrossRef] [Green Version]
- Iliopoulos, D.; Jaeger, S.A.; Hirsch, H.A.; Bulyk, M.L.; Struhl, K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol. Cell 2010, 39, 493–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Kappil, M.A.; Li, A.; Dassanayake, P.S.; Darrah, T.H.; Friedman, A.E.; Friedman, M.; Lambertini, L.; Landrigan, P.; Stodgell, C.J.; et al. Exploring the associations between microRNA expression profiles and environmental pollutants in human placenta from the National Children’s Study (NCS). Epigenetics 2015, 10, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, N.; Xu, Y.; Hua, L.; Zhou, D.; Zheng, M.; Deng, X. Effects of chronic PM2.5 exposure on pulmonary epithelia: Transcriptome analysis of mRNA-exosomal miRNA interactions. Toxicol. Lett. 2019, 316, 49–59. [Google Scholar] [CrossRef]
- Anzalone, G.; Moscato, M.; Montalbano, A.M.; Albano, G.D.; Gagliardo, R.; Marchese, R.; Fucarino, A.; Nigro, C.L.; Drago, G.; Profita, M. PBDEs affect inflammatory and oncosuppressive mechanisms via the EZH2 methyltransferase in airway epithelial cells. Life Sci. 2021, 282, 119827. [Google Scholar] [CrossRef] [PubMed]
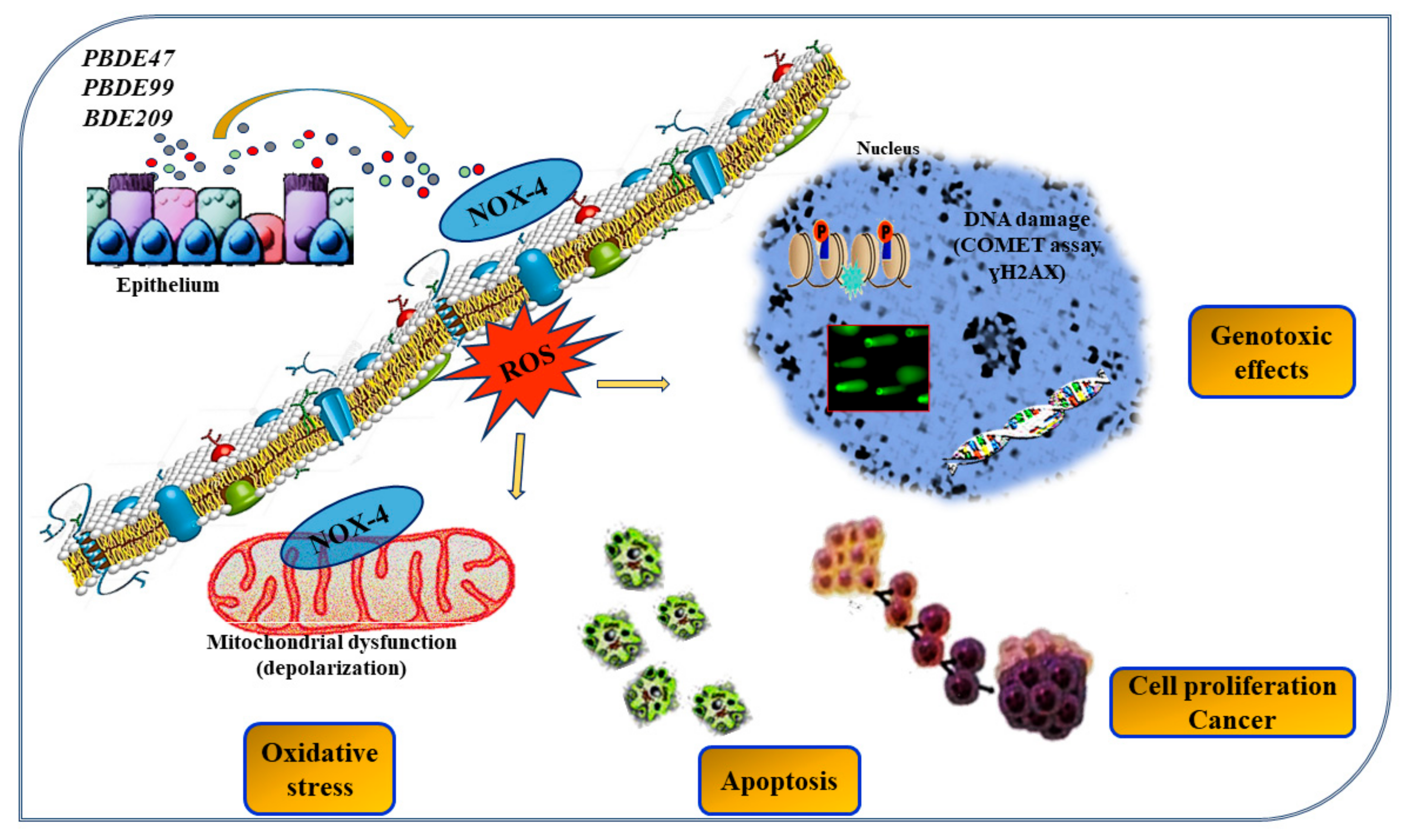
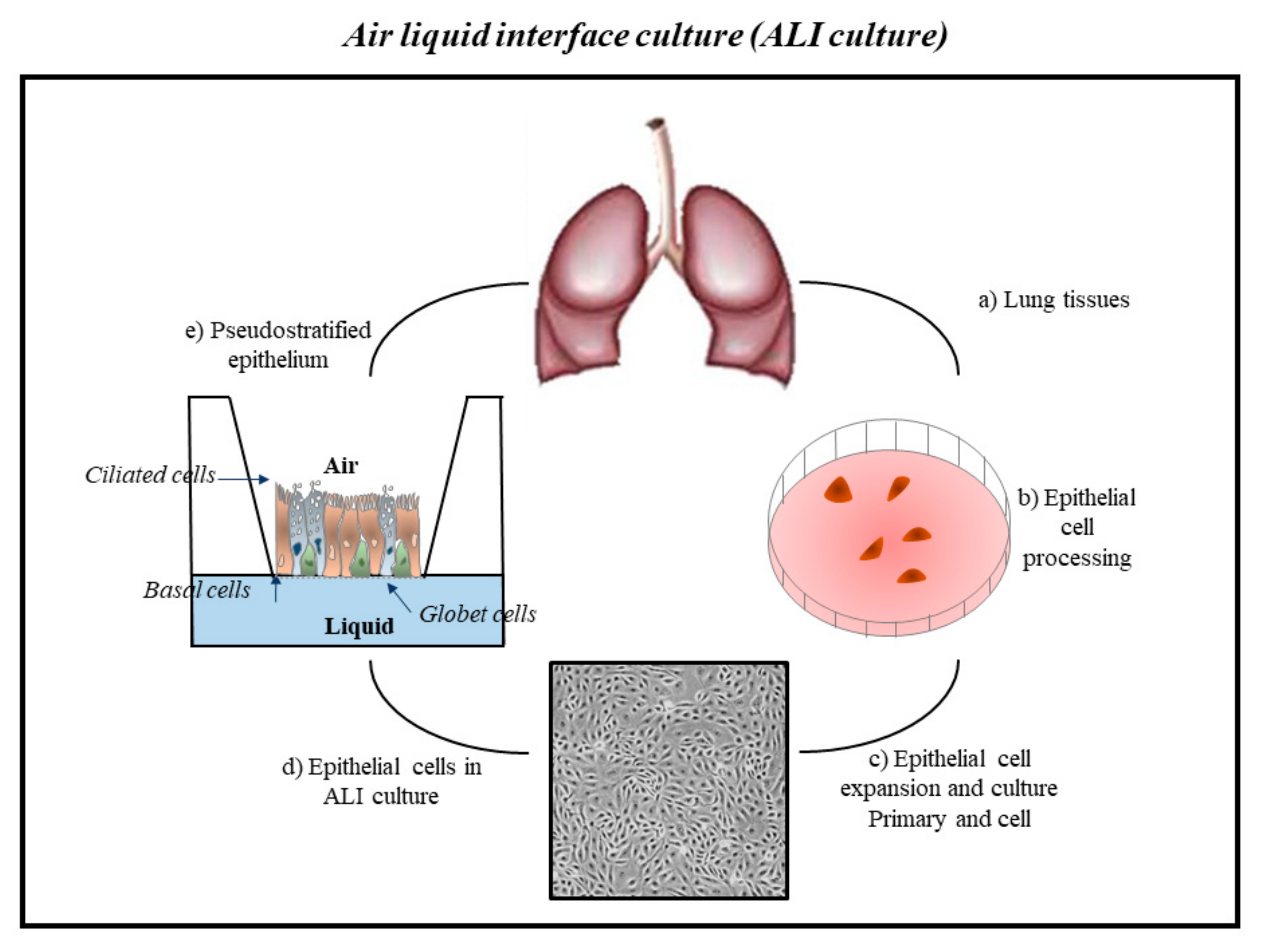




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albano, G.D.; Montalbano, A.M.; Gagliardo, R.; Anzalone, G.; Profita, M. Impact of Air Pollution in Airway Diseases: Role of the Epithelial Cells (Cell Models and Biomarkers). Int. J. Mol. Sci. 2022, 23, 2799. https://doi.org/10.3390/ijms23052799
Albano GD, Montalbano AM, Gagliardo R, Anzalone G, Profita M. Impact of Air Pollution in Airway Diseases: Role of the Epithelial Cells (Cell Models and Biomarkers). International Journal of Molecular Sciences. 2022; 23(5):2799. https://doi.org/10.3390/ijms23052799
Chicago/Turabian StyleAlbano, Giusy Daniela, Angela Marina Montalbano, Rosalia Gagliardo, Giulia Anzalone, and Mirella Profita. 2022. "Impact of Air Pollution in Airway Diseases: Role of the Epithelial Cells (Cell Models and Biomarkers)" International Journal of Molecular Sciences 23, no. 5: 2799. https://doi.org/10.3390/ijms23052799